
Why Are Some People with Lower Urinary Tract Symptoms (LUTS) Depressed? New Evidence That Peripheral Inflammation in the Bladder Causes Central Inflammation and Mood Disorders

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. There Is a Strong Clinical Association between LUTS and Depression/Anxiety
3. The Correlation between LUTS and Depression/Anxiety Is Bidirectional
4. Interventional Studies Support This Correlation
5. Interstitial Cystitis/Bladder Pain Syndrome (IC/BPS)
6. Inflammation as a Possible Common Mechanism between LUTS and Depression
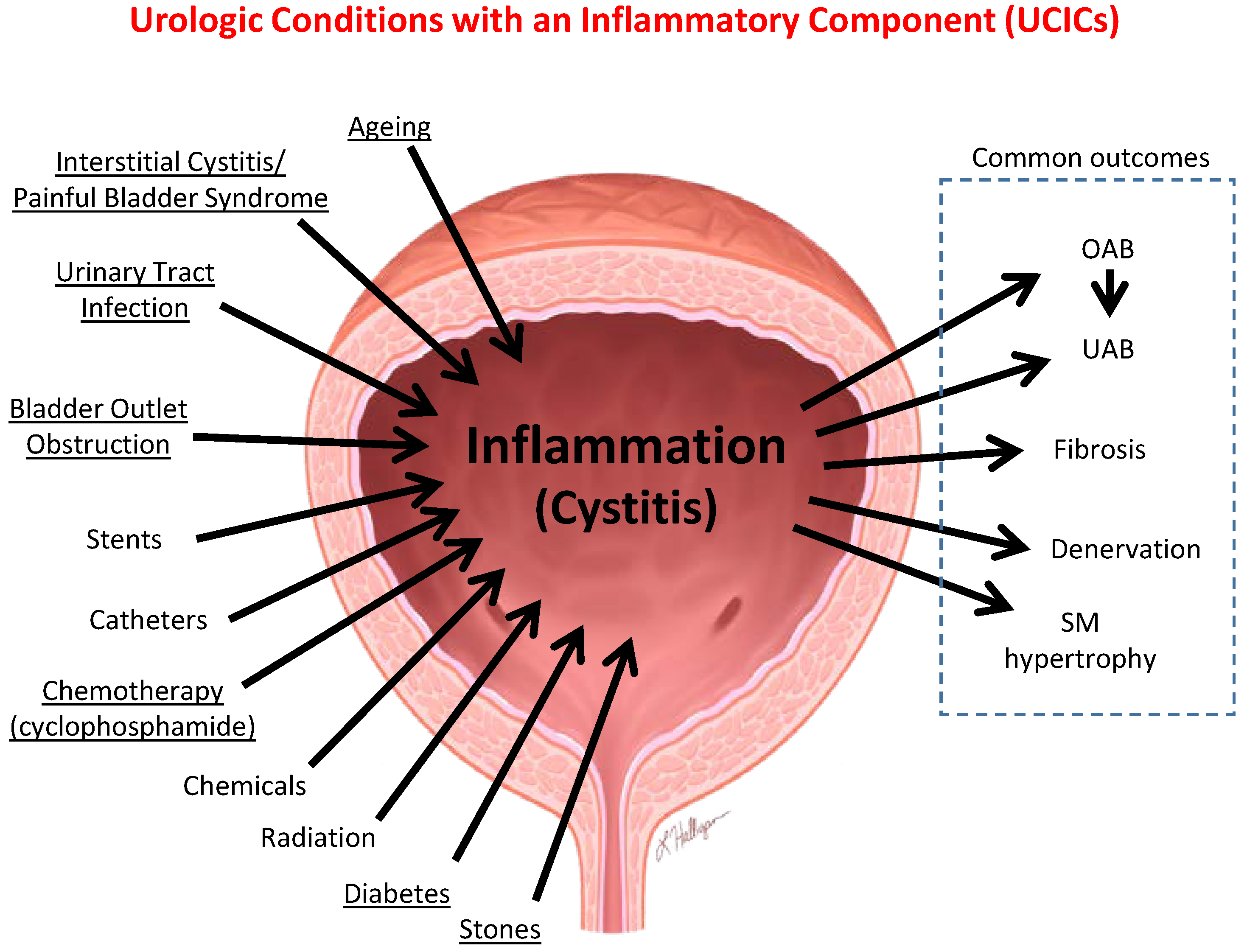
6.1. Inflammation Is a Well-Established Component of Many Benign Urological Disorders That Cause LUTS
6.2. Neuroinflammation Is Involved in the Etiology of Depression
7. Modern Theories of Depression and the Role of Neuroinflammation
7.1. Serotonin
7.2. The HPA Axis
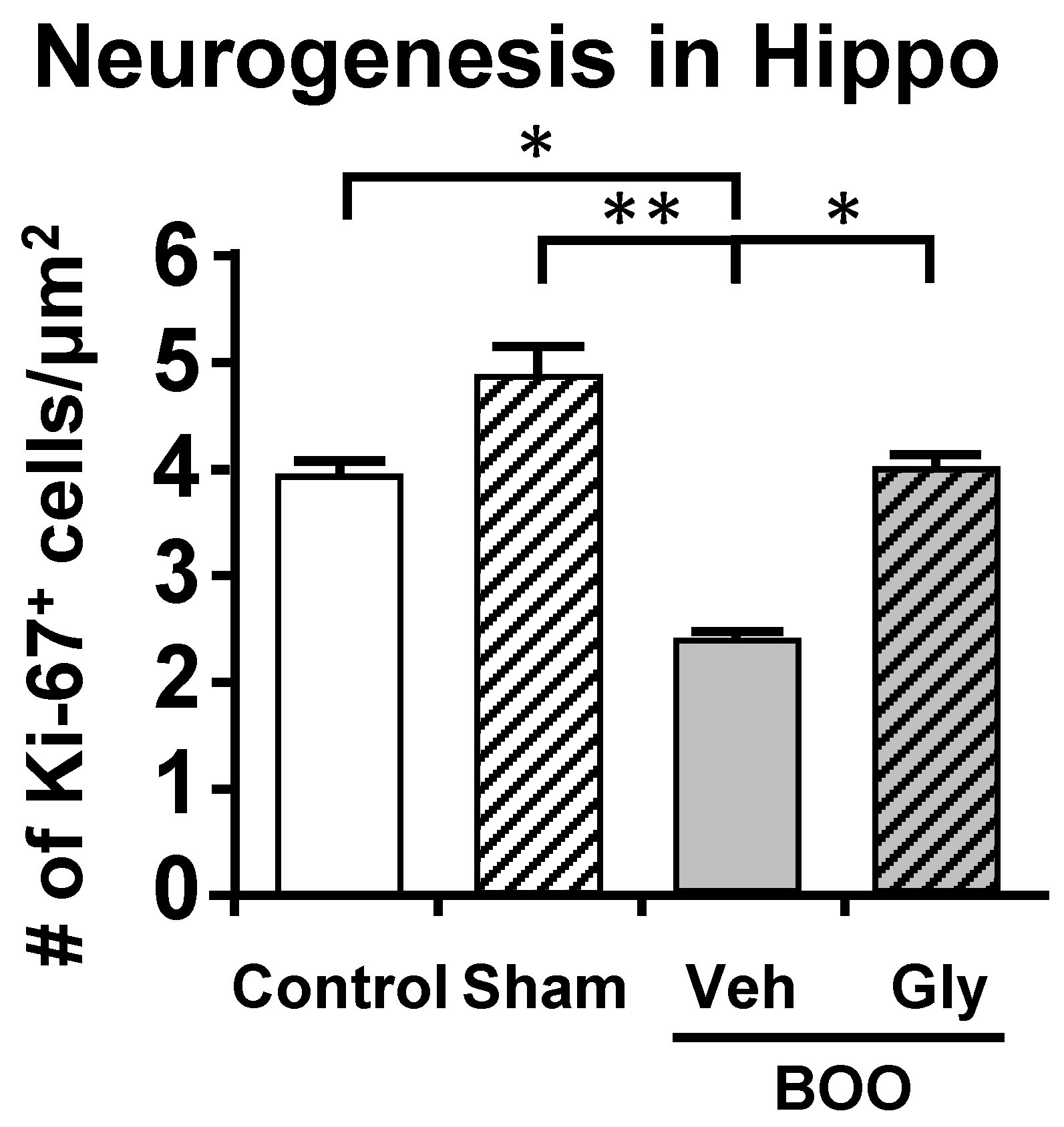
7.3. Neurogenesis
7.4. Towards a Unified Model of Depression
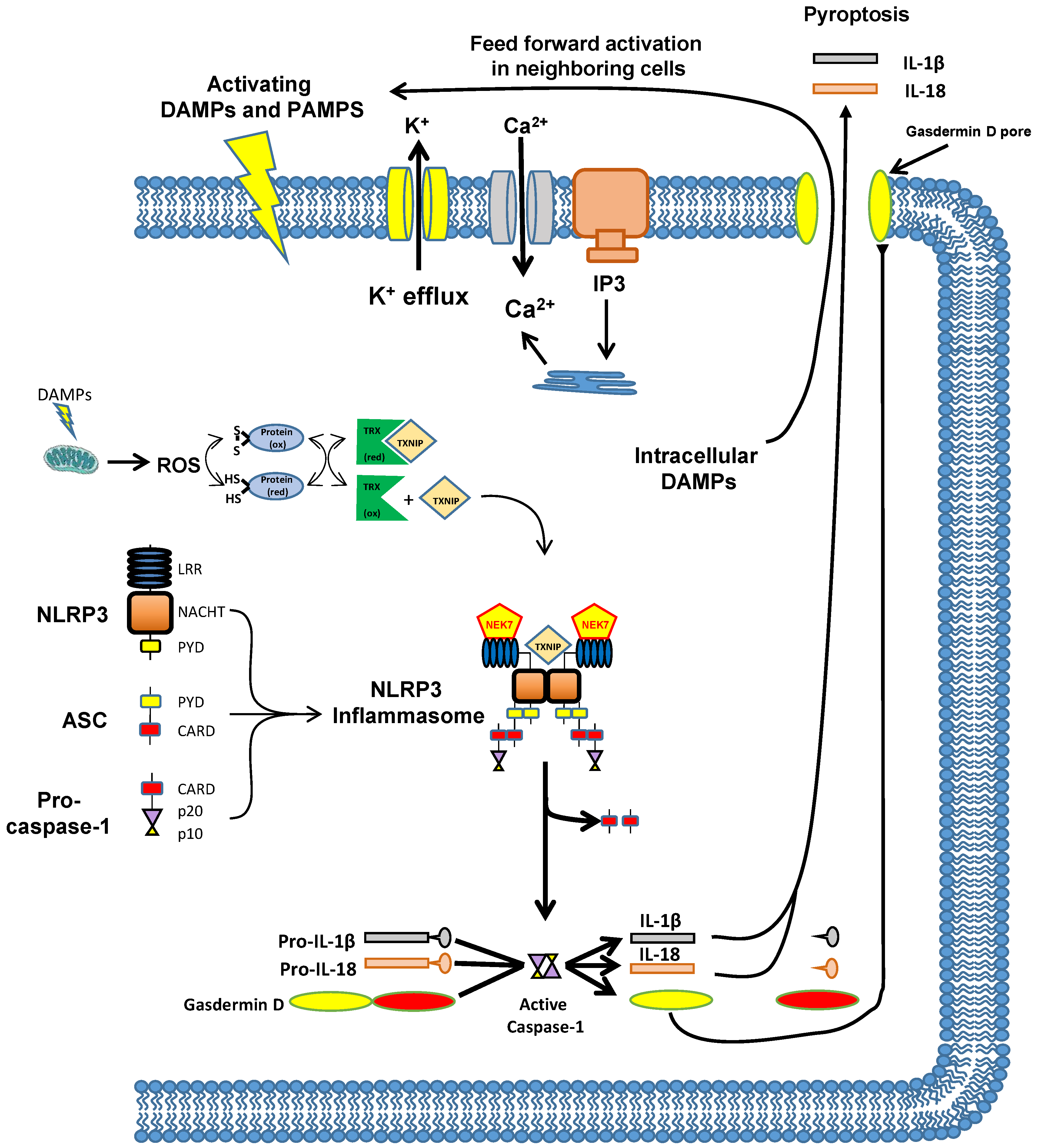
8. The Mechanism by Which Inflammation Is Triggered—The NLRP3 Inflammasome
9. Pro-Inflammatory Events in Peripheral Organs or Tissues Result in Concurrent Inflammatory Changes in the CNS
9.1. Inflammatory Bowel Disease (IBD)
9.2. Rheumatoid Arthritis
9.3. Sepsis
10. Animal Models Confirm the Connection between Bladder Inflammation, Neuroinflammation and Mood Disorders
10.1. The Cyclophosphamide-Induced Hemorrhagic Cystitis Model
10.2. The Bladder Outlet Obstruction (BOO) Model
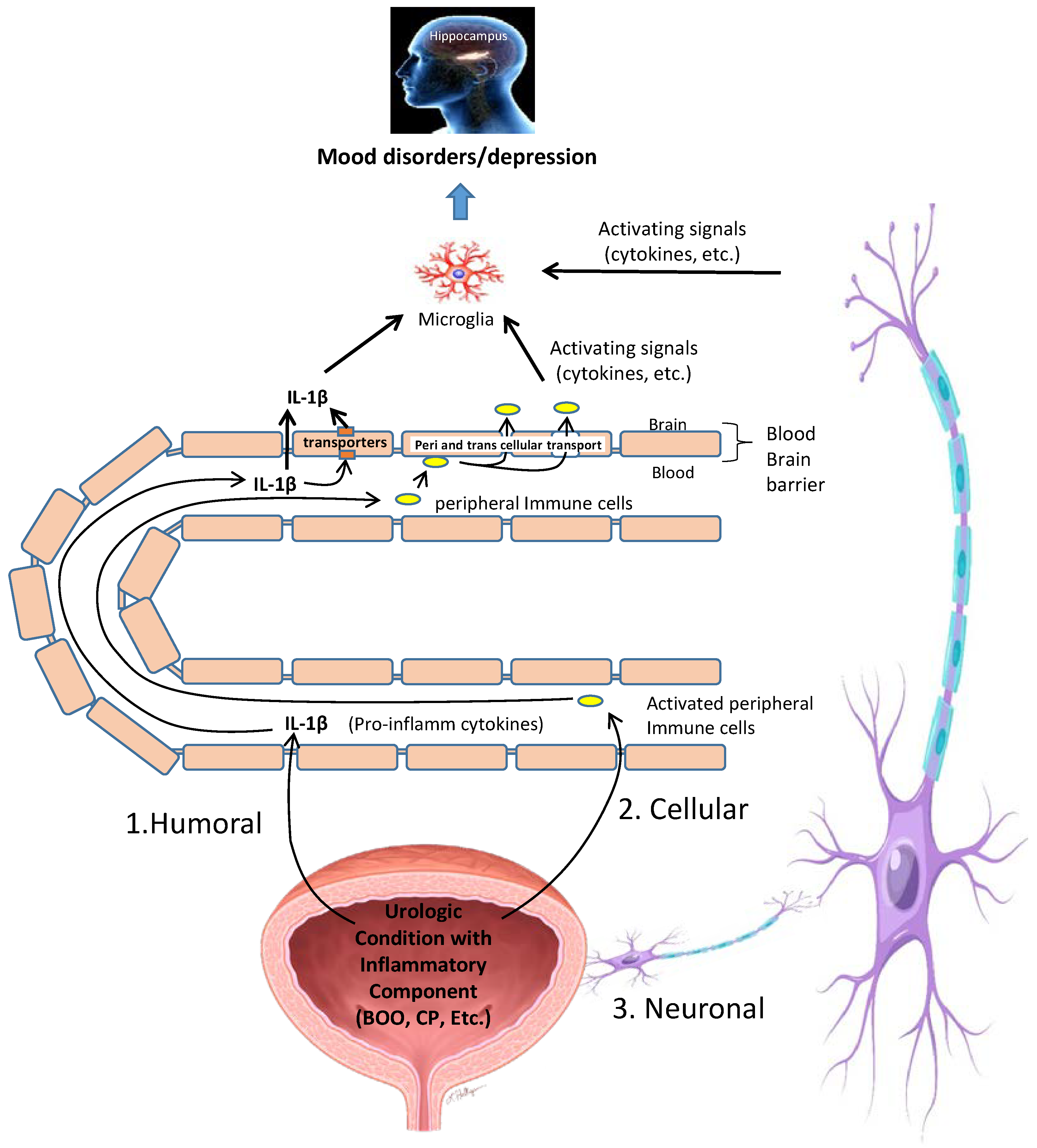
11. Possible Mechanisms by Which Inflammation in the Bladder Is Converted into Neuroinflammation in the CNS
12. Translational Potential
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hollander, A.; Gordon, J.R.; Sloan, B.S. Infravesical Obstruction and Its Relationship to Edema and Psychotic Behavior. South. Med. J. 1964, 57, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Coyne, K.S.; Wein, A.J.; Tubaro, A.; Sexton, C.C.; Thompson, C.L.; Kopp, Z.S.; Aiyer, L.P. The burden of lower urinary tract symptoms: Evaluating the effect of LUTS on health-related quality of life, anxiety and depression: EpiLUTS. BJU Int. 2009, 103, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.-M.; Lee, S.R.; Choi, E.J.; Jeong, K.; Chung, H.W. Urinary incontinence is strongly associated with depression in middle-aged and older Korean women: Data from the Korean longitudinal study of ageing. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 220, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, C.; Laor, L.; Te, A.; Kaplan, S.; Chughtai, B. Relationship Between Depression and Lower Urinary Tract Symptoms Secondary to Benign Prostatic Hyperplasia. Rev. Urol. 2015, 17, 51–57. [Google Scholar] [CrossRef]
- Huang, C.L.-C.; Wu, M.-P.; Ho, C.-H.; Wang, J.-J. The bidirectional relationship between anxiety, depression, and lower urinary track symptoms: A nationwide population-based cohort study. J. Psychosom. Res. 2017, 100, 77–82. [Google Scholar] [CrossRef]
- Drake, M. Nocturia and depressive symptoms in older men. BJU Int. 2017, 120, 159. [Google Scholar] [CrossRef]
- Golabek, T.; Skalski, M.; Przydacz, M.; Świerkosz, A.; Siwek, M.; Golabek, K.; Stangel-Wójcikiewicz, K.; Dudek, D.; Chlosta, P. Lower urinary tract symptoms, nocturia and overactive bladder in patients with depression and anxiety. Psychiatr. Pol. 2016, 50, 417–430. [Google Scholar] [CrossRef]
- Vrijens, D.; Drossaerts, J.; van Koeveringe, G.; Van Kerrebroeck, P.; van Os, J.; Leue, C. Affective symptoms and the over-active bladder—A systematic review. J. Psychosom. Res. 2015, 78, 95–108. [Google Scholar] [CrossRef]
- Hughes, F.M., Jr.; Hirshman, N.; Malick, H.; White, S.; Jin, H.; Harper, S.; Purves, J. A possible mechanism un-derlying mood disorders associated with LUTS: Chronic bladder outlet obstruction causes NLRP3-dependent inflammation in the hippocampus and depressive behavior in rats. Neurourol. Urodyn. 2020, 39, 1700–1707. [Google Scholar] [CrossRef]
- Hirshman, N.A.; Hughes, F., Jr.; Jin, H.; Harrison, W.; White, S.; Doan, I.; Harper, S.; Leidig, P.; Purves, J. Cyclo-phosphamide-induced cystitis results in NLRP3-mediated inflammation in the hippocampus and depression in rats. Am. J. Physiol. Ren. Physiol. 2020, 318, F354–F362. [Google Scholar] [CrossRef]
- Milsom, I.; Kaplan, S.A.; Coyne, K.S.; Sexton, C.C.; Kopp, Z.S. Effect of bothersome overactive bladder symptoms on health-related quality of life, anxiety, depression, and treatment seeking in the united states: Results from EpiLUTS. Urology 2012, 80, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, N.-S.; Chang, H.-A.; Chung, C.-H.; Kao, Y.-C.; Yeh, H.-W.; Yeh, C.-B.; Chiang, W.-S.; Huang, S.-Y.; Lu, R.-B.; Chien, W.-C. Risk of Psychiatric Disorders in Overactive Bladder Syndrome: A Nationwide Cohort Study in Taiwan. J. Investig. Med. 2019, 67, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Tully, P.J.; Kahokehr, A.A.; Jay, A.; Wittert, G.A. The bidirectional association between depression and lower urinary tract symptoms (LUTS) in men: A systematic review and meta-analysis of observational studies. Neurourol. Urodyn. 2022, 41, 552–561. [Google Scholar] [CrossRef]
- Wong, S.Y.; Hong, A.; Leung, J.; Kwok, T.; Leung, P.C.; Woo, J. Lower urinary tract symptoms and depressive symptoms in elderly men. J. Affect. Disord. 2006, 96, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Litman, H.J.; Steers, W.D.; Wei, J.T.; Kupelian, V.; Link, C.L.; McKinlay, J.B. Relationship of lifestyle and clinical factors to lower urinary tract symptoms: Results from Boston area community health survey. Urology 2007, 70, 916–921. [Google Scholar] [CrossRef]
- Mahjani, B.; Koskela, L.R.; Batuure, A.; Mahjani, C.G.; Janecka, M.; Hultman, C.M.; Reichenberg, A.; Buxbaum, J.D.; Akre, O.; Grice, D.E. Systematic review and meta-analysis identify significant relationships between clinical anxiety and lower urinary tract symptoms. Brain Behav. 2021, 11, e2268. [Google Scholar] [CrossRef]
- Johnson, T.V.; Abbasi, A.; Ehrlich, S.S.; Kleris, R.S.; Owen-Smith, A.; Raison, C.L.; Master, V.A. IPSS quality of life question: A possible indicator of depression among patients with lower urinary tract symptoms. Can. J. Urol. 2012, 19, 6100. [Google Scholar]
- Staskin, D.R.; Rosenberg, M.T.; Dahl, N.V.; Polishuk, P.V.; Zinner, N.R. Effects of oxybutynin transdermal system on health-related quality of life and safety in men with overactive bladder and prostate conditions. Int. J. Clin. Pract. 2008, 62, 27–38. [Google Scholar] [CrossRef]
- Renard, J.; Ballarini, S.; Mascarenhas, T.; Zahran, M.; Quimper, E.; Choucair, J.; Iselin, C.E. Recurrent Lower Urinary Tract Infections Have a Detrimental Effect on Patient Quality of Life: A Prospective, Observational Study. Infect. Dis. Ther. 2014, 4, 125–135. [Google Scholar] [CrossRef]
- Quek, K.; Low, W.; Razack, A.; Loh, C. The psychological effects of treatments for lower urinary tract symptoms. BJU Int. 2000, 86, 630–633. [Google Scholar] [CrossRef]
- Clemens, J.Q.; Brown, S.O.; Calhoun, E.A. Mental health diagnoses in patients with interstitial cystitis/painful bladder syndrome and chronic prostatitis/chronic pelvic pain syndrome: A case/control study. J. Urol. 2008, 180, 1378–1382. [Google Scholar] [CrossRef]
- Goldstein, H.B.; Safaeian, P.; Garrod, K.; Finamore, P.; Kellogg-Spadt, S.; Whitmore, K. Depression, abuse and its rela-tionship to interstitial cystitis. Int. Urogynecol. J. 2008, 19, 1683–1686. [Google Scholar] [CrossRef] [PubMed]
- Koziol, J.A.; Clark, D.C.; Gittes, R.F.; Tan, E.M. The natural history of interstitial cystitis: A survey of 374 patients. J. Urol. 1993, 149, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Rothrock, N.E.; Lutgendorf, S.; Hoffman, A.; Kreder, K. Depressive symptoms and quality of life in patients with in-terstitial cystitis. J. Urol. 2002, 167, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Hepner, K.A.; Watkins, K.E.; Elliott, M.N.; Clemens, J.Q.; Hilton, L.G.; Berry, S.H. Suicidal ideation among patients with bladder pain syndrome/interstitial cystitis. Urology 2012, 80, 280–285. [Google Scholar] [CrossRef]
- Michaelides, A.; Zis, P. Depression, anxiety and acute pain: Links and management challenges. Postgrad. Med. 2019, 131, 438–444. [Google Scholar] [CrossRef]
- Gacci, M.; Sebastianelli, A.; Salvi, M.; De Nunzio, C.; Tubaro, A.; Gravas, S.; Moncada, I.; Serni, S.; Maggi, M.; Vignozzi, L. The Impact of Central Obesity on Storage Luts and Urinary Incontinence After Prostatic Surgery. Curr. Urol. Rep. 2016, 17, 61. [Google Scholar] [CrossRef]
- Daneshgari, F.; Liu, G.; Hanna-Mitchell, A.T. Path of translational discovery of urological complications of obesity and diabetes. Am. J. Physiol. Physiol. 2017, 312, F887–F896. [Google Scholar] [CrossRef]
- Sarma, A.V.; Townsend, M.; Grodstein, F.; Breyer, B.; Brown, J. Urologic Diseases and Sexual Dysfunction in Diabetes. In Diabetes in America; Cowie, C.C., Ed.; National Library of Medicine: Bethesda, MD, USA, 2018. [Google Scholar]
- Xu, Z.; Elrashidy, R.A.; Li, B.; Liu, G. Oxidative Stress: A Putative Link Between Lower Urinary Tract Symptoms and Aging and Major Chronic Diseases. Front. Med. 2022, 9, 568. [Google Scholar] [CrossRef]
- Kupelian, V.; McVary, K.T.; Barry, M.J.; Link, C.L.; Rosen, R.C.; Aiyer, L.P.; Mollon, P.; McKinlay, J.B. Association of C-reactive protein and lower urinary tract symptoms in men and women: Results from Boston area community health survey. Urology 2009, 73, 950–957. [Google Scholar] [CrossRef]
- Martin, S.; Vincent, A.; Taylor, A.W.; Atlantis, E.; Jenkins, A.; Januszewski, A.; O’Loughlin, P.; Wittert, G. Lower Urinary Tract Symptoms, Depression, Anxiety and Systemic Inflammatory Factors in Men: A Population-Based Cohort Study. PLoS ONE 2015, 10, e0137903. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, R.; Hughes, F., Jr.; Inouye, B.; Jin, H.; McMains, J.; Pak, E.; Hannan, J.; Purves, J. NLRP3/IL-1β mediates denervation during bladder outlet obstruction in rats. Neurourol. Urodyn. 2018, 37, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Hughes, F.M., Jr.; Sexton, S.; Jin, H.; Govada, V.; Purves, J. Bladder fibrosis during outlet obstruction is triggered through the NLRP3 inflammasome and the production of IL-1beta. Am. J. Physiol. Ren. Physiol. 2017, 313, F603–F610. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Dru, C.; Choudhury, D.; Mishra, R.; Fernandez, A.; Biondi, S.; Liu, Z.; Shimada, K.; Arditi, M.; Bhowmick, N. Inflammation and pyroptosis mediate muscle expansion in an interleukin-1beta (IL-1beta)-dependent manner. J. Biol. Chem. 2015, 290, 6574–6583. [Google Scholar] [CrossRef]
- Fusco, F.; Creta, M.; De Nunzio, C.; Iacovelli, V.; Mangiapia, F.; Marzi, V.L.; Agrò, E.F. Progressive bladder remodeling due to bladder outlet obstruction: A systematic review of morphological and molecular evidences in humans. BMC Urol. 2018, 18, 15. [Google Scholar] [CrossRef]
- Troubat, R.; Barone, P.; Leman, S.; DeSmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef]
- Felger, J.C.; Lotrich, F.E. Inflammatory cytokines in depression: Neurobiological mechanisms and therapeutic implications. Neuroscience 2013, 246, 199–229. [Google Scholar] [CrossRef]
- Richards, E.M.; Zanotti-Fregonara, P.; Fujita, M.; Newman, L.; Farmer, C.; Ballard, E.D.; Machado-Vieira, R.; Yuan, P.; Niciu, M.J.; Lyoo, C.H.; et al. PET radioligand binding to translocator protein (TSPO) is increased in unmedicated depressed subjects. EJNMMI Res. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Coyne, K.S.; Kaplan, S.A.; Chapple, C.; Sexton, C.C.; Kopp, Z.; Bush, E.N.; Aiyer, L.P. EpiLUTS The EpiLUTS Team Risk factors and comorbid conditions associated with lower urinary tract symptoms: EpiLUTS. BJU Int. 2009, 103, 24–32. [Google Scholar] [CrossRef]
- Cabrera-Pastor, A.; Llansola, M.; Montoliu, C.; Malaguarnera, M.; Balzano, T.; Taoro-Gonzalez, L.; Garcia-Garcia, R.; Man-gas-Losada, A.; Izquierdo-Altarejos, P.; Arenas, Y.; et al. Peripheral inflammation induces neuroinflammation that alters neu-rotransmission and cognitive and motor function in hepatic encephalopathy: Underlying mechanisms and therapeutic im-plications. Acta Physiol. 2019, 226, e13270. [Google Scholar] [CrossRef]
- Maes, M.; Berk, M.; Goehler, L.; Song, C.; Anderson, G.; Galecki, P.; Leonard, B. Depression and sickness behavior are Ja-nus-faced responses to shared inflammatory pathways. BMC Med. 2012, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Maes, M. Evidence for an immune response in major depression: A review and hypothesis. Prog. Neuro Psychopharmacol. Biol. Psychiatry 1995, 19, 11–38. [Google Scholar] [CrossRef]
- Dantzer, R. Cytokine-Induced Sickness Behavior: Mechanisms and Implications. Ann. N. Y. Acad. Sci. 2001, 933, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Girardet, C.; Bonnet, M.S.; Jdir, R.; Sadoud, M.; Thirion, S.; Tardivel, C.; Roux, J.; Lebrun, B.; Mounien, L.; Trouslard, J.; et al. Central Inflammation and Sickness-Like Behavior Induced by the Food Contaminant Deoxynivalenol: A PGE2-Independent Mechanism. Toxicol. Sci. 2011, 124, 179–191. [Google Scholar] [CrossRef]
- Konsman, J.P.; Parnet, P.; Dantzer, R. Cytokine-induced sickness behaviour: Mechanisms and implications. Trends Neurosci. 2002, 25, 154–159. [Google Scholar] [CrossRef]
- Kappelmann, N.; Lewis, G.; Dantzer, R.; Jones, P.; Khandaker, G. Antidepressant activity of anti-cytokine treatment: A systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol. Psychiatry 2018, 23, 335–343. [Google Scholar] [CrossRef]
- Köhler-Forsberg, O.; Lydholm, C.N.; Hjorthøj, C.; Nordentoft, M.; Mors, O.; Benros, M. Efficacy of anti-inflammatory treatment on major depressive disorder or depressive symptoms: Meta-analysis of clinical trials. Acta Psychiatr. Scand. 2019, 139, 404–419. [Google Scholar] [CrossRef]
- Bai, S.; Guo, W.; Feng, Y.; Deng, H.; Li, G.; Nie, H.; Guo, G.; Yu, H.; Ma, Y.; Wang, J.; et al. Efficacy and safety of anti-inflammatory agents for the treatment of major depressive disorder: A systematic review and meta-analysis of randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2019, 91, 21–32. [Google Scholar] [CrossRef]
- Caiaffo, V.; Oliveira, B.D.R.; Sá, F.B.; Neto, J.E. Anti-inflammatory, antiapoptotic, and antioxidant activity of fluoxetine. Pharmacol. Res. Perspect. 2016, 4, e00231. [Google Scholar] [CrossRef]
- Pereira, V.S.; Hiroaki-Sato, V.A. A brief history of antidepressant drug development: From tricyclics to beyond ketamine. Acta Neuropsychiatr. 2018, 30, 307–322. [Google Scholar] [CrossRef]
- Dean, J.; Keshavan, M. The neurobiology of depression: An integrated view. Asian J. Psychiatry 2017, 27, 101–111. [Google Scholar] [CrossRef]
- Pfefferkorn, E.R. Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc. Natl. Acad. Sci. USA 1984, 81, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, E.R.; Eckel, M.; Rebhun, S. Interferon-gamma suppresses the growth of Toxoplasma gondii in human fibroblasts through starvation for tryptophan. Mol. Biochem. Parasitol. 1986, 20, 215–224. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.C.; André, C.; Wang, Y.; Lawson, M.A.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Interferon- and tumor necrosis factor- mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J. Neurosci. 2009, 29, 4200–4209. [Google Scholar] [CrossRef]
- Schwarcz, R.; Stone, T.W. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology 2017, 112, 237–247. [Google Scholar] [CrossRef]
- Meier, T.B.; Drevets, W.C.; Wurfel, B.E.; Ford, B.N.; Morris, H.M.; Victor, T.A.; Bodurka, J.; Teague, T.K.; Dantzer, R.; Savitz, J. Relationship between neurotoxic kynurenine metabolites and reductions in right medial prefrontal cortical thickness in major depressive disorder. Brain Behav. Immun. 2016, 53, 39–48. [Google Scholar] [CrossRef]
- Savitz, J. Role of Kynurenine Metabolism Pathway Activation in Major Depressive Disorders. Inflamm. Assoc. Depress. Evid. Mech. Implic. 2017, 31, 249–267. [Google Scholar] [CrossRef]
- Pawlowski, T.; Pawlak, D.; Inglot, M.; Zalewska, M.; Marciniak, D.; Bugajska, J.; Janocha-Litwin, J.; Malyszczak, K. The role of anthranilic acid in the increase of depressive symptoms and major depressive disorder during treatment for hepatitis C with pegylated interferon-α2a and oral ribavirin. J. Psychiatry Neurosci. 2021, 46, E166–E175. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, C.B.; Widerlöv, E.; Bissette, G.; Walléus, H.; Karlsson, I.; Eklund, K.; Kilts, C.D.; Loosen, P.T.; Vale, W. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science 1984, 226, 1342–1344. [Google Scholar] [CrossRef]
- Muehlenbein, M.P.; Hirschtick, J.L.; Bonner, J.Z.; Swartz, A.M. Toward quantifying the usage costs of human immunity: Altered metabolic rates and hormone levels during acute immune activation in men. Am. J. Hum. Biol. 2010, 22, 546–556. [Google Scholar] [CrossRef]
- Straub, R.H.; Cutolo, M.; Buttgereit, F.; Pongratz, G. Review: Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J. Intern. Med. 2010, 267, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Swaab, D.F.; Bao, A.-M.; Lucassen, P.J. The stress system in the human brain in depression and neurodegeneration. Ageing Res. Rev. 2005, 4, 141–194. [Google Scholar] [CrossRef] [PubMed]
- Pariante, C.M. Why are depressed patients inflamed? A reflection on 20 years of research on depression, glucocorticoid resistance and inflammation. Eur. Neuropsychopharmacol. 2017, 27, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Topete, D.; Cidlowski, J.A. One hormone, two actions: Anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Chinenov, Y.; Rogatsky, I. Glucocorticoids and the innate immune system: Crosstalk with the Toll-like receptor signaling network. Mol. Cell. Endocrinol. 2007, 275, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Gao, Z.-G.; Jacobson, K.A.; Suffredini, A.F. Dexamethasone enhances ATP-induced inflammatory responses in endothelial cells. Experiment 2010, 335, 693–702. [Google Scholar] [CrossRef]
- Di Virgilio, F. The Therapeutic Potential of Modifying Inflammasomes and NOD-Like Receptors. Pharmacol. Rev. 2013, 65, 872–905. [Google Scholar] [CrossRef]
- Busillo, J.M.; Azzam, K.M.; Cidlowski, J.A. Glucocorticoids Sensitize the Innate Immune System through Regulation of the NLRP3 Inflammasome. J. Biol. Chem. 2011, 286, 38703–38713. [Google Scholar] [CrossRef]
- Cattaneo, A.; Gennarelli, M.; Uher, R.; Breen, G.; Farmer, A.; Aitchison, K.; Craig, I.; Anacker, C.; Zunsztain, P.; McGuffin, P.; et al. Candidate genes expression profile associated with antidepressants response in the GENDEP study: Differentiating be-tween baseline ‘predictors’ and longitudinal ‘targets’. Neuropsychopharmacology 2013, 38, 377–385. [Google Scholar] [CrossRef]
- Peterson, J.C. The Adaptive Neuroplasticity Hypothesis of Behavioral Maintenance. Neural Plast. 2012, 2012, 1–12. [Google Scholar] [CrossRef]
- Abdissa, D.; Hamba, N.; Gerbi, A. Review Article on adult neurogenesis in humans. Transl. Res. Anat. 2020, 20, 100074. [Google Scholar] [CrossRef]
- Eliwa, H.; Belzung, C.; Surget, A. Adult hippocampal neurogenesis: Is it the alpha and omega of antidepressant action? Biochem. Pharmacol. 2017, 141, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Planchez, B.; Lagunas, N.; Le Guisquet, A.-M.; Legrand, M.; Surget, A.; Hen, R.; Belzung, C. Increasing Adult Hippocampal Neurogenesis Promotes Resilience in a Mouse Model of Depression. Cells 2021, 10, 972. [Google Scholar] [CrossRef]
- Snyder, J.S.; Soumier, A.; Brewer, M.; Pickel, J.; Cameron, H.A. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 2011, 476, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Re-quirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 1, 805–809. [Google Scholar] [CrossRef]
- Ekdahl, C.T.; Claasen, J.-H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar] [CrossRef]
- Goshen, I.; Kreisel, T.; Ben-Menachem-Zidon, O.; Licht, T.; Weidenfeld, J.; Ben-Hur, T.; Yirmiya, R. Brain interleukin-1 me-diates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol. Psychiatry 2008, 13, 717–728. [Google Scholar] [CrossRef]
- Hughes, F.M., Jr.; Odom, M.R.; Cervantes, A.; Purves, J.T. Inflammation triggered by the NLRP3 inflammasome is a critical driver of diabetic bladder dysfunction. Front. Physiol. 2022, 13, 2465. [Google Scholar] [CrossRef]
- Inouye, B.M.; Hughes, F., Jr.; Sexton, S.; Purves, J. The Emerging Role of Inflammasomes as Central Mediators in Inflammatory Bladder Pathology. Curr. Urol. 2018, 11, 57–72. [Google Scholar] [CrossRef]
- Lu, F.; Lan, Z.; Xin, Z.; He, C.; Guo, Z.; Xia, X.; Hu, T. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. J. Cell. Physiol. 2020, 235, 3207–3221. [Google Scholar] [CrossRef]
- Lin, J.; Cheng, A.; Cheng, K.; Deng, Q.; Zhang, S.; Lan, Z.; Wang, W.; Chen, J. New Insights into the Mechanisms of Pyroptosis and Implications for Diabetic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 7057. [Google Scholar] [CrossRef] [PubMed]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Asp. Med. 2020, 76, 100924. [Google Scholar] [CrossRef]
- Feng, Y.; Li, M.; Yangzhong, X.; Zhang, X.; Zu, A.; Hou, Y.; Li, L.; Sun, S. Pyroptosis in inflammation-related respiratory disease. J. Physiol. Biochem. 2022, 78, 721–737. [Google Scholar] [CrossRef] [PubMed]
- Pizzuto, M.; Pelegrin, P.; Ruysschaert, J.-M. Lipid-protein interactions regulating the canonical and the non-canonical NLRP3 inflammasome. Prog. Lipid Res. 2022, 87, 101182. [Google Scholar] [CrossRef] [PubMed]
- Craig, C.F.; Filippone, R.T.; Stavely, R.; Bornstein, J.C.; Apostolopoulos, V.; Nurgali, K. Neuroinflammation as an etiological trigger for depression comorbid with inflammatory bowel disease. J. Neuroinflamm. 2022, 19, 4. [Google Scholar] [CrossRef]
- Gao, X.; Tang, Y.; Lei, N.; Luo, Y.; Chen, P.; Liang, C.; Duan, S.; Zhang, Y. Symptoms of anxiety/depression is associated with more aggressive inflammatory bowel disease. Sci. Rep. 2021, 11, 1440. [Google Scholar] [CrossRef]
- Kim, M.C.; Jung, Y.S.; Song, Y.S.; Lee, J.I.; Park, J.H.; Sohn, C.I.; Choi, K.Y.; Park, D.I. Factors Associated with Anxiety and Depression in Korean Patients with Inactive Inflammatory Bowel Disease. Gut Liver 2016, 10, 399–405. [Google Scholar] [CrossRef]
- Sroor, H.M.; Hassan, A.M.; Zenz, G.; Valadez-Cosmes, P.; Farzi, A.; Holzer, P.; El-Sharif, A.; Gomaa, F.A.-Z.M.; Kargl, J.; Reichmann, F. Experimental colitis reduces microglial cell activation in the mouse brain without affecting microglial cell numbers. Sci. Rep. 2019, 9, 20217. [Google Scholar] [CrossRef]
- Han, Y.; Zhao, T.; Cheng, X.; Zhao, M.; Gong, S.-H.; Zhao, Y.-Q.; Wu, H.-T.; Fan, M.; Zhu, L.-L. Cortical Inflammation is Increased in a DSS-Induced Colitis Mouse Model. Neurosci. Bull. 2018, 34, 1058–1066. [Google Scholar] [CrossRef]
- He, X.-F.; Li, L.-L.; Xian, W.-B.; Li, M.-Y.; Zhang, L.-Y.; Xu, J.-H.; Pei, Z.; Zheng, H.-Q.; Hu, X.-Q. Chronic colitis exacerbates NLRP3-dependent neuroinflammation and cognitive impairment in middle-aged brain. J. Neuroinflamm. 2021, 18, 153. [Google Scholar] [CrossRef] [PubMed]
- Covic, T.; Cumming, S.R.; Pallant, J.F.; Manolios, N.; Emery, P.; Conaghan, P.G.; Tennant, A. Depression and anxiety in patients with rheumatoid arthritis: Prevalence rates based on a comparison of the Depression, Anxiety and Stress Scale (DASS) and the hospital, Anxiety and Depression Scale (HADS). BMC Psychiatry 2012, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.-C.; Guo, H.-R.; Lin, M.-C.; Livneh, H.; Lai, N.-S.; Tsai, T.-Y. Bidirectional associations between rheumatoid arthritis and depression: A nationwide longitudinal study. Sci. Rep. 2016, 6, 20647. [Google Scholar] [CrossRef] [PubMed]
- Matcham, F.; Rayner, L.; Steer, S.; Hotopf, M. The prevalence of depression in rheumatoid arthritis: A systematic review and meta-analysis. Rheumatology 2013, 52, 2136–2148. [Google Scholar] [CrossRef]
- Lampa, J.; Westman, M.; Kadetoff, D.; Agréus, A.N.; Le Maître, E.; Gillis-Haegerstrand, C.; Andersson, M.; Khademi, M.; Corr, M.; Christianson, C.A.; et al. Peripheral inflammatory disease associated with centrally activated IL-1 system in humans and mice. Proc. Natl. Acad. Sci. USA 2012, 109, 12728–12733. [Google Scholar] [CrossRef]
- Süß, P.; Rothe, T.; Hoffmann, A.; Schlachetzki, J.C.M.; Winkler, J. The Joint-Brain Axis: Insights from Rheumatoid Arthritis on the Crosstalk Between Chronic Peripheral Inflammation and the Brain. Front. Immunol. 2020, 11, 612104. [Google Scholar] [CrossRef]
- Nishioku, T.; Yamauchi, A.; Takata, F.; Watanabe, T.; Furusho, K.; Shuto, H.; Dohgu, S.; Kataoka, Y. Disruption of the blood–brain barrier in collagen-induced arthritic mice. Neurosci. Lett. 2010, 482, 208–211. [Google Scholar] [CrossRef]
- Park, S.M.; Shin, J.; Moon, G.; Cho, S.; Lee, Y.; Gwag, B. Effects of collagen-induced rheumatoid arthritis on amy-loidosis and microvascular pathology in APP/PS1 mice. BMC Neurosci. 2011, 12, 106. [Google Scholar] [CrossRef]
- Sandiego, C.M.; Gallezot, J.-D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.-F.; Matuskey, D.; Lee, J.-Y.; O’Connor, K.C.; Huang, Y.; et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc. Natl. Acad. Sci. USA 2015, 112, 12468–12473. [Google Scholar] [CrossRef]
- Hirotsu, A.; Miyao, M.; Tatsumi, K.; Tanaka, T. Sepsis-associated neuroinflammation in the spinal cord. PLoS ONE 2022, 17, e0269924. [Google Scholar] [CrossRef]
- Birder, L.; Andersson, K.-E. Animal Modelling of Interstitial Cystitis/Bladder Pain Syndrome. Int. Neurourol. J. 2018, 22, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Genka, S.; Deutsch, J.; Stahle, P.; Shetty, U.; John, V.; Robinson, C.; Rapoport, S.; Greig, N. Brain and plasma phar-macokinetics and anticancer activities of cyclophosphamide and phosphoramide mustard in the rat. Cancer Chemother. Pharmacol. 1990, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Muldoon, L.L.; Dickey, D.T.; Lewin, S.J.; Varallyay, C.G.; Neuwelt, E.A. Cyclophosphamide Enhances human tumor growth in nude rat xenografted tumor models. Neoplasia 2009, 11, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Barta, Z.; Toth, L.; Zeher, M. Pulse cyclophosphamide therapy for inflammatory bowel disease. World J. Gastroenterol. 2006, 12, 1278–1280. [Google Scholar] [CrossRef] [PubMed]
- Hughes, F.; Vivar, N.P.; Kennis, J.G.; Pratt-Thomas, J.D.; Lowe, D.W.; Shaner, B.E.; Nietert, P.J.; Spruill, L.S.; Purves, J.T. Inflammasomes are important mediators of cyclophosphamide-induced bladder inflammation. Am. J. Physiol. Physiol. 2014, 306, F299–F308. [Google Scholar] [CrossRef] [PubMed]
- Krzyzanowska, A.; Martin, Y.; Avendaño, C.; Piedras, M.J.; Krzyzanowska, A. Evaluation of Evans Blue extravasation as a measure of peripheral inflammation. Protoc. Exch. 2010. [Google Scholar] [CrossRef]
- Hughes, F.M.; McKeithan, P.; Ellett, J.; Armeson, K.E.; Purves, J.T. Simvastatin suppresses cyclophosphamide-induced changes in urodynamics and bladder inflammation. Urology 2013, 81, 209.e9–209.e14. [Google Scholar] [CrossRef]
- Hughes, F.; Hill, H.M.; Wood, C.M.; Edmondson, A.T.; Dumas, A.; Foo, W.-C.; Oelsen, J.M.; Rac, G.; Purves, J.T. The NLRP3 Inflammasome Mediates Inflammation Produced by Bladder Outlet Obstruction. J. Urol. 2016, 195, 1598–1605. [Google Scholar] [CrossRef]
- Belayev, L.; Busto, R.; Zhao, W.; Ginsberg, M.D. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996, 739, 88–96. [Google Scholar] [CrossRef]
- Michels, M.; Vieira, A.S.; Vuolo, F.; Zapelini, H.G.; Mendonça, B.; Mina, F.; Dominguini, D.; Steckert, A.; Schuck, P.F.; Quevedo, J.; et al. The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav. Immun. 2015, 43, 54–59. [Google Scholar] [CrossRef]
- Chen, J.L.; Zhou, X.; Liu, B.; Wei, X.; Ding, H.; Lin, Z.; Zhan, H.; Yang, F.; Li, W.; Xie, J.; et al. Normalization of magnesium deficiency attenuated mechanical allodynia, depressive-like behaviors, and memory deficits associated with cy-clophosphamide-induced cystitis by inhibiting TNF-alpha/NF-kappaB signaling in female rats. J. Neuroinflamm. 2020, 17, 99. [Google Scholar] [CrossRef]
- Liu, B.; Su, M.; Tang, S.; Zhou, X.; Zhan, H.; Yang, F.; Li, W.; Li, T.; Xie, J. Spinal astrocytic activation contributes to mechanical allodynia in a rat model of cyclophosphamide-induced cystitis. Mol. Pain 2016, 12, 1744806916674479. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, X.; Ding, H.; Zhan, H.; Yang, F.; Li, W.; Xie, J.; Liu, X.; Xu, Y.; Su, M.; et al. Neuregulin-1-ErbB signaling promotes microglia activation contributing to mechanical allodynia of cyclophosphamide-induced cystitis. Neurourol. Urodyn. 2019, 38, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Su, M.; Zhang, C.; Zhan, H.; Yang, F.; Gao, Z.; Zhou, X.; Liu, B. Activation of translocator protein alleviates mechanical allodynia and bladder dysfunction in cyclophosphamide-induced cystitis through repression of BDNF-mediated neuroinflammation. Eur. J. Pain 2022, 26, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Bagnall-Moreau, C.; Chaudhry, S.; Salas-Ramirez, K.; Ahles, T.; Hubbard, K. Chemotherapy-Induced Cognitive Impairment Is Associated with Increased Inflammation and Oxidative Damage in the Hippocampus. Mol. Neurobiol. 2019, 56, 7159–7172. [Google Scholar] [CrossRef] [PubMed]
- Turrina, S.; Gibelli, F.; De Leo, D. Chemotherapy-induced cognitive impairment from the forensic medicine perspective: A review of the updated literature. J. Forensic Leg. Med. 2020, 76, 102070. [Google Scholar] [CrossRef] [PubMed]
- Saigal, C.S.; Joyce, G. Economic Costs of Benign Prostatic Hyperplasia in the Private Sector. J. Urol. 2005, 173, 1309–1313. [Google Scholar] [CrossRef]
- Liu, W.; Ge, T.; Leng, Y.; Pan, Z.; Fan, J.; Yang, W.; Cui, R. The Role of Neural Plasticity in Depression: From Hippocampus to Prefrontal Cortex. Neural Plast. 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Hughes, F.M.; Sexton, S.J.; Ledig, P.D.; Yun, C.E.; Jin, H.; Purves, J.T. Bladder decompensation and reduction in nerve density in a rat model of chronic bladder outlet obstruction are attenuated with the NLRP3 inhibitor glyburide. Am. J. Physiol. Physiol. 2019, 316, F113–F120. [Google Scholar] [CrossRef]
- Xia, C.-Y.; Guo, Y.-X.; Lian, W.-W.; Yan, Y.; Ma, B.-Z.; Cheng, Y.-C.; Xu, J.-K.; He, J.; Zhang, W.-K. The NLRP3 Inflammasome in Depression: Potential Mechanisms and Therapies. Pharmacol. Res. 2022, 187, 106625. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, I.; Ko, J.; Moon, C.; Kim, S.; Shin, I.; Seo, Y.; Kim, H.; Kim, J. Diallyl Disulfide Prevents Cy-clophosphamide-Induced Hemorrhagic Cystitis in Rats through the Inhibition of Oxidative Damage, MAPKs, and NF-kappaB Pathways. Biomol. Ther. 2015, 23, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Mitsui, T.; Kitta, T.; Moriya, K.; Tsukiyama, T.; Hatakeyama, S.; Nonomura, K. The inflammatory cytokine IL-1beta is involved in bladder remodeling after bladder outlet obstruction in mice. Neurourol. Urodyn. 2016, 35, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Engelhardt, B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroin-flammation. Vasc. Biol. 2020, 2, H1–H18. [Google Scholar] [CrossRef]
- Wolburg, H.; Wolburg-Buchholz, K.; Engelhardt, B. Diapedesis of mononuclear cells across cerebral venules during ex-perimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol. 2005, 109, 181–190. [Google Scholar] [CrossRef]
- D’Mello, C.; Le, T.; Swain, M. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis fac-toralpha signaling during peripheral organ inflammation. J. Neurosci. 2009, 29, 2089–2102. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef]
- Kohler, O.; Krogh, J.; Mors, O.; Benros, M. Inflammation in Depression and the Potential for Anti-Inflammatory Treat-ment. Curr. Neuropharmacol. 2016, 14, 732–742. [Google Scholar] [CrossRef]
- Kohler, O.; Benros, M.; Nordentoft, M.; Farkouh, M.; Iyengar, R.; Mors, O.; Krogh, J. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: A systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef]
- Husain, M.I.; Chaudhry, I.B.; Khoso, A.B.; Hodsoll, J.; Ansari, M.A.; Naqvi, H.A.; Minhas, F.A.; Carvalho, A.F.; Meyer, J.H.; Deakin, J.; et al. Minocycline and celecoxib as adjunctive treatments for bipolar depression: A multicentre, factorial design randomised controlled trial. Lancet Psychiatry 2020, 7, 515–527. [Google Scholar] [CrossRef]
- Ghanizadeh, A.; Hedayati, A. Augmentation of fluoxetine with lovastatin for treating major depressive disorder, a randomized double-blind placebo controlled-clinical trial. Depress. Anxiety 2013, 30, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Gougol, A.; Zareh-Mohammadi, N.; Raheb, S.; Farokhnia, M.; Salimi, S.; Iranpour, N.; Yekehtaz, H.; Akhondzadeh, S. Simvastatin as an adjuvant therapy to fluoxetine in patients with moderate to severe major depression: A double-blind pla-cebo-controlled trial. J. Psychopharmacol. 2015, 29, 575–581. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Lu, W.; Zhao, S.; Hu, Z.; Zhang, J. The relationship between statins and depression: A review of the literature. Expert Opin. Pharmacother. 2013, 14, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Arana, G.W.; Santos, A.B.; Laraia, M.T.; McLeod-Bryant, S.; Beale, M.D.; Rames, L.J.; Roberts, J.M.; Dias, J.K.; Molloy, M. Dexamethasone for the treatment of depression: A randomized, placebo- controlled, double-blind trial. Am. J. Psychiatry 1995, 152, 265–267. [Google Scholar] [CrossRef]
- Debattista, C.; Posener, J.A.; Kalehzan, B.M.; Schatzberg, A.F. Acute antidepressant effects of intravenous hydrocortisone and CRH in depressed patients: A double-blind, placebo-controlled study. Am. J. Psychiatry 2000, 157, 1334–1337. [Google Scholar] [CrossRef] [PubMed]
- Sepanjnia, K.; Modabbernia, A.; Ashrafi, M.; Modabbernia, M.-J.; Akhondzadeh, S. Pioglitazone adjunctive therapy for moderate-to-severe major depressive disorder: Randomized double-blind placebo-controlled trial. Neuropsychopharmacology 2012, 37, 2093–2100. [Google Scholar] [CrossRef]
- Kashani, L.; Omidvar, T.; Farazmand, B.; Modabbernia, A.; Ramzanzadeh, F.; Tehraninejad, E.S.; Ashrafi, M.; Tabrizi, M.; Akhondzadeh, S. Does pioglitazone improve depression through insulin-sensitization? Results of a randomized double-blind metformin-controlled trial in patients with polycystic ovarian syndrome and comorbid depression. Psychoneuroendocrinology 2013, 38, 767–776. [Google Scholar] [CrossRef]
- Tomás-Camardiel, M.; Rite, I.; Herrera, A.J.; de Pablos, R.M.; Cano, J.; Machado, A.; Venero, J.L. Minocycline reduces the lipopolysaccharide-induced inflammatory reaction, peroxynitrite-mediated nitration of proteins, disruption of the blood–brain barrier, and damage in the nigral dopaminergic system. Neurobiol. Dis. 2004, 16, 190–201. [Google Scholar] [CrossRef]
- Abolfazli, R.; Hosseini, M.; Ghanizadeh, A.; Ghaleiha, A.; Tabrizi, M.; Raznahan, M.; Golalizadeh, M.; Akhondzadeh, S. Double-blind randomized parallel-group clinical trial of efficacy of the combination fluoxetine plus modafinil versus fluoxetine plus placebo in the treatment of major depression. Depress. Anxiety 2011, 28, 297–302. [Google Scholar] [CrossRef]
- Goss, A.J.; Kaser, M.; Costafreda, S.; Sahakian, B.; Fu, C. Modafinil augmentation therapy in unipolar and bipolar depression: A systematic review and meta-analysis of randomized controlled trials. J. Clin. Psychiatry 2013, 74, 1101–1107. [Google Scholar] [CrossRef]
- Reale, M.; Costantini, E. Cholinergic Modulation of the Immune System in Neuroinflammatory Diseases. Diseases 2021, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.H.; Chavan, S.S.; Pavlov, V.A. Cholinergic Control of Inflammation, Metabolic Dysfunction, and Cognitive Impairment in Obesity-Associated Disorders: Mechanisms and Novel Therapeutic Opportunities. Front. Neurosci. 2019, 13, 263. [Google Scholar] [CrossRef] [PubMed]
- Alen, N.V. The Cholinergic Anti-inflammatory Pathway in Humans: State-of-the-Art Review and Future Directions. Neurosci. Biobehav. Rev. 2022, 136, 104622. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.J.; Breathnach, C.; Tracey, K.J.; Donnelly, S.C. Manipulation of the inflammatory reflex as a therapeutic strategy. Cell Rep. Med. 2022, 3, 100696. [Google Scholar] [CrossRef] [PubMed]
- Zorbaz, T.; Madrer, N.; Soreq, H. Cholinergic blockade of neuroinflammation: From tissue to RNA regulators. Neuronal Signal. 2022, 6, NS20210035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.C.; Yao, W.; Ren, Q.; Yang, C.; Dong, C.; Ma, M.; Wu, J.; Hashimoto, K. Depression-like phenotype by deletion of alpha7 nicotinic acetylcholine receptor: Role of BDNF-TrkB in nucleus accumbens. Sci. Rep. 2016, 6, 36705. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Chang, D.; Zhang, X.; Lian, H.; Du, X.; Gao, L. Low-Dose Ketamine Improves LPS-Induced Depression-like Behavior in Rats by Activating Cholinergic Anti-inflammatory Pathways. ACS Chem. Neurosci. 2020, 11, 752–762. [Google Scholar] [CrossRef]
- Jope, R.S.; Walter-Ryan, W.G.; Alarcon, R.D.; Lally, K.M. Cholinergic processes in blood samples from patients with major psychiatric disorders. Biol. Psychiatry 1985, 20, 1258–1266. [Google Scholar] [CrossRef]
- Mineur, Y.S.; Fote, G.; Blakeman, S.; Cahuzac, E.; Newbold, S.; Picciotto, M. Multiple Nicotinic Acetylcholine Receptor Subtypes in the Mouse Amygdala Regulate Affective Behaviors and Response to Social Stress. Neuropsychopharmacology 2016, 41, 1579–1587. [Google Scholar] [CrossRef]
- Mineur, Y.S.; Mose, T.; Blakeman, S.; Picciotto, M. Hippocampal alpha7 nicotinic ACh receptors contribute to modu-lation of depression-like behaviour in C57BL/6J mice. Br. J. Pharmacol. 2018, 175, 1903–1914. [Google Scholar] [CrossRef]
- Mineur, Y.S.; Picciotto, M.R. Nicotine receptors and depression: Revisiting and revising the cholinergic hypothesis. Trends Pharmacol. Sci. 2010, 31, 580–586. [Google Scholar] [CrossRef]
- Mitic, M.; Lazarevic-Pasti, T. Does the application of acetylcholinesterase inhibitors in the treatment of Alzheimer’s disease lead to depression? Expert Opin. Drug Metab. Toxicol. 2021, 17, 841–856. [Google Scholar] [CrossRef] [PubMed]
- Deyama, S.; Kaneda, K.; Minami, M. Resolution of depression: Antidepressant actions of resolvins. Neurosci. Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2015, 16, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Hamberg, M.; Samuelsson, B. Trihydroxytetraenes: A novel series of compounds formed from arachidonic acid in human leukocytes. Biochem. Biophys. Res. Commun. 1984, 118, 943–949. [Google Scholar] [CrossRef]
- Brennan, E.; Kantharidis, P.; Cooper, M.E.; Godson, C. Pro-resolving lipid mediators: Regulators of inflammation, metabolism and kidney function. Nat. Rev. Nephrol. 2021, 17, 725–739. [Google Scholar] [CrossRef]
- Chiang, N.; Serhan, C.N. Specialized pro-resolving mediator network: An update on production and actions. Essays Biochem. 2020, 64, 443–462. [Google Scholar] [CrossRef]
- Fattori, V.; Zaninelli, T.H.; Rasquel-Oliveira, F.S.; Casagrande, R.; Verri, W.A. Specialized pro-resolving lipid mediators: A new class of non-immunosuppressive and non-opioid analgesic drugs. Pharmacol. Res. 2020, 151, 104549. [Google Scholar] [CrossRef]
- Perretti, M.; Dalli, J. Resolution Pharmacology: Focus on Pro-Resolving Annexin A1 and Lipid Mediators for Therapeutic In-novation in Inflammation. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 449–469. [Google Scholar] [CrossRef]
- Dalli, J.; Colas, R.; Arnardottir, H.; Serhan, C. Vagal Regulation of Group 3 Innate Lymphoid Cells and the Immu-noresolvent PCTR1 Controls Infection Resolution. Immunity 2017, 46, 92–105. [Google Scholar] [CrossRef]
- Mirakaj, V.; Dalli, J.; Granja, T.; Rosenberger, P.; Serhan, C.N. Vagus nerve controls resolution and pro-resolving mediators of inflammation. J. Exp. Med. 2014, 211, 1037–1048. [Google Scholar] [CrossRef]
- Serhan, C.N.; de la Rosa, X.; Jouvene, C. Novel mediators and mechanisms in the resolution of infectious inflammation: Evidence for vagus regulation. J. Intern. Med. 2019, 286, 240–258. [Google Scholar] [CrossRef] [PubMed]
- Hughes, F.M., Jr.; Allkanjari, A.; Odom, M.; Jin, H.; Purves, J. Specialized pro-resolution mediators in the bladder: Re-ceptor expression and recovery of bladder function from cystitis. Exp. Biol. Med. 2022, 247, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Hughes, F.M., Jr.; Harper, S.; Nose, B.; Allkanjari, A.; Sheng, M.; Jin, H.; Purves, J. Specialized Pro-resolution Me-diators in the bladder; Annexin-A1 normalizes inflammation and bladder dysfunction during bladder outlet obstruction. Am. J. Physiol. Renal Physiol. 2021, 321, F443–F454. [Google Scholar] [CrossRef] [PubMed]
- Borsini, A.; Nicolaou, A.; Camacho-Muñoz, D.; Kendall, A.C.; Di Benedetto, M.G.; Giacobbe, J.; Su, K.-P.; Pariante, C.M. Omega-3 polyunsaturated fatty acids protect against inflammation through production of LOX and CYP450 lipid mediators: Relevance for major depression and for human hippocampal neurogenesis. Mol. Psychiatry 2021, 26, 6773–6788. [Google Scholar] [CrossRef]
- Casimir, G.J.A.; Duchateau, J. Gender Differences in Inflammatory Processes Could Explain Poorer Prognosis for Males. J. Clin. Microbiol. 2011, 49, 478–479. [Google Scholar] [CrossRef]
- Derry, H.M.; Padin, A.; Kuo, J.; Hughes, S.; Kiecolt-Glaser, J. Sex Differences in Depression: Does Inflammation Play a Role? Curr. Psychiatry Rep. 2015, 17, 78. [Google Scholar] [CrossRef]
- Seney, M.L.; Sibille, E. Sex differences in mood disorders: Perspectives from humans and rodent models. Biol. Sex Differ. 2014, 5, 17. [Google Scholar] [CrossRef]
- Kessler, R.C.; Berglund, P.A.; Foster, C.L.; Saunders, W.B.; Stang, P.E.; Walters, E.E. Social consequences of psychiatric disorders, II: Teenage parenthood. Am. J. Psychiatry 1997, 154, 1405–1411. [Google Scholar] [CrossRef]
- Gater, R.; Tansella, M.; Korten, A.; Tiemens, B.; Mavreas, V.; Olatawura, M. Sex differences in the prevalence and detection of depressive and anxiety disorders in general health care settings: Report from the World Health Organization Collaborative Study on Psychological Problems in General Health Care. Arch. Gen. Psychiatry 1998, 55, 405–413. [Google Scholar] [CrossRef]
- Bangasser, D.A.; Cuarenta, A. Sex differences in anxiety and depression: Circuits and mechanisms. Nat. Rev. Neurosci. 2021, 22, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Rainville, J.R.; Hodes, G.E. Inflaming sex differences in mood disorders. Neuropsychopharmacology 2019, 44, 184–199. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hughes, F.M., Jr.; Odom, M.R.; Cervantes, A.; Livingston, A.J.; Purves, J.T. Why Are Some People with Lower Urinary Tract Symptoms (LUTS) Depressed? New Evidence That Peripheral Inflammation in the Bladder Causes Central Inflammation and Mood Disorders. Int. J. Mol. Sci. 2023, 24, 2821. https://doi.org/10.3390/ijms24032821
Hughes FM Jr., Odom MR, Cervantes A, Livingston AJ, Purves JT. Why Are Some People with Lower Urinary Tract Symptoms (LUTS) Depressed? New Evidence That Peripheral Inflammation in the Bladder Causes Central Inflammation and Mood Disorders. International Journal of Molecular Sciences. 2023; 24(3):2821. https://doi.org/10.3390/ijms24032821
Chicago/Turabian StyleHughes, Francis M., Jr., Michael R. Odom, Anissa Cervantes, Austin J. Livingston, and J. Todd Purves. 2023. "Why Are Some People with Lower Urinary Tract Symptoms (LUTS) Depressed? New Evidence That Peripheral Inflammation in the Bladder Causes Central Inflammation and Mood Disorders" International Journal of Molecular Sciences 24, no. 3: 2821. https://doi.org/10.3390/ijms24032821
APA StyleHughes, F. M., Jr., Odom, M. R., Cervantes, A., Livingston, A. J., & Purves, J. T. (2023). Why Are Some People with Lower Urinary Tract Symptoms (LUTS) Depressed? New Evidence That Peripheral Inflammation in the Bladder Causes Central Inflammation and Mood Disorders. International Journal of Molecular Sciences, 24(3), 2821. https://doi.org/10.3390/ijms24032821