

p27Kip1 Deficiency Impairs Brown Adipose Tissue Function Favouring Fat Accumulation in Mice

 , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Results
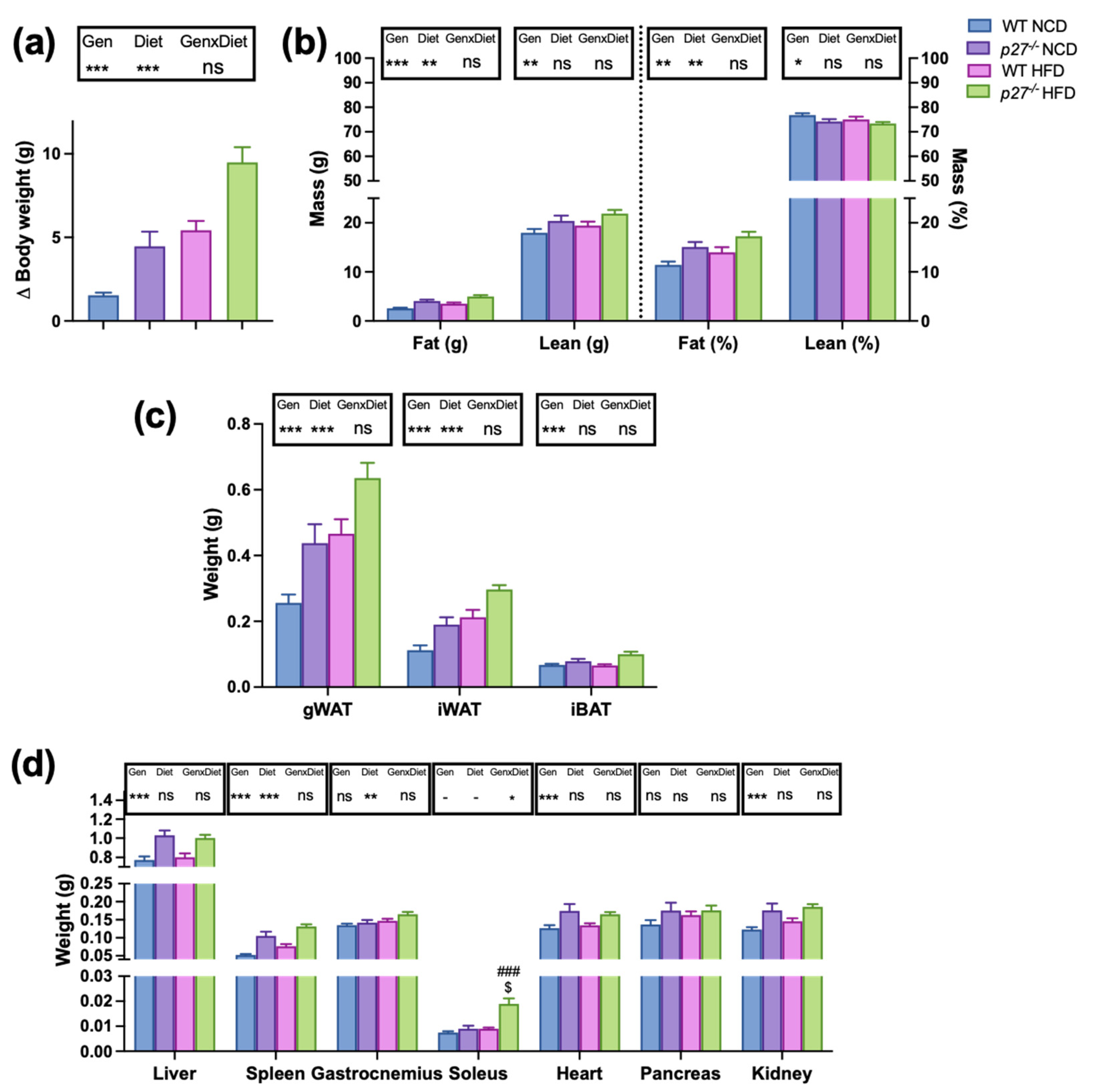
2.1. p27−/− Mice are More Prone to Develop Obesity Than WT Mice
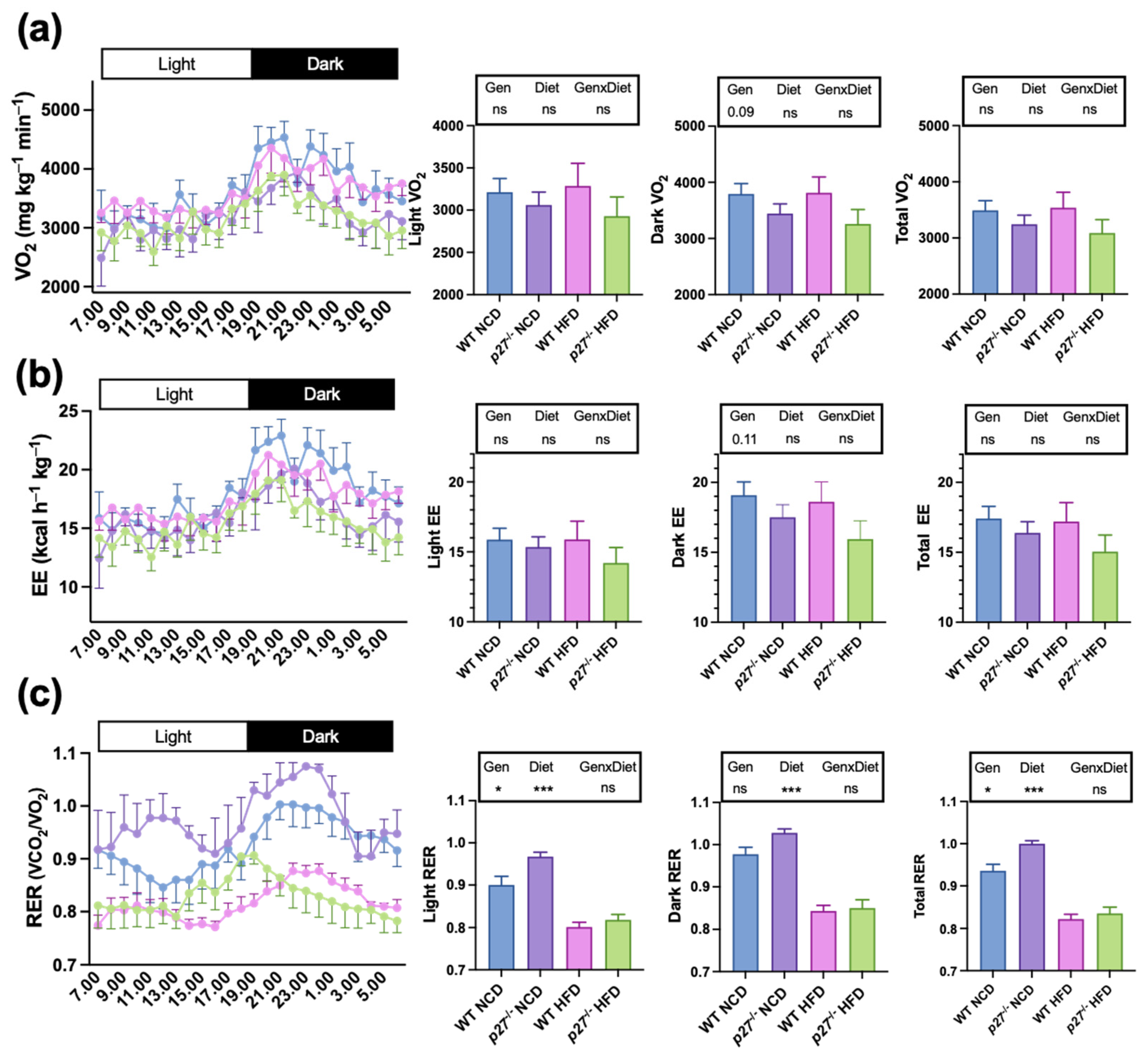
2.2. p27−/− Mice Exhibit Increased Respiratory Exchange Ratio
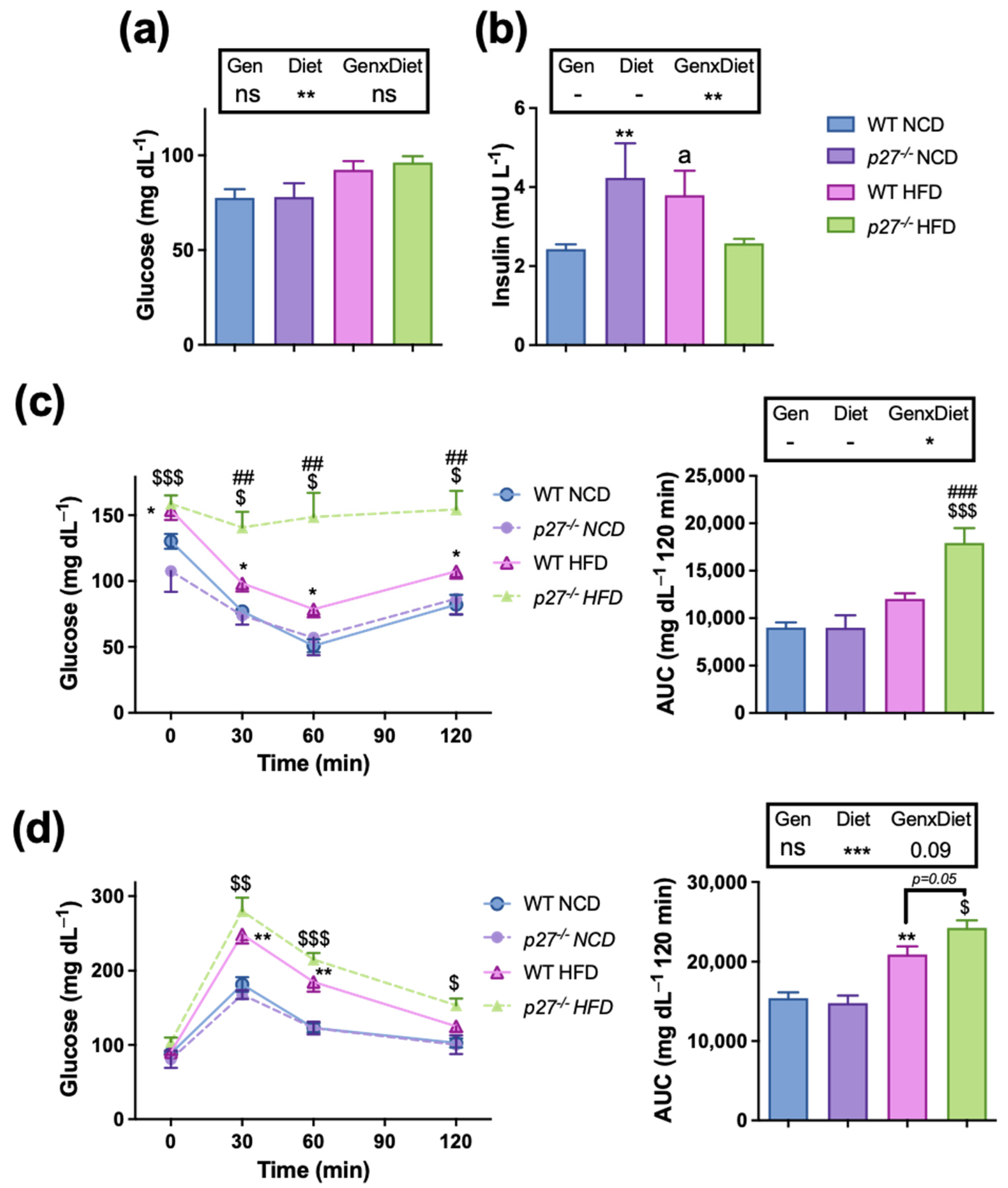
2.3. Mice Lacking p27 Are More Susceptible to HFD-Induced Insulin Resistance
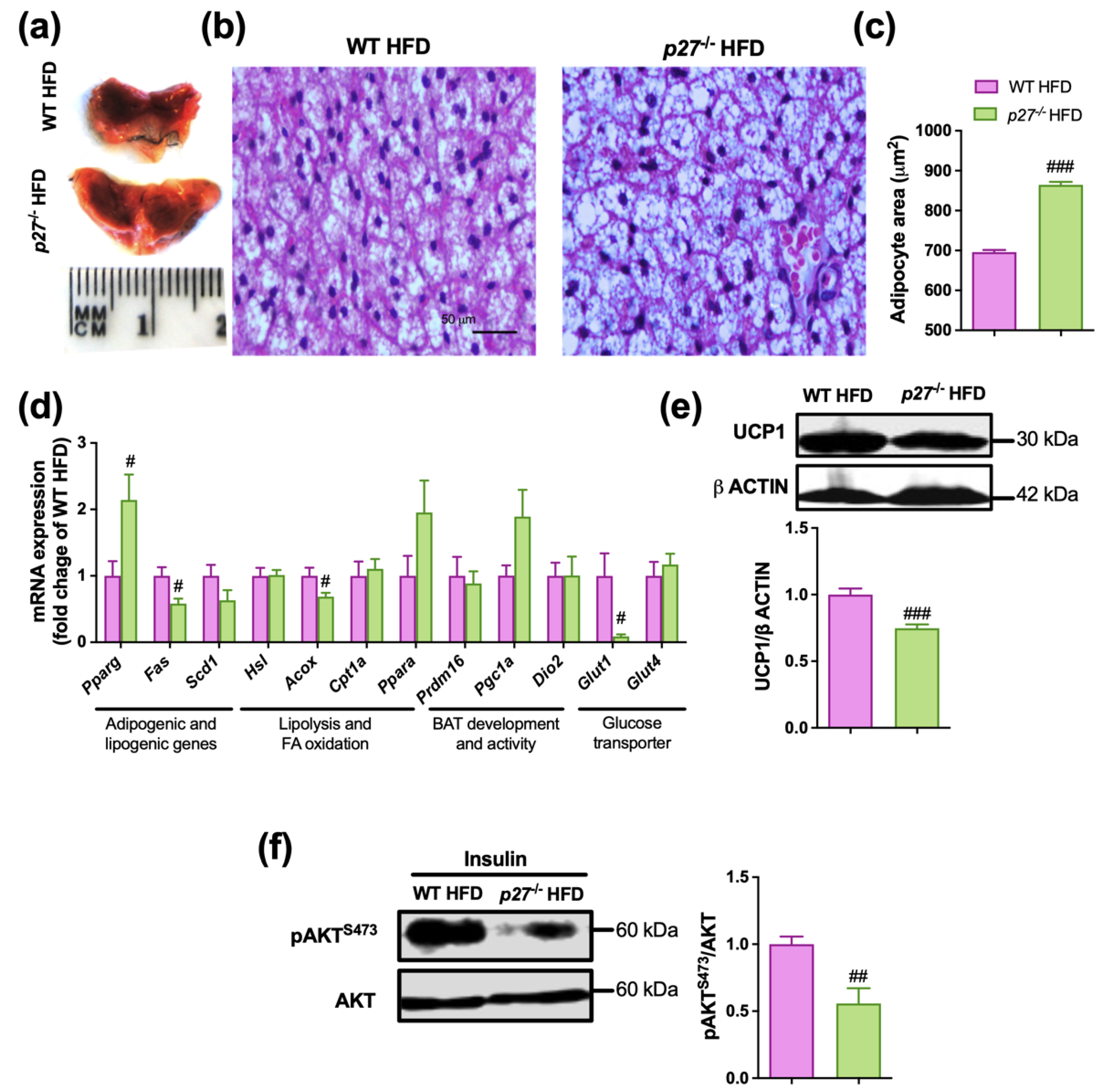
2.4. iBAT of p27−/− HFD Mice Exhibits Whitening and an Impaired Insulin Signalling
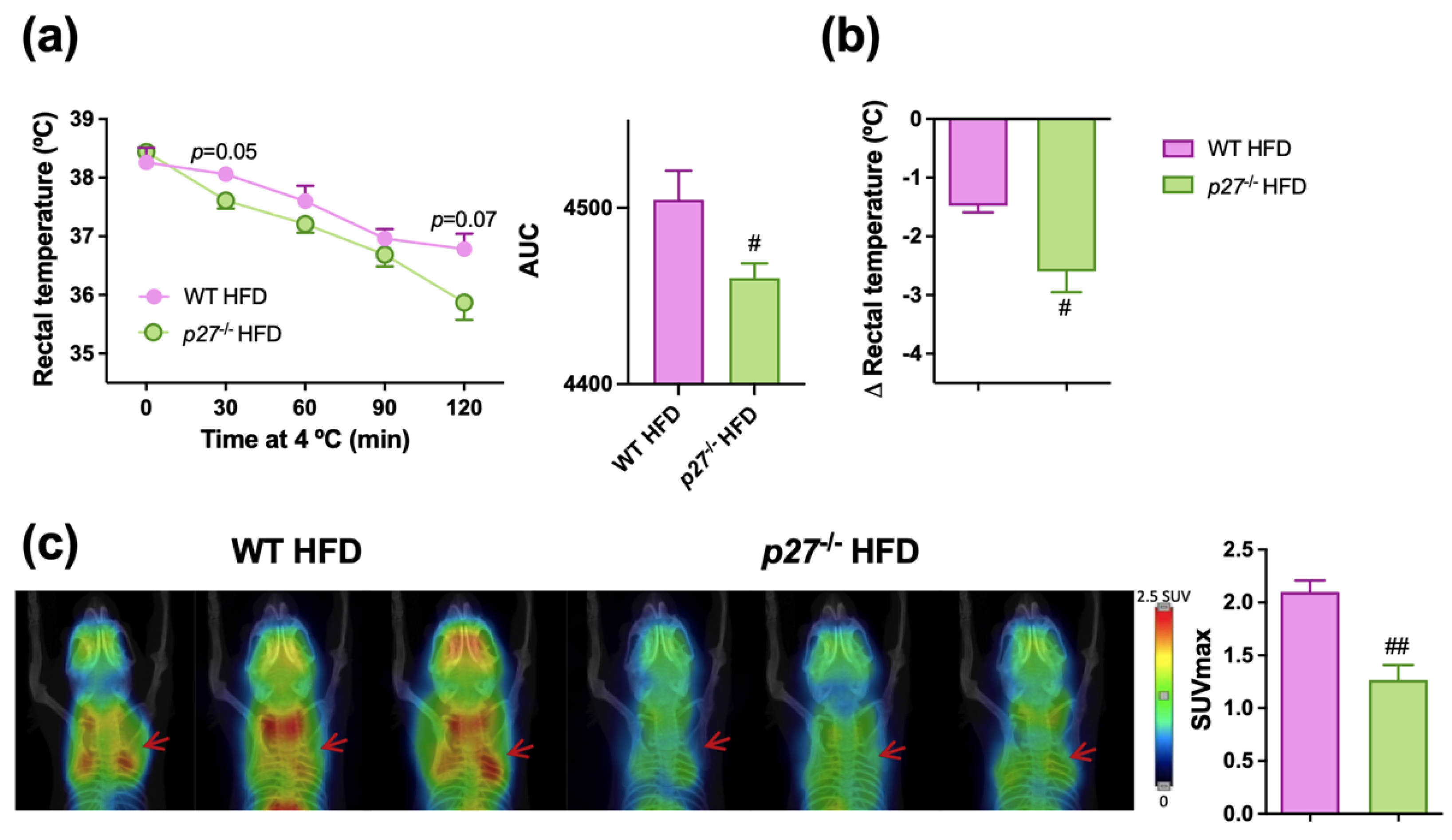
2.5. p27−/− HFD Mice Have a Less Functional BAT and a Diminished Thermogenic Capacity
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Insulin and Glucose Tolerance Test
4.3. Biochemical Analysis
4.4. Insulin Signalling Assessment
4.5. Real-Time Quantitative PCR
4.6. Protein Extraction and Western Blot
4.7. Acute Cold Exposure Challenge
4.8. In Vivo Study of iBAT Activation by Positron Emission Tomography (PET)
4.9. Indirect Calorimetry and Activity Measurements
4.10. Histological Analysis
4.11. Liver Triglyceride Content Assay
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, Regional, and National Prevalence of Overweight and Obesity in Children and Adults during 1980–2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef]
- Thaker, V.V. Genetic and Epigenetic Causes of Obesity. Adolesc. Med. State Art Rev. 2017, 28, 379–405. [Google Scholar] [PubMed]
- Vinci, L.; Krieger, J.-P.; Braun, J.; Pestoni, G.; Bender, N.; Rohrmann, S.; Faeh, D.; Staub, K. Clustering of Sociodemographic and Lifestyle Factors among Adults with Excess Weight in a Multilingual Country. Nutrition 2019, 62, 177–185. [Google Scholar] [CrossRef]
- Pi-Sunyer, X. The Medical Risks of Obesity. Postgrad. Med. 2009, 121, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, J.; Farr, O.; Perakakis, N.; Ghaly, W.; Mantzoros, C. Obesity as a Disease. Med. Clin. N. Am. 2018, 102, 13–33. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Fenzl, A.; Kiefer, F.W. Brown Adipose Tissue and Thermogenesis. Horm. Mol. Biol. Clin. Investig. 2014, 19, 25–37. [Google Scholar] [CrossRef]
- Scheja, L.; Heeren, J. Metabolic Interplay between White, Beige, Brown Adipocytes and the Liver. J. Hepatol. 2016, 64, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Kahn, C.R. Brown Fat as a Therapy for Obesity and Diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 143–149. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and Metabolic Health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Bukowiecki, L.; Collet, A.J.; Follea, N.; Guay, G.; Jahjah, L. Brown Adipose Tissue Hyperplasia: A Fundamental Mechanism of Adaptation to Cold and Hyperphagia. Am. J. Physiol. 1982, 242, E353–E359. [Google Scholar] [CrossRef] [PubMed]
- Fukano, K.; Okamatsu-Ogura, Y.; Tsubota, A.; Nio-Kobayashi, J.; Kimura, K. Cold Exposure Induces Proliferation of Mature Brown Adipocyte in a SS3-Adrenergic Receptor-Mediated Pathway. PLoS ONE 2016, 11, e0166579. [Google Scholar] [CrossRef]
- Solaki, M.; Ewald, J.C. Fueling the Cycle: CDKs in Carbon and Energy Metabolism. Front. Cell Dev. Biol. 2018, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Kaldis, P.; Cheng, A.; Solomon, M.J. The Effects of Changing the Site of Activating Phosphorylation in CDK2 from Threonine to Serine. J. Biol. Chem. 2000, 275, 32578–32584. [Google Scholar] [CrossRef] [PubMed]
- Viallard, J.F.; Lacombe, F.; Belloc, F.; Pellegrin, J.L.; Reiffers, J. Molecular mechanisms controlling the cell cycle: Fundamental aspects and implications for oncology. Cancer Radiother. 2001, 5, 109–129. [Google Scholar] [CrossRef]
- Polyak, K.; Lee, M.H.; Erdjument-Bromage, H.; Koff, A.; Roberts, J.M.; Tempst, P.; Massagué, J. Cloning of P27Kip1, a Cyclin-Dependent Kinase Inhibitor and a Potential Mediator of Extracellular Antimitogenic Signals. Cell 1994, 78, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, H.; Hunter, T. P27, a Novel Inhibitor of G1 Cyclin-Cdk Protein Kinase Activity, Is Related to P21. Cell 1994, 78, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Carvalhal, A.V.; Marcelino, I.; Carrondo, M.J.T. Metabolic Changes during Cell Growth Inhibition by P27 Overexpression. Appl. Microbiol. Biotechnol. 2003, 63, 164–173. [Google Scholar] [CrossRef]
- Colón-Mesa, I.; Fernández-Galilea, M.; Sáinz, N.; Lopez-Yus, M.; Artigas, J.M.; Arbonés-Mainar, J.M.; Félix-Soriano, E.; Escoté, X.; Moreno-Aliaga, M.J. Regulation of P27 and Cdk2 Expression in Different Adipose Tissue Depots in Aging and Obesity. Int. J. Mol. Sci. 2021, 22, 11745. [Google Scholar] [CrossRef]
- Naaz, A.; Holsberger, D.R.; Iwamoto, G.A.; Nelson, A.; Kiyokawa, H.; Cooke, P.S. Loss of Cyclin-Dependent Kinase Inhibitors Produces Adipocyte Hyperplasia and Obesity. FASEB J. 2004, 18, 1925–1927. [Google Scholar] [CrossRef]
- Auld, C.A.; Fernandes, K.M.; Morrison, R.F. Skp2-Mediated P27(Kip1) Degradation during S/G2 Phase Progression of Adipocyte Hyperplasia. J. Cell. Physiol. 2007, 211, 101–111. [Google Scholar] [CrossRef]
- Okamatsu-Ogura, Y.; Fukano, K.; Tsubota, A.; Nio-Kobayashi, J.; Nakamura, K.; Morimatsu, M.; Sakaue, H.; Saito, M.; Kimura, K. Cell-Cycle Arrest in Mature Adipocytes Impairs BAT Development but Not WAT Browning, and Reduces Adaptive Thermogenesis in Mice. Sci. Rep. 2017, 7, 6648. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, J.; Dai, H.; Duan, Y.; An, Y.; Shi, L.; Lv, Y.; Li, H.; Wang, C.; Ma, Q.; et al. Brown and Beige Adipose Tissue: A Novel Therapeutic Strategy for Obesity and Type 2 Diabetes Mellitus. Adipocyte 2021, 10, 48–65. [Google Scholar] [CrossRef]
- Kim, S.Y.; Lee, J.-H.; Merrins, M.J.; Gavrilova, O.; Bisteau, X.; Kaldis, P.; Satin, L.S.; Rane, S.G. Loss of Cyclin-Dependent Kinase 2 in the Pancreas Links Primary β-Cell Dysfunction to Progressive Depletion of β-Cell Mass and Diabetes. J. Biol. Chem. 2017, 292, 3841–3853. [Google Scholar] [CrossRef]
- Berthet, C.; Aleem, E.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cdk2 Knockout Mice Are Viable. Curr. Biol. 2003, 13, 1775–1785. [Google Scholar] [CrossRef]
- Lagarrigue, S.; Lopez-Mejia, I.C.; Denechaud, P.-D.; Escoté, X.; Castillo-Armengol, J.; Jimenez, V.; Chavey, C.; Giralt, A.; Lai, Q.; Zhang, L.; et al. CDK4 Is an Essential Insulin Effector in Adipocytes. J. Clin. Investig. 2016, 126, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a Link between Obesity, Metabolic Syndrome and Type 2 Diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Bartelt, A.; Heeren, J. Adipose Tissue Browning and Metabolic Health. Nat. Rev. Endocrinol. 2014, 10, 24–36. [Google Scholar] [CrossRef]
- Lowell, B.B.; S-Susulic, V.; Hamann, A.; Lawitts, J.A.; Himms-Hagen, J.; Boyer, B.B.; Kozak, L.P.; Flier, J.S. Development of Obesity in Transgenic Mice after Genetic Ablation of Brown Adipose Tissue. Nature 1993, 366, 740–742. [Google Scholar] [CrossRef]
- Stanford, K.I.; Middelbeek, R.J.W.; Townsend, K.L.; An, D.; Nygaard, E.B.; Hitchcox, K.M.; Markan, K.R.; Nakano, K.; Hirshman, M.F.; Tseng, Y.-H.; et al. Brown Adipose Tissue Regulates Glucose Homeostasis and Insulin Sensitivity. J. Clin. Investig. 2013, 123, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Lee, M.W.; Yoo, K.H.; Lee, T.-H.; Kim, H.J.; Jang, I.K.; Chun, Y.H.; Kim, H.J.; Park, S.J.; Lee, S.H.; et al. Gene Expression Profiles of Human Adipose Tissue-Derived Mesenchymal Stem Cells Are Modified by Cell Culture Density. PLoS ONE 2014, 9, e83363. [Google Scholar] [CrossRef]
- Nakayama, K.; Ishida, N.; Shirane, M.; Inomata, A.; Inoue, T.; Shishido, N.; Horii, I.; Loh, D.Y.; Nakayama, K. Mice Lacking P27(Kip1) Display Increased Body Size, Multiple Organ Hyperplasia, Retinal Dysplasia, and Pituitary Tumors. Cell 1996, 85, 707–720. [Google Scholar] [CrossRef]
- Choi, M.-S.; Kim, Y.-J.; Kwon, E.-Y.; Ryoo, J.Y.; Kim, S.R.; Jung, U.J. High-Fat Diet Decreases Energy Expenditure and Expression of Genes Controlling Lipid Metabolism, Mitochondrial Function and Skeletal System Development in the Adipose Tissue, along with Increased Expression of Extracellular Matrix Remodelling- and Inflammation-Related Genes. Br. J. Nutr. 2015, 113, 867–877. [Google Scholar] [CrossRef]
- Ellis, A.C.; Hyatt, T.C.; Hunter, G.R.; Gower, B.A. Respiratory Quotient Predicts Fat Mass Gain in Premenopausal Women. Obesity 2010, 18, 2255–2259. [Google Scholar] [CrossRef]
- Shook, R.P.; Hand, G.A.; Paluch, A.E.; Wang, X.; Moran, R.; Hébert, J.R.; Jakicic, J.M.; Blair, S.N. High Respiratory Quotient Is Associated with Increases in Body Weight and Fat Mass in Young Adults. Eur. J. Clin. Nutr. 2016, 70, 1197–1202. [Google Scholar] [CrossRef]
- Villarroya, F.; Cereijo, R.; Villarroya, J.; Giralt, M. Brown Adipose Tissue as a Secretory Organ. Nat. Rev. Endocrinol. 2017, 13, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, H.M.; Golozoubova, V.; Cannon, B.; Nedergaard, J. UCP1 Ablation Induces Obesity and Abolishes Diet-Induced Thermogenesis in Mice Exempt from Thermal Stress by Living at Thermoneutrality. Cell Metab. 2009, 9, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Bentley, R.A.; Ross, C.N.; O’Brien, M.J. Obesity, Metabolism, and Aging: A Multiscalar Approach. Prog. Mol. Biol. Transl. Sci. 2018, 155, 25–42. [Google Scholar] [CrossRef]
- Kotzbeck, P.; Giordano, A.; Mondini, E.; Murano, I.; Severi, I.; Venema, W.; Cecchini, M.P.; Kershaw, E.E.; Barbatelli, G.; Haemmerle, G.; et al. Brown Adipose Tissue Whitening Leads to Brown Adipocyte Death and Adipose Tissue Inflammation. J. Lipid Res. 2018, 59, 784–794. [Google Scholar] [CrossRef]
- Moreno-Fernandez, M.E.; Giles, D.A.; Stankiewicz, T.E.; Sheridan, R.; Karns, R.; Cappelletti, M.; Lampe, K.; Mukherjee, R.; Sina, C.; Sallese, A.; et al. Peroxisomal β-Oxidation Regulates Whole Body Metabolism, Inflammatory Vigor, and Pathogenesis of Nonalcoholic Fatty Liver Disease. JCI Insight 2018, 3, 93626. [Google Scholar] [CrossRef]
- Olsen, J.M.; Sato, M.; Dallner, O.S.; Sandström, A.L.; Pisani, D.F.; Chambard, J.-C.; Amri, E.-Z.; Hutchinson, D.S.; Bengtsson, T. Glucose Uptake in Brown Fat Cells Is Dependent on MTOR Complex 2-Promoted GLUT1 Translocation. J. Cell Biol. 2014, 207, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Townsend, K.L.; Tseng, Y.-H. Brown Fat Fuel Utilization and Thermogenesis. Trends Endocrinol. Metab. 2014, 25, 168–177. [Google Scholar] [CrossRef]
- Roberts-Toler, C.; O’Neill, B.T.; Cypess, A.M. Diet-Induced Obesity Causes Insulin Resistance in Mouse Brown Adipose Tissue. Obesity 2015, 23, 1765–1770. [Google Scholar] [CrossRef]
- Bryant, N.J.; Gould, G.W. Insulin Stimulated GLUT4 Translocation—Size Is Not Everything! Curr. Opin. Cell Biol. 2020, 65, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Rachdi, L.; Balcazar, N.; Elghazi, L.; Barker, D.J.; Krits, I.; Kiyokawa, H.; Bernal-Mizrachi, E. Differential Effects of P27 in Regulation of Beta-Cell Mass during Development, Neonatal Period, and Adult Life. Diabetes 2006, 55, 3520–3528. [Google Scholar] [CrossRef] [PubMed]
- Georgia, S.; Bhushan, A. P27 Regulates the Transition of Beta-Cells from Quiescence to Proliferation. Diabetes 2006, 55, 2950–2956. [Google Scholar] [CrossRef]
- Cerf, M.E. Beta Cell Dysfunction and Insulin Resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef]
- Nedergaard, J.; Golozoubova, V.; Matthias, A.; Asadi, A.; Jacobsson, A.; Cannon, B. UCP1: The Only Protein Able to Mediate Adaptive Non-Shivering Thermogenesis and Metabolic Inefficiency. Biochim. Biophys. Acta 2001, 1504, 82–106. [Google Scholar] [CrossRef]
- Saito, M. Brown Adipose Tissue as a Regulator of Energy Expenditure and Body Fat in Humans. Diabetes Metab. J. 2013, 37, 22–29. [Google Scholar] [CrossRef]
- Bartness, T.J.; Vaughan, C.H.; Song, C.K. Sympathetic and Sensory Innervation of Brown Adipose Tissue. Int. J. Obes. 2010, 34 (Suppl. S1), S36–S42. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Armengol, J.; Barquissau, V.; Geller, S.; Ji, H.; Severi, I.; Venema, W.; Fenandez, E.A.; Moret, C.; Huber, K.; Leal-Esteban, L.C.; et al. Hypothalamic CDK4 Regulates Thermogenesis by Modulating Sympathetic Innervation of Adipose Tissues. EMBO Rep. 2020, 21, e49807. [Google Scholar] [CrossRef]
- Sav, A.; Rotondo, F.; Syro, L.V.; Scheithauer, B.W.; Kovacs, K. Biomarkers of Pituitary Neoplasms. Anticancer Res. 2012, 32, 4639–4654. [Google Scholar]
- Shen, A.-G.; Shi, S.-X.; Chen, M.-L.; Qin, J.; Gao, S.-F.; Cheng, C. Dynamic Changes of P27(Kip1) and Skp2 Expression in Injured Rat Sciatic Nerve. Cell Mol. Neurobiol. 2008, 28, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Fero, M.L.; Rivkin, M.; Tasch, M.; Porter, P.; Carow, C.E.; Firpo, E.; Polyak, K.; Tsai, L.H.; Broudy, V.; Perlmutter, R.M.; et al. A Syndrome of Multiorgan Hyperplasia with Features of Gigantism, Tumorigenesis, and Female Sterility in P27(Kip1)-Deficient Mice. Cell 1996, 85, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Rajareddy, S.; Reddy, P.; Du, C.; Liu, L.; Jagarlamudi, K.; Tang, W.; Shen, Y.; Berthet, C.; Peng, S.L.; Kaldis, P.; et al. P27kip1 (Cyclin-Dependent Kinase Inhibitor 1B) Controls Ovarian Development by Suppressing Follicle Endowment and Activation and Promoting Follicle Atresia in Mice. Mol. Endocrinol. 2007, 21, 2189–2202. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Aliaga, M.J.; Pérez-Echarri, N.; Marcos-Gómez, B.; Larequi, E.; Gil-Bea, F.J.; Viollet, B.; Gimenez, I.; Martínez, J.A.; Prieto, J.; Bustos, M. Cardiotrophin-1 Is a Key Regulator of Glucose and Lipid Metabolism. Cell Metab. 2011, 14, 242–253. [Google Scholar] [CrossRef]
- Félix-Soriano, E.; Sáinz, N.; Gil-Iturbe, E.; Collantes, M.; Fernández-Galilea, M.; Castilla-Madrigal, R.; Ly, L.; Dalli, J.; Moreno-Aliaga, M.J. Changes in Brown Adipose Tissue Lipid Mediator Signatures with Aging, Obesity, and DHA Supplementation in Female Mice. FASEB J. 2021, 35, e21592. [Google Scholar] [CrossRef]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the Concentration of Low-Density Lipoprotein Cholesterol in Plasma, without Use of the Preparative Ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.L.; Nga, M.E.; Ho, H.K. In Vivo Demonstration of a Novel Non-Invasive Model for Inducing Localized Hypothermia to Ameliorate Hepatotoxicity. Sci. Rep. 2021, 11, 18620. [Google Scholar] [CrossRef]
- Wu, C.; Cheng, W.; Xing, H.; Dang, Y.; Li, F.; Zhu, Z. Brown Adipose Tissue Can Be Activated or Inhibited within an Hour before 18F-FDG Injection: A Preliminary Study with MicroPET. J. Biomed. Biotechnol. 2011, 2011, 159834. [Google Scholar] [CrossRef]
- Corrales, P.; Vivas, Y.; Izquierdo-Lahuerta, A.; Horrillo, D.; Seoane-Collazo, P.; Velasco, I.; Torres, L.; Lopez, Y.; Martínez, C.; López, M.; et al. Long-Term Caloric Restriction Ameliorates Deleterious Effects of Aging on White and Brown Adipose Tissue Plasticity. Aging Cell 2019, 18, e12948. [Google Scholar] [CrossRef]
- Parlee, S.D.; Lentz, S.I.; Mori, H.; MacDougald, O.A. Quantifying Size and Number of Adipocytes in Adipose Tissue. Methods Enzymol. 2014, 537, 93–122. [Google Scholar] [CrossRef]
- McGowan, M.W.; Artiss, J.D.; Strandbergh, D.R.; Zak, B. A Peroxidase-Coupled Method for the Colorimetric Determination of Serum Triglycerides. Clin. Chem. 1983, 29, 538–542. [Google Scholar] [CrossRef]
- Fossati, P.; Prencipe, L. Serum Triglycerides Determined Colorimetrically with an Enzyme That Produces Hydrogen Peroxide. Clin. Chem. 1982, 28, 2077–2080. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT NCD | p27−/− NCD | WT HFD | p27−/− HFD | Gen | Diet | GenxDiet | |
|---|---|---|---|---|---|---|---|
| Total cholesterol (mg/dL) | 112.40 ± 5.61 | 102.80 ± 7.77 | 122.00 ± 7.20 | 121.20 ± 7.11 | ns | ns | ns |
| LDL-cholesterol (mg/dL) | 42.90 ± 3.13 | 43.34 ± 7.63 | 47.34 ± 3.57 | 47.57 ± 4.22 | ns | ns | ns |
| HDL-cholesterol (mg/dL) | 57.27 ± 3.00 | 43.94 ± 2.23 | 56.45 ± 3.86 | 54.54 ± 3.61 | ns | 0.07 | ns |
| β Hydroxybutyrate (mg/dL) | 1.92 ± 0.27 | 1.67 ± 0.47 | 1.38 ± 0.31 | 0.83 ± 0.13 | ns | * | ns |
| Triglycerides (mg/dL) | 91.25 ± 7.30 | 77.60 ± 8.37 | 95.56 ± 6.83 | 93.56 ± 5.61 | ns | ns | ns |
| ALT (U/L) | 41.36 ± 2.75 | 40.44 ± 1.36 | 46.50 ± 3.50 | 55.28 ± 4.91 | ns | * | ns |
| AST (U/L) | 278.20 ± 30.16 | 240.00 ± 20.12 | 397.90 ± 45.81 | 478.10 ± 49.82 | ns | * | ns |
| Gene | Species | Sequence/Reference |
|---|---|---|
| Pparg | Mus musculus | Mm00440940_m1 |
| Fas | Mus musculus | Fw: GCTGCGGAAACTTCAGGAAAT |
| Rv: AGAGACGTGTCACTCCTGGACTT | ||
| Scd1 | Mus musculus | Mm00772290_m1 |
| Hsl | Mus musculus | Fw: CTGCTTCTCCCTCTCGTCTG |
| Rv: CAAAATGGTCCTCTGCCTCT | ||
| Acox | Mus musculus | Fw: CTATGGGATCAGCCAGAAAG |
| Rv: AGTCAAAGGCATCCACCAA | ||
| Cpt1a | Mus musculus | Fw: CACCAACGGGCTCATCTTCTA |
| Rv: CAAAATGACCTAGCCTTCTATCGAA | ||
| Ppara | Mus musculus | Rv: TCAGGGTACCACTACGGAGT |
| Fw: CTTGGCATTCTTCCAAAGCG | ||
| Prdm16 | Mus musculus | Rv: CAGCACGGTGAAGCCATTC |
| Fw: GCGTGCATCCGCTTGTG | ||
| Pgc1a | Mus musculus | Fw: CTAGCCATGGATGGCCTATTT |
| Rv: GTCTCGACACGGAGAGTTAAAG | ||
| Dio2 | Mus musculus | Mm00515664_m1 |
| Glut1 | Mus musculus | Fw: TCAACACGGCCTTCACTG |
| Rv: CACGATGCTCAGATAGGACATC | ||
| Glut4 | Mus musculus | Fw: AAAAGTGCCTGAAACCAGAG |
| Rv: TCACCTCCTGCTCTAAAAGG | ||
| 36b4 | Mus musculus | Fw: CACTGGTCTAGGACCCGAGAAG |
| Rv: GGTGCCTCTGGAGATTTTCG | ||
| Cdk2 | Mus musculus | Fw: GGGTCCATCAAGCTGGCAGA |
| Rv: CCACAGGGTCACCACCTCAT | ||
| Ccne | Mus musculus | Fw: GCATCAGTATGAGATTAGGAATTG |
| Rv: CAGAATGCAGAACTTGAAAATGT | ||
| Ccna | Mus musculus | Rv: CAAGACTCGACGGGTTGCTC |
| Fw: GCTGGCCTCTTCTGAGTCTC | ||
| MKi67 | Mus musculus | Rv: AATCCAACTCAAGTAAACGGGG |
| Fw: TTGGCTTGCTTCCATCCTCA | ||
| Tpx2 | Mus musculus | Fw: CCGAGTGCCCATCAAAGATC |
| Rv: ATGGTGTCAAAATGGGGCAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colon-Mesa, I.; Sainz, N.; Corrales, P.; Collantes, M.; Kaldis, P.; Martinez, J.A.; Medina-Gómez, G.; Moreno-Aliaga, M.J.; Escoté, X. p27Kip1 Deficiency Impairs Brown Adipose Tissue Function Favouring Fat Accumulation in Mice. Int. J. Mol. Sci. 2023, 24, 2664. https://doi.org/10.3390/ijms24032664
Colon-Mesa I, Sainz N, Corrales P, Collantes M, Kaldis P, Martinez JA, Medina-Gómez G, Moreno-Aliaga MJ, Escoté X. p27Kip1 Deficiency Impairs Brown Adipose Tissue Function Favouring Fat Accumulation in Mice. International Journal of Molecular Sciences. 2023; 24(3):2664. https://doi.org/10.3390/ijms24032664
Chicago/Turabian StyleColon-Mesa, Ignacio, Neira Sainz, Patricia Corrales, María Collantes, Philipp Kaldis, José Alfredo Martinez, Gema Medina-Gómez, María Jesús Moreno-Aliaga, and Xavier Escoté. 2023. "p27Kip1 Deficiency Impairs Brown Adipose Tissue Function Favouring Fat Accumulation in Mice" International Journal of Molecular Sciences 24, no. 3: 2664. https://doi.org/10.3390/ijms24032664
APA StyleColon-Mesa, I., Sainz, N., Corrales, P., Collantes, M., Kaldis, P., Martinez, J. A., Medina-Gómez, G., Moreno-Aliaga, M. J., & Escoté, X. (2023). p27Kip1 Deficiency Impairs Brown Adipose Tissue Function Favouring Fat Accumulation in Mice. International Journal of Molecular Sciences, 24(3), 2664. https://doi.org/10.3390/ijms24032664