

Targeting the Brain with Single-Domain Antibodies: Greater Potential Than Stated So Far?

 ,
, 
Abstract
1. Introduction
2. Application of Antibodies in Brain Diseases
2.1. Multiple Sclerosis
2.2. Migraine
2.3. Brain Tumors

{kind=link}
| Antibody | Target | Clinical Status | Dose | Key Findings/Mode of Action | References |
|---|---|---|---|---|---|
| Bevacizumab (Avastin®) Humanized IgG1 mAb | VEGF | Approved by FDA and EMA | i.v. injection 15 mg/kg every 3 weeks In combination with other antibodies | Binds to circulating VEGF and inhibits its binding to VEGFR | [59,60] |
| C-C7 sdAb | Dynactin-1-p150Glued | Preclinical stages | / | Targets selectively a subpopulation of tumor vessels | [61] |
| VH-9.7 VH fragment | Human GSC xenografts. Specific target unknown | Preclinical stages | i.v. injection of 300 pmol in a mouse model with intracerebral xenograft | Localizes local tumor accumulation | [63] |
| Rituximab (Rituxan™) Chimeric mAb | CD20 | Phase III | Infusion of 500 or 1000 mg every 6–12 months | Blocks B lymphocytes’ extravasation | [64,65] |
| Trastuzumab (Herceptin®) Humanized IgG1 mAb | HER2 receptor | Approved by FDA and EMA | Loading dose of 8 mg/kg and 6 mg/kg every 3 weeks for 52 weeks (infusion) | Delays the onset of brain symptoms of metastasis from breast cancer | [66] |
2.4. Alzheimer’s Disease
| Antibody | Target | Clinical Status | Dose | Key Findings/Mode of Action | References |
|---|---|---|---|---|---|
| Bapineuzumab (AAB-001) Humanized IgG1 mAb | N-terminal region of Aβ Targets fibrillar and soluble monomeric forms | Phase III—discontinued | 0.5, 1.5, or 5 mg/kg through i.v. injection, every 13 weeks for 78 weeks | No differences observed compared to placebo groups. Slight clearance of fibrillar cerebral Aβ | [72,81,82,83] |
| Solanezumab (LY2062430) Humanized IgG1 mAb | Mid-domain of Aβ Targets soluble monomeric forms | Phase III—discontinued | i.v. infusion of 400 mg once a month for 80 weeks | No reduced cognitive decline in mild AD patients versus placebo | [73] |
| Crenezumab (MABT5102A) Humanized IgG4 mAb | Aggregated Aβ forms (oligomeric, fibrillar and plaques) | Phase III—discontinued | i.v. infusion of 60 mg/kg every 4 weeks for 100 weeks | Lowering of 20% of cognitive decline. Stabilization of Aβ42 and rise of Aβ40 levels | [74,75] |
| Gantenerumab (RO4909832) Human IgG1 mAb | N-terminal and mid-domain of Aβ. Targets Aβ fibrils | Phase III | s.c. injection of 255 mg or 510 mg every week | Reduces Aβ plaques, CSF total Tau, and phospho-Tau181 | [76,77] |
| Aducanumab (AlduhemTM) Human IgG1 mAb | Aggregated Aβ forms | Approved by FDA—Phase IV confirmatory trial ongoing | Monthly i.v. infusion. Titrated dosing reaching 10 mg/kg at the 7th infusion | Reduction in cognitive decline, Aβ and Tau levels | [78,79,88,89,92,93] |
| Lecanemab (BAN2401) Humanized IgG1 mAb | Large and soluble Aβ protofibrils | Approved by FDA | i.v. infusion of 5 mg/kg or 10 mg/kg every 2 or 4 weeks for 18 months | Reduction of brain Aβ and decreased cognitive decline | [80,94] |
| Gosuranemab (BIIB092) Humanized IgG4 mAb | N-terminal domain of Tau | Phase II—discontinued | i.v. infusion of 2100 mg every 4 weeks for 1.5 years | Increased cognitive decline | [95] |
| Tilavonemab (ABBV-8E12) Humanized IgG4 mAb | N-terminal domain of extracellular aggregated Tau | Phase II—discontinued | i.v. infusion of 2000 or 4000 mg at days 1, 15 and 29, and every 28 days for 1 year | No benefit of antibody over placebo | [96,99] |
| Semorinemab (RO7105705) Humanized IgG4 mAb | Extracellular aggregated Tau | Phase II | i.v. infusion of 1500 mg, 4500 mg, or 8100 mg every 2 weeks for the 3 first infusions, and every 4 weeks for 73 weeks | No changes observed between antibody-treated and placebo patients | [97,98] |
| Zagotenemab (LY3303560) Humanized mAb | N-terminal domain of Tau | Phase II—discontinued | i.v. infusion of 1400 or 5600 mg every 4 weeks for 100 weeks | No benefit of antibody over placebo | [70] |
| R3VQ sdAb | Aβ brain aggregates | Preclinical stages | i.v. infusion of 50 mg/kg to PS2APP mouse model | Crosses the BBB and binds to amyloid aggregates | [103] |
| A2 sdAb | Tau inclusions | Preclinical stages | i.v. infusion of 10 mg/kg to Tg4510 mouse model | Crosses the BBB and binds to NFTs | [103] |
2.5. Parkinson’s Disease
2.6. Creutzfeldt-Jacob Disease
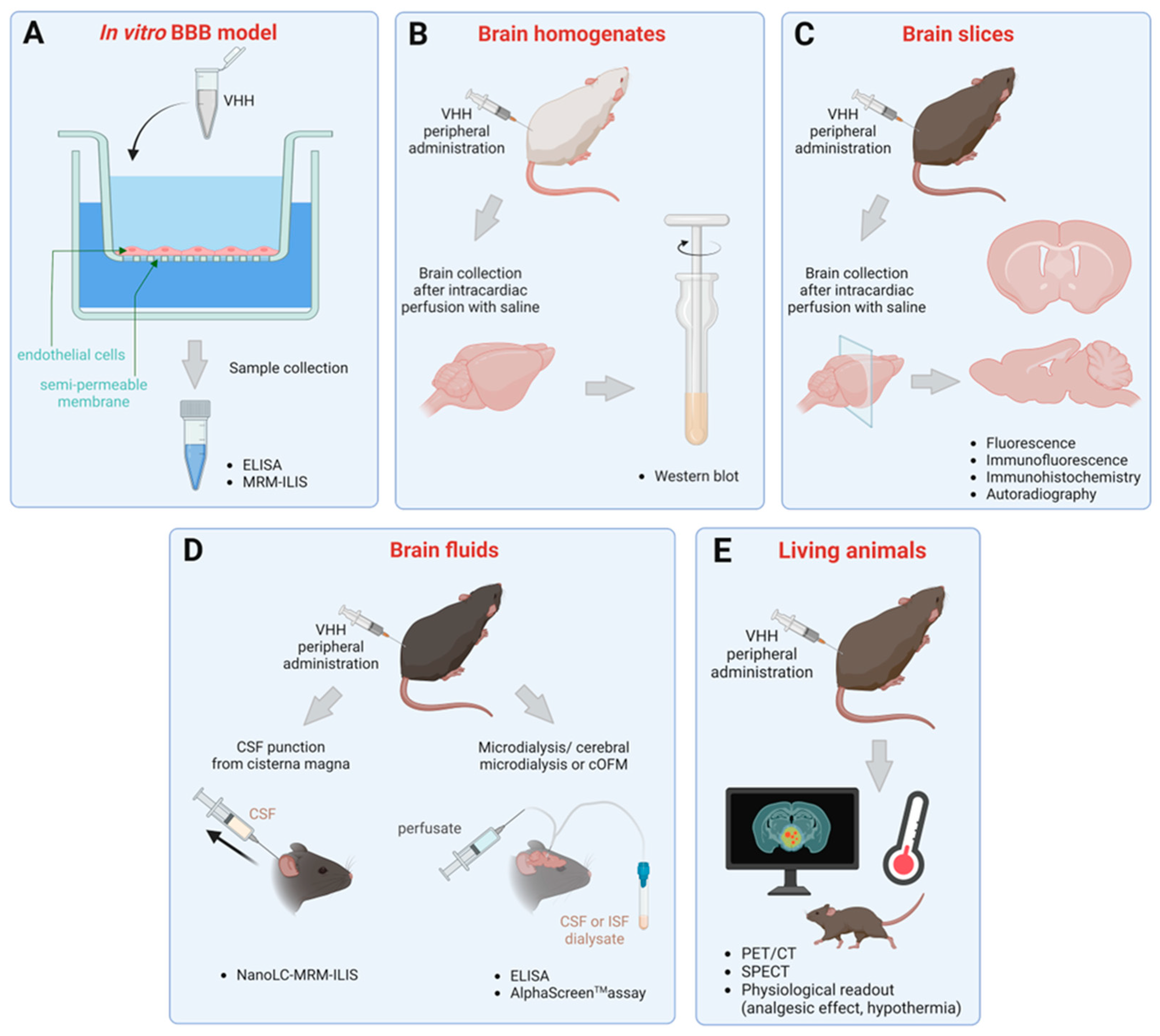
3. Methods to Assess Brain Penetration of sdAbs
3.1. Transmigration across In Vitro BBB Models
3.2. Detection in Brain Homogenates and Slices
3.3. Quantification in Brain Fluids
3.4. Evaluation in Living Animals
4. Conclusions
| Evaluation Method | Protocol | Assay Used | sdAb Evaluated | sdAb Format | Target | Key Findings | References |
|---|---|---|---|---|---|---|---|
| Transmigration across in vitro BBB model | HCEC + medium conditioned by fetal human astrocytes | ELISA | * FC5 | VHH | TMEM-30A | Both FC5 and FC44 cross the in vitro BBB in an energy-dependent manner | [135] |
| ** FC44 | HCEC proteins | ||||||
| SV-ARBEC | Mass spectrometry (MRM-ILIS) | FC5 | TMEM-30A | FC5 crosses more rapidly than FC44 and A20.1. A20.1 did not accumulate over time while FC5 reached the highest accumulation level at 60 min time point | [136] | ||
| FC44 | HCEC proteins | ||||||
| A20.1 | Clostridium difficile toxin A | ||||||
| SV-ARBEC | IGF1R3, IGF1R4, IGF1R5 | Extracellular domain of the human IGF-1R | The three VHHs cross the in vitro BBB, while A20.1 cannot. IGF1R3 shows a 3-fold higher apparent permeability value compared to FC5 | [137,156] | |||
| HCMEC/D3 | ELISA | E9 | Cytosolic human GFAP | 7.8% of the applied quantity of E9 was detected at 60 min time point | [138] | ||
| BCEC + newborn rat astrocytes | ni3a, pa2H | Aβ brain aggregates | VHHs cross the in vitro BBB in an energy-dependent manner and with a higher transmigration velocity than FC5 | [139,140] | |||
| Ex vivo detection after peripheral administration | i.v. administration + brain homogenization | Western blot | FC5 | VHH | TMEM-30A | Both FC5 and FC44 could be detected by western blot after their extraction from brain homogenates by ion affinity chromatography | [135] |
| FC44 | HCEC proteins | ||||||
| i.v. administration + optical tomography sectioning of brain | Fluorescence imaging | FC5 | TMEM-30A | FC5-injected animals showed higher fluorescence in the brain compared to control VHHs (EG2 and A20.1) | [136] | ||
| intracarotid infusion or i.v. administration + brain slicing | Immunostaining | E9 | Cytosolic human GFAP | Astrocytes were immunostained at 1 h post-intracarotid injection (4 and 25 mg/kg). An administration by i.v. route showed immunostaining in the proximity of ventricular regions | [138] | ||
| i.v. administration in P2APP mice + brain slicing | Fluorescence imaging | R3VQ | Aβ brain aggregates | Amyloid plaques were labeled throughout the brain 4 h post-injection | [103] | ||
| i.v. administration in Tg4510 + brain slicing | A2 | Tau inclusions | Neurofibrillary tangle-like structures were stained throughout the brain 4 h post-injection | [103] | |||
| i.p. administration in FVB/N mice + brain homogenization or brain slicing | ELISA and immunohistochemistry | PrioV3 | Isoform scrapie prion protein PrPSc | The immunodetection showed a biphasic pattern in brain homogenates. PrioV3 accumulated in the hippocampus, the alveus, and the cerebellar cortex from 4 to 24 h post-injection | [134] | ||
| i.v. administration in wild type and and ArcSwe mice | Autoradiography | E9 | Cytosolic human GFAP | The radiolabeling signal was more intense throughout the brain of ArcSwe mice at 8 h, but did not show a clear association with the target (GFAP) staining | [141] | ||
| i.v. administration + brain homogenization or brain slicing | ELISA and immunohistochemistry | *** TXB2 fused with human Fc domain | Fc-fused VNAR | Transferrin receptor | A brain concentration of 6 nM was found at 18 h. Immunoreactivity was observed in endothelial cells, choroid plexus epithelial cells and neurons in different regions of the brain | [157] | |
| i.v. administration in a mouse model of brain cancer + brain slicing | Fluorescence imaging | VH-9.7 | VH | Glioblastoma stem-like cells (GSC) | VH-9.7 was localized to the human 22 GSC orthotopic xenografts | [62,64] | |
| Detection in brain fluids after peripheral administration | i.v. administration in rats + CSF sampling | Mass spectrometry (NanoLC -MRM-ILIS) | FC5 | VHH | TMEM-30A | The unlabeled VHHs could be detected at very low amount (1.7 ng/mL) without removing proteins naturally occurring in the matrix. | [136] |
| FC44 | HCEC proteins | ||||||
| EG.2 | EGFR | ||||||
| A20.1 | Clostridium difficile toxin A | ||||||
| i.v. administration in both healthy and rats with encephalitis + hippocampal microdialysate sampling | ELISA + radioactivity measurement | An-33 | Trypanosoma brucei brucei variant-specific surface glycoprotein | Approximately 0.0005% of the administered dose was detected, which is below the therapeutic concentration | [145] | ||
| i.p. administration in mice + followed by microdialysate collection | Alpha-Screen | Nb105 | bi-VHH | Transferrin receptor and green fluorescent protein | A peak concentration was observed at 150 min time point | [146] | |
| i.v. administration or intracarotid infusion in healthy C3HeB/FeJ mice | PET/CT | Nb11 | No specific target in mouse brain | The nanobody showed a higher brain-uptake via intracarotid route but did not accumulate in the controlateral side. Half of the nanobody still remained at 24 h | [151] | ||
| i.v. administration in both healthy and rats with encephalitis | μ-SPECT | An-33 | Trypanosoma brucei brucei variant-specific surface glycoprotein | Only a small portion of the VHH reaches the brain parenchyma in healthy animals (∼0.0005% of the initial dose). This passage is increased in the pathological condition | [145] | ||
| i.v. administration in wild-type and ArcSwe mice | PET | E9 | Cytosolic human GFAP | VHH E9 displayed a brain average concentration of 0.15% ID/g at 2 h post-injection in wild-type mice. The radiolabeling was detected in the brains of both wild-type and ArcSwe mice up to 24 h | [141] | ||
| i.v. administration in P2APP mice | in vivo two-photon imaging | R3VQ | Aβ brain aggregates | Both plaques and vascular Aβ were stained in the cortical surface up to 350 μm deep at 30 min post-injection. It persisted up to 4 h post-injection | [103] | ||
| i.v. administration in Tg4510 | A2 | Tau inclusions | Neurofibrillary tangle-like structures were stained in the cortical surface up to 350 μm at 2 h post-injection. It persisted up to 4 h post-injection | [133] | |||
| i.v. bolus injection in wild-type and APP/PS1 transgenic mice | PET/SPECT | ni3a, pa2H | Aβ brain aggregates | Both ni3a and pa2H show lower brain uptake than FC5 | [140] | ||
| Physiological readout | i.v. administration of the sdAb fused with galanin in the Hargreaves model | Analgesic effect | FC5 | VHH | TMEM-30A | IGF1R4-galanin exhibited a more pronounced analgesic effect than FC5-galanin. This suggests a better uptake of IGF1R4 | [137] |
| IGF1R4 | IGF-1R | ||||||
| i.v. administration of the sdAb fused with galanin or neuropeptide Y in the Hargreaves model | FC5 fused with human Fc domain | FC-fused VHH | TMEM-30A | Systemic administration of FC5-dalagrin induced an analgesic response with a maximal effect obtained after three injections of 7 mg/kg separated 1h apart. Systemic dosing of FC5-neuropeptide Y suppressed thermal hyperalgesia | [152] | ||
| i.v., i.p. and s.c. administration of the sdAb fused with neurotensin in TLR4-/- mice | Hypothermia | Nb62 | VHH | Transferrin receptor | A peak effect was obtained at 100–110 min and 120–180 min after i.v. and i.p. injections, respectively. The drop amplitude is doubled by the i.p. route (–6 °C) and the hypothermic effect lasted 7 h compared to 3 h by i.v. route. The subcutaneous route showed the more prolonged effect | [155] | |
| i.v. administration of the sdAb fused with neurotensin in mice | Hypothermia | *** TXB2 fused with human Fc domain | Fc-fused VNAR | Transferrin receptor | TXB2-hFc induces a drop in temperature in a dose-dependent manner at 2 h time point and returned to normal by 6 h. The minimal dose required to produce hypothermic effect is 10 nmol/kg (0.75 mg/kg) | [157] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| %ID/cc | Percentage of injected dose per cubic centimeter of tissue |
| AAV | Adeno-associated virus |
| AD | Alzheimer’s disease |
| ARIA | Amyloid-related imaging abnormalities |
| Aβ | Amyloid-β peptides |
| BBB | Blood-brain barrier |
| BCEC | Brain capillary endothelial cells |
| CD20 | B-lymphocyte antigen named “cluster of differentiate 20” |
| CD52 | B-lymphocyte antigen named “cluster of differentiate 52” |
| CGRP | Calcitonin gene-related protein |
| CJD | Creutzfeldt-Jakob’s disease |
| CNS | Central nervous system |
| cOFM | Cerebral open flow microperfusion |
| CSF | Cerebrospinal fluid |
| CSFCM | Cerebrospinal fluid of cisterna magma |
| CSFLV | Cerebrospinal fluid of lateral ventricle |
| ELISA | Enzyme-linked immunosorbent assay |
| EMA | European medicines agency |
| Fc | Fragment crystallizable of classical antibodies |
| FDA | Food and drug administration |
| GFAP | Glial fibrillary acidic protein |
| GPCR | G protein-coupled receptor |
| GSC | Glioblastoma stem-like cells |
| HCEC | Human cerebromicroventricular endothelial cells |
| hCMEC/D3 | Human cerebral microvascular endothelial cell line hCMEC/D3 |
| HER2 | Human epidermal growth factor recptor-2 |
| i.p. | Intraperitoneal |
| i.v. | Intravenous |
| IGF1R | Insulin-like growth factor 1 receptor |
| IgG | Immunoglobulin G |
| ILIS | Isotype labeled internal standards |
| ISF | Interstitial fluid |
| ISFST | Interstitial fluid of striatum |
| LINGO-1 | Leucine-rich repeat and immunoglobulin domain-containing protein 1 |
| LRRK2 | Leucine rich-repeat kinase 2 |
| mAb | Monoclonal antibody |
| MRM | Multiple reaction monitoring (also known as SRM) |
| MS | Multiple sclerosis |
| MSRV-Env protein | Multiple sclerosis associated retrovirus envelope protein |
| NanoLC-SRM-ILIS | Nano-liquid chromatography coupled with selective reaction monitoring and isotype labeled internal standards |
| NFT | Neurofibrillary tangles |
| PCNSL | Primary central nervous system lymphoma |
| PD | Parkinson’s disease |
| PET/CT | Positron emission tomography/computed tomography |
| PET | Positron emission tomography |
| PrPC | Cellular isoform of the prion protein |
| PrPSc | Scrapie isoform of the prion protein |
| RMGa | Repulsive guidance molecule A |
| s.c. | Subcutaneous |
| sdAb | Single-domain antibody |
| SPECT | Single-photon emission computed tomography |
| SRM | Selective reaction monitoring (also known as MRM) |
| SV-ARBEC | SV40-immortalized adult rat brain endothelial cells |
| TLR4 | Toll-like receptor 4 |
| TMEM-30A | Transmembrane protein 30A |
| VCAM | Vascular cell adhesion molecule |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| VH | Heavy chain variable domain of mammalian antibody |
| VHH | Heavy chain variable domain of camelid’s antibody |
| VLA-4 | Very late antigen-4 |
| VNAR | Heavy chain variable domain of sharks’ antibody, also known as variable domain of new antigen receptor |
References
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. Neurotherapeutics 2005, 2, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRX 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kouhi, A.; Pachipulusu, V.; Kapenstein, T.; Hu, P.; Epstein, A.L.; Khawli, L.A. Brain disposition of antibody-based therapeutics: Dogma, approaches and perspectives. Int. J. Mol. Sci. 2021, 22, 6442. [Google Scholar] [CrossRef]
- Yu, Y.J.; Watts, R.J. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 2013, 10, 459–472. [Google Scholar] [CrossRef]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, C.; Muyldermans, S. Nanobody-based delivery systems for diagnosis and targeted tumor therapy. Front. Immunol. 2017, 8, 1442. [Google Scholar] [CrossRef]
- Jovčevska, I.; Muyldermans, S. The therapeutic potential of nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef]
- Menzel, S.; Schwarz, N.; Haag, F.; Koch-Nolte, F. Nanobody-based biologics for modulating purinergic signaling in inflammation and immunity. Front. Pharmacol. 2018, 9, 266. [Google Scholar] [CrossRef]
- Uchański, T.; Pardon, E.; Steyaert, J. Nanobodies to study protein conformational states. Curr. Opin. Struct. Biol. 2020, 60, 117–123. [Google Scholar] [CrossRef]
- Wu, Y.; Jiang, S.; Ying, T. Single-domain antibodies as therapeutics against human viral diseases. Front. Immunol. 2017, 8, 1802. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Avila, D.; Hughes, M.; Hughes, A.; McKinney, E.C.; Flajnik, M.F. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature 1995, 374, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Pothin, E.; Lesuisse, D.; Lafaye, P. Brain delivery of single-domain antibodies: A focus on VHH and VNAR. Pharmaceutics 2020, 12, 937. [Google Scholar] [CrossRef]
- Vu, K.B.; Ghahroudi, M.A.; Wyns, L.; Muyldermans, S. Comparison of llama VH sequences from conventional and heavy chain antibodies. Mol. Immunol. 1997, 34, 1121–1131. [Google Scholar] [CrossRef]
- Cheong, W.S.; Leow, C.Y.; Abdul Majeed, A.B.; Leow, C.H. Diagnostic and therapeutic potential of shark variable new antigen receptor (VNAR) single domain antibody. Int. J. Biol. Macromol. 2020, 147, 369–375. [Google Scholar] [CrossRef]
- Desmyter, A.; Transue, T.R.; Ghahroudi, M.A.; Dao Thi, M.-H.; Poortmans, F.; Hamers, R.; Muyldermans, S.; Wyns, L. Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat. Struct. Mol. Biol. 1996, 3, 803–811. [Google Scholar] [CrossRef]
- Stanfield, R.L.; Dooley, H.; Flajnik, M.F.; Wilson, I.A. Crystal Structure of a shark single-domain antibody V region in complex with lysozyme. Science 2004, 305, 1770–1773. [Google Scholar] [CrossRef]
- Streltsov, V.A.; Carmichael, J.A.; Nuttall, S.D. Structure of a shark IgNAR antibody variable domain and modeling of an early-developmental isotype. Protein Sci. 2005, 14, 2901–2909. [Google Scholar] [CrossRef]
- Dang, V.C.; Christie, M.J. Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons. Br. J. Pharmacol. 2012, 165, 1704–1716. [Google Scholar] [CrossRef]
- Rao, M.S.; Gupta, R.; Liguori, M.J.; Hu, M.; Huang, X.; Mantena, S.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Van Vleet, T.R. Novel computational approach to predict off-target interactions for small molecules. Front. Big Data 2019, 2, 25. [Google Scholar] [CrossRef] [PubMed]
- Scholler, P.; Nevoltris, D.; de Bundel, D.; Bossi, S.; Moreno-Delgado, D.; Rovira, X.; Møller, T.C.; El Moustaine, D.; Mathieu, M.; Blanc, E.; et al. Allosteric Nanobodies uncover a role of hippocampal mGlu2 receptor homodimers in contextual fear consolidation. Nat. Commun. 2017, 8, 1967. [Google Scholar] [CrossRef]
- Haubrich, J.; Font, J.; Quast, R.B.; Goupil-Lamy, A.; Scholler, P.; Nevoltris, D.; Acher, F.; Chames, P.; Rondard, P.; Prézeau, L.; et al. A nanobody activating metabotropic glutamate receptor 4 discriminates between homo- and heterodimers. Proc. Natl. Acad. Sci. USA 2021, 118, e2105848118. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Xu, C.; Lafon, P.-A.; Roux, S.; Mathieu, M.; Zhou, R.; Scholler, P.; Blanc, E.; Becker, J.A.J.; Le Merrer, J.; et al. Nanobody-based sensors reveal a high proportion of mGlu heterodimers in the brain. Nat. Chem. Biol. 2022, 18, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Nickols, H.H.; Conn, P.J. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol. Dis. 2014, 61, 55–71. [Google Scholar] [CrossRef]
- McCormack, P.L. Natalizumab: A review of its use in the management of relapsing-remitting multiple sclerosis. Drugs 2013, 73, 1463–1481. [Google Scholar] [CrossRef]
- Voge, N.V.; Alvarez, E. Monoclonal antibodies in multiple sclerosis: Present and future. Biomedicines 2019, 7, 20. [Google Scholar] [CrossRef]
- He, D.; Guo, R.; Zhang, F.; Zhang, C.; Dong, S.; Zhou, H. Rituximab for relapsing-remitting multiple sclerosis. Cochrane Database Syst. Rev. 2013, 2, CD009130. [Google Scholar] [CrossRef]
- Di Gaetano, N.; Cittera, E.; Nota, R.; Vecchi, A.; Grieco, V.; Scanziani, E.; Botto, M.; Introna, M.; Golay, J. Complement activation determines the therapeutic activity of Rituximab in vivo. J. Immunol. 2003, 171, 1581–1587. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef]
- Fox, E.; Lovett-Racke, A.E.; Gormley, M.; Liu, Y.; Petracca, M.; Cocozza, S.; Shubin, R.; Wray, S.; Weiss, M.S.; Bosco, J.A.; et al. A phase 2 multicenter study of Ublituximab, a novel glycoengineered anti-CD20 monoclonal antibody, in patients with relapsing forms of multiple sclerosis. Mult. Scler. 2021, 27, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.-P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.A. B-cell depletion—A frontier in monoclonal antibodies for multiple sclerosis. N. Engl. J. Med. 2017, 376, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, H.V.; Rosebraugh, M.R.; Misko, T.P.; Ziemann, A.; Liu, W.; Cree, B.A.C. Phase 1 evaluation of Elezanumab (anti–repulsive guidance molecule monoclonal antibody) in healthy and multiple sclerosis participants. Ann. Neurol. 2022, 93, 285–296. [Google Scholar] [CrossRef]
- Curtin, F.; Lang, A.B.; Perron, H.; Laumonier, M.; Vidal, V.; Porchet, H.C.; Hartung, H.-P. GNbAC1, a humanized monoclonal antibody against the envelope protein of multiple sclerosis—Associated endogenous retrovirus: A first-in-humans randomized clinical study. Clin. Ther. 2012, 34, 2268–2278. [Google Scholar] [CrossRef]
- Mi, S.; Lee, X.; Shao, Z.; Thill, G.; Ji, B.; Relton, J.; Levesque, M.; Allaire, N.; Perrin, S.; Sands, B.; et al. LINGO-1 is a component of the Nogo-66 receptor/P75 signaling complex. Nat. Neurosci. 2004, 7, 221–228. [Google Scholar] [CrossRef]
- Zhu, S.; Huang, A.-G.; Luo, F.; Li, J.; Li, J.; Zhu, L.; Zhao, L.; Zhu, B.; Ling, F.; Wang, G.-X. Application of virus targeting nanocarrier drug delivery system in virus-induced central nervous system disease treatment. ACS Appl. Mater. Interfaces 2019, 11, 19006–19016. [Google Scholar] [CrossRef]
- Cadavid, D.; Mellion, M.; Hupperts, R.; Edwards, K.R.; Calabresi, P.A.; Drulović, J.; Giovannoni, G.; Hartung, H.-P.; Arnold, D.L.; Fisher, E.; et al. Safety and efficacy of Opicinumab in patients with relapsing multiple sclerosis (SYNERGY): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2019, 18, 845–856. [Google Scholar] [CrossRef]
- Ahmed, Z.; Fulton, D.; Douglas, M.R. Opicinumab: Is it a potential treatment for multiple sclerosis? Ann. Transl. Med. 2020, 8, 892. [Google Scholar] [CrossRef]
- Bar-Or, A.; Grove, R.A.; Austin, D.J.; Tolson, J.M.; VanMeter, S.A.; Lewis, E.W.; Derosier, F.J.; Lopez, M.C.; Kavanagh, S.T.; Miller, A.E.; et al. Subcutaneous Ofatumumab in patients with relapsing-remitting multiple sclerosis: The MIRROR study. Neurology 2018, 90, e1805–e1814. [Google Scholar] [CrossRef]
- Lipton, R.B.; Bigal, M.E.; Diamond, M.; Freitag, F.; Reed, M.L.; Stewart, W.F. AMPP Advisory group migraine prevalence, disease burden, and the need for preventive therapy. Neurology 2007, 68, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Lipton, R.B.; Stewart, W.F.; Diamond, S.; Diamond, M.L.; Reed, M. Prevalence and burden of migraine in the United States: Data from the American migraine study II. Headache 2001, 41, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: A systematic analysis for the global burden of disease study 2013. Lancet 2015, 386, 743–800. [Google Scholar] [CrossRef] [PubMed]
- Stewart, W.F.; Ricci, J.A.; Chee, E.; Morganstein, D.; Lipton, R. Lost productive time and cost due to common pain conditions in the US workforce. JAMA 2003, 290, 2443–2454. [Google Scholar] [CrossRef]
- Gustavsson, A.; Svensson, M.; Jacobi, F.; Allgulander, C.; Alonso, J.; Beghi, E.; Dodel, R.; Ekman, M.; Faravelli, C.; Fratiglioni, L.; et al. Cost of disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 718–779. [Google Scholar] [CrossRef]
- Ho, T.W.; Edvinsson, L.; Goadsby, P.J. CGRP and its receptors provide new insights into migraine pathophysiology. Nat. Rev. Neurol. 2010, 6, 573–582. [Google Scholar] [CrossRef]
- Cavaco, M.; Gaspar, D.; ARB Castanho, M.; Neves, V. Antibodies for the treatment of brain metastases, a dream or a reality? Pharmaceutics 2020, 12, 62. [Google Scholar] [CrossRef]
- Dodick, D.W. A phase-by-phase review of migraine pathophysiology. J. Head Face Pain 2018, 58, 4–16. [Google Scholar] [CrossRef]
- Ashina, M.; Saper, J.; Cady, R.; Schaeffler, B.A.; Biondi, D.M.; Hirman, J.; Pederson, S.; Allan, B.; Smith, J. Eptinezumab in episodic migraine: A randomized, double-blind, placebo-controlled study (PROMISE-1). Cephalalgia 2020, 40, 241–254. [Google Scholar] [CrossRef]
- Detke, H.C.; Goadsby, P.J.; Wang, S.; Friedman, D.I.; Selzler, K.J.; Aurora, S.K. Galcanezumab in chronic migraine. Neurology 2018, 91, e2211–e2221. [Google Scholar] [CrossRef]
- Dodick, D.W.; Silberstein, S.D.; Bigal, M.E.; Yeung, P.P.; Goadsby, P.J.; Blankenbiller, T.; Grozinski-Wolff, M.; Yang, R.; Ma, Y.; Aycardi, E. Effect of Fremanezumab compared with placebo for prevention of episodic migraine. JAMA 2018, 319, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Gklinos, P.; Papadopoulou, M.; Stanulovic, V.; Mitsikostas, D.D.; Papadopoulos, D. Monoclonal antibodies as neurological therapeutics. Pharmaceuticals 2021, 14, 92. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J.; Reuter, U.; Hallström, Y.; Broessner, G.; Bonner, J.H.; Zhang, F.; Sapra, S.; Picard, H.; Mikol, D.D.; Lenz, R.A. A controlled trial of Erenumab for episodic migraine. N. Engl. J. Med. 2017, 377, 2123–2132. [Google Scholar] [CrossRef] [PubMed]
- Skljarevski, V.; Matharu, M.; Millen, B.A.; Ossipov, M.H.; Kim, B.-K.; Yang, J.Y. Efficacy and safety of Galcanezumab for the prevention of episodic migraine: Results of the EVOLVE-2 phase 3 randomized controlled clinical trial. Cephalalgia 2018, 38, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, V.L.; Dodick, D.W.; Zhang, Q.; Carter, J.N.; Ailani, J.; Conley, R.R. Evaluation of Galcanezumab for the prevention of episodic migraine. JAMA Neurol. 2018, 75, 1080–1088. [Google Scholar] [CrossRef]
- Dhillon, S. Eptinezumab: First approval. Drugs 2020, 80, 733–739. [Google Scholar] [CrossRef]
- Reuter, U. A review of monoclonal antibody therapies and other preventative treatments in migraine. J. Head Face Pain 2018, 58, 48–59. [Google Scholar] [CrossRef]
- Ray, J.C.; Allen, P.; Bacsi, A.; Bosco, J.J.; Chen, L.; Eller, M.; Kua, H.; Lim, L.L.; Matharu, M.S.; Monif, M.; et al. Inflammatory complications of CGRP monoclonal antibodies: A case series. J. Headache Pain 2021, 22, 121. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in progressive glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- van Lith, S.A.M.; Roodink, I.; Verhoeff, J.J.C.; Mäkinen, P.I.; Lappalainen, J.P.; Ylä-Herttuala, S.; Raats, J.; van Wijk, E.; Roepman, R.; Letteboer, S.J.; et al. In vivo phage display screening for tumor vascular targets in glioblastoma identifies a llama nanobody against dynactin-1-P150Glued. Oncotarget 2016, 7, 71594–71607. [Google Scholar] [CrossRef] [PubMed]
- Zorniak, M.; Clark, P.A.; Cho, Y.P.; Umlauf, B.J.; Shusta, E.V.; Huo, J.S.-S. Antibodies Targeting Glioblastoma Stem-Like Cells and Methods of Use Thereof. Patent WO2019074892A1, 18 April 2019. [Google Scholar]
- Zorniak, M.; Clark, P.A.; Umlauf, B.J.; Cho, Y.; Shusta, E.V.; Kuo, J.S. Yeast display biopanning identifies human antibodies targeting glioblastoma stem-like cells. Sci. Rep. 2017, 7, 15840. [Google Scholar] [CrossRef]
- Grommes, C.; Rubenstein, J.L.; DeAngelis, L.M.; Ferreri, A.J.M.; Batchelor, T.T. Comprehensive approach to diagnosis and treatment of newly diagnosed primary CNS lymphoma. Neuro. Oncol. 2019, 21, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Lampson, L.A. Monoclonal antibodies in neuro-oncology. mAbs 2011, 3, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Duchnowska, R.; Loibl, S.; Jassem, J. Tyrosine kinase inhibitors for brain metastases in HER2-positive breast cancer. Cancer Treat. Rev. 2018, 67, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Barker, W.; Luis, C.; Kashuba, A.; Luis, M.; Harwood, D.; Loewenstein, D.; Waters, C.; Jimison, P.; Shepherd, E.; Sevush, S.; et al. Relative frequencies of Alzheimer disease, lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida brain bank. Alz. Dis. Assoc. Disord. 2002, 16, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease International. World Alzheimer Report 2022. Available online: https://www.alzint.org/u/World-Alzheimer-Report-2022.pdf (accessed on 22 November 2021).
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid hypothesis in Alzheimer disease: New insights from new therapeutics. Nat. Rev. Drug Discov. 2022, 21, 306–318. [Google Scholar] [CrossRef]
- Hu, C.; Adedokun, O.; Ito, K.; Raje, S.; Lu, M. Confirmatory population pharmacokinetic analysis for Bapineuzumab phase 3 studies in patients with mild to moderate Alzheimer’s disease. J. Clin. Pharmacol. 2015, 55, 221–229. [Google Scholar] [CrossRef]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for mild dementia due to Alzheimer’s disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef]
- Yoshida, K.; Moein, A.; Bittner, T.; Ostrowitzki, S.; Lin, H.; Honigberg, L.; Jin, J.Y.; Quartino, A. Pharmacokinetics and pharmacodynamic effect of Crenezumab on plasma and cerebrospinal fluid beta-amyloid in patients with mild-to-moderate Alzheimer’s disease. Alz. Res. Ther. 2020, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Ostrowitzki, S.; Bittner, T.; Sink, K.M.; Mackey, H.; Rabe, C.; Honig, L.S.; Cassetta, E.; Woodward, M.; Boada, M.; van Dyck, C.H.; et al. Evaluating the safety and efficacy of Crenezumab vs placebo in adults with early Alzheimer disease: Two phase 3 randomized placebo-controlled trials. JAMA Neurol. 2022, 79, 1113. [Google Scholar] [CrossRef]
- Salloway, S.; Farlow, M.; McDade, E.; Clifford, D.B.; Wang, G.; Llibre-Guerra, J.J.; Hitchcock, J.M.; Mills, S.L.; Santacruz, A.M.; Aschenbrenner, A.J.; et al. A trial of Gantenerumab or Solanezumab in dominantly inherited Alzheimer’s disease. Nat. Med. 2021, 27, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Cummings, J.; Schobel, S.; Salloway, S.; Vellas, B.; Boada, M.; Black, S.E.; Blennow, K.; Fontoura, P.; Klein, G.; et al. Gantenerumab: An anti-amyloid monoclonal antibody with potential disease-modifying effects in early Alzheimer’s disease. Alz. Res. Ther. 2022, 14, 178. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Banerjee, D. A primer on the evolution of Aducanumab: The first antibody approved for treatment of Alzheimer’s disease. J. Alzeimer’s Dis. 2021, 83, 1537–1552. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, J.; Williams, L.; Stella, H.; Leitermann, K.; Mikulskis, A.; O’Gorman, J.; Sevigny, J. First-in-human, double-blind, placebo-controlled, single-dose escalation study of Aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimer’s Dement. 2016, 2, 169–176. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with Lecanemab, an anti-Aβ protofibril antibody. Alz. Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Salloway, S.; Marshall, G.A.; Lu, M.; Brashear, H.R. Long-Term safety and efficacy of Bapineuzumab in patients with mild-to-moderate Alzheimer’s disease: A phase 2, open-label extension study. Curr. Alz. Res. 2018, 15, 1231–1243. [Google Scholar] [CrossRef]
- Sperling, R.A.; Jack, C.R.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s association research roundtable workgroup. Alzheimer’s Dement. 2011, 7, 367–385. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D.; et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alz. Res. Ther. 2016, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Roche. Roche to Discontinue Phase III CREAD 1 and 2 Clinical Studies of Crenezumab in Early Alzheimer’s Disease (AD). Available online: https://www.roche.com/media/releases/med-cor-2019-01-30 (accessed on 14 April 2022).
- ClinicalTrials.gov. A Phase III, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group, Efficacy, and Safety Study of Gantenerumab in Patients with Early (Prodromal to Mild) Alzheimer’s Disease. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03444870 (accessed on 13 April 2022).
- ClinicalTrials.gov. A Placebo-Controlled, Double-Blind, Parallel-Group, 18-Month Study with an Open-Label Extension Phase to Confirm Safety and Efficacy of BAN2401 in Subjects with Early Alzheimer’s Disease. 2022. Available online: clinicaltrials.gov (accessed on 13 April 2022).
- ClinicalTrials.gov. A Phase III, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group, Safety, and Efficacy Study of Gantenerumab in Patients with Early (Prodromal to Mild) Alzheimer’s Disease. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03443973 (accessed on 13 April 2022).
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody Aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Controversial Alzheimer’s drug approval could affect other diseases. Nature 2021, 595, 162–163. [Google Scholar] [CrossRef] [PubMed]
- EMA. Aduhelm: Withdrawn Application. Available online: https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/aduhelm (accessed on 26 June 2022).
- FDA. Aducanumab (Marketed as Aduhelm) Information; FDA: Silver Spring, MD, USA, 2021. [Google Scholar]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating Aducanumab in patients with early Alzheimer disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef]
- Silvestro, S.; Valeri, A.; Mazzon, E. Aducanumab and its effects on Tau pathology: Is this the turning point of amyloid hypothesis? Int. J. Mol. Sci. 2022, 23, 2011. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Dam, T.; Boxer, A.L.; Golbe, L.I.; Höglinger, G.U.; Morris, H.R.; Litvan, I.; Lang, A.E.; Corvol, J.-C.; Aiba, I.; Grundman, M.; et al. Safety and efficacy of anti-Tau monoclonal antibody Gosuranemab in progressive supranuclear palsy: A phase 2, randomized, placebo-controlled trial. Nat. Med. 2021, 27, 1451–1457. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Litvan, I.; Mendonca, N.; Wang, D.; Zheng, H.; Rendenbach-Mueller, B.; Lon, H.-K.; Jin, Z.; Fisseha, N.; Budur, K.; et al. Safety and efficacy of Tilavonemab in progressive supranuclear palsy: A phase 2, randomised, placebo-controlled trial. Lancet Neurol. 2021, 20, 182–192. [Google Scholar] [CrossRef]
- Slomski, A. Anti-Tau antibody Semorinemab fails to slow Alzheimer disease. JAMA 2022, 328, 415. [Google Scholar] [CrossRef]
- Ayalon, G.; Lee, S.-H.; Adolfsson, O.; Foo-Atkins, C.; Atwal, J.K.; Blendstrup, M.; Booler, H.; Bravo, J.; Brendza, R.; Brunstein, F.; et al. Antibody Semorinemab reduces Tau pathology in a transgenic mouse model and engages Tau in patients with Alzheimer’s disease. Sci. Transl. Med. 2021, 13, eabb2639. [Google Scholar] [CrossRef]
- Clinical Trials. A Phase 2 Multiple Dose, Multicenter, Randomized, Double-blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of ABBV-8E12 in Subjects With Early Alzheimer’s Disease. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT02880956 (accessed on 8 September 2022).
- Clinical Trials. Assessment of Safety, Tolerability, and Efficacy of LY3303560 in Early Symptomatic Alzheimer’s Disease. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03518073 (accessed on 23 June 2022).
- Ackaert, C.; Smiejkowska, N.; Xavier, C.; Sterckx, Y.G.J.; Denies, S.; Stijlemans, B.; Elkrim, Y.; Devoogdt, N.; Caveliers, V.; Lahoutte, T.; et al. Immunogenicity risk profile of nanobodies. Front. Immunol. 2021, 12, 632687. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, K.; Iqbal, U.; Tanha, J.; MacKenzie, R.; Moreno, M.; Stanimirovic, D. Single-domain antibodies as therapeutic and imaging agents for the treatment of CNS diseases. Antibodies 2019, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Vandesquille, M.; Koukouli, F.; Dudeffant, C.; Youssef, I.; Lenormand, P.; Ganneau, C.; Maskos, U.; Czech, C.; Grueninger, F.; et al. Camelid single-domain antibodies: A versatile tool for in vivo imaging of extracellular and intracellular brain targets. J. Control Release 2016, 243, 1–10. [Google Scholar] [CrossRef]
- Gibb, W.R.; Lees, A.J. The relevance of the lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. 1988, 51, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Castonguay, A.-M.; Gravel, C.; Lévesque, M. Treating Parkinson’s disease with antibodies: Previous studies and future directions. J. Parkinsons Dis. 2021, 11, 71–92. [Google Scholar] [CrossRef]
- Lee, H.-J.; Bae, E.-J.; Lee, S.-J. Extracellular α-Synuclein—A novel and crucial factor in lewy body diseases. Nat. Rev. Neurol. 2014, 10, 92–98. [Google Scholar] [CrossRef]
- Lang, A.E.; Siderowf, A.D.; Macklin, E.A.; Poewe, W.; Brooks, D.J.; Fernandez, H.H.; Rascol, O.; Giladi, N.; Stocchi, F.; Tanner, C.M.; et al. Trial of Cinpanemab in early Parkinson’s disease. N. Engl. J. Med. 2022, 387, 408–420. [Google Scholar] [CrossRef]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti–α-synuclein monoclonal antibody, in patients with Parkinson disease. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef]
- Hoffmann-La Roche. A Randomized, Double-Blind, Placebo-Controlled, 52-Week Phase II Study to Evaluate the Efficacy of Intravenous RO7046015/Prasinezumab (PRX002) in Participants with Early Parkinson’s Disease with a 6-Year All-Participants-On-Treatment Extension. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03100149 (accessed on 8 September 2022).
- Hoffmann-La Roche. A Phase IIB, Randomized, Double-Blind, Placebo-Controlled, Multicenter Study to Evaluate the Efficacy and Safety of Intravenous Prasinezumab in Participants with Early Parkinson’s Disease. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04777331 (accessed on 8 September 2022).
- AstraZeneca. A Randomized, Double-Blind, Placebo-Controlled Study of the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Multiple Ascending Doses of MEDI1341 in Subjects with Parkinson’s Disease. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04449484 (accessed on 8 September 2022).
- Butler, Y.R.; Liu, Y.; Kumbhar, R.; Zhao, P.; Gadhave, K.; Wang, N.; Li, Y.; Mao, X.; Wang, W. α-synuclein fibril-specific nanobody reduces prion-like α-synuclein spreading in mice. Nat. Commun. 2022, 13, 4060. [Google Scholar] [CrossRef]
- De Genst, E.J.; Guilliams, T.; Wellens, J.; O’Day, E.M.; Waudby, C.A.; Meehan, S.; Dumoulin, M.; Hsu, S.-T.D.; Cremades, N.; Verschueren, K.H.G.; et al. Structure and properties of a complex of α-synuclein and a single-domain camelid antibody. J. Mol. Biol. 2010, 402, 326–343. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, T.; El-Turk, F.; Buell, A.K.; O’Day, E.M.; Aprile, F.A.; Esbjörner, E.K.; Vendruscolo, M.; Cremades, N.; Pardon, E.; Wyns, L.; et al. Nanobodies raised against monomeric α-synuclein distinguish between fibrils at different maturation stages. J. Mol. Biol. 2013, 425, 2397–2411. [Google Scholar] [CrossRef]
- Chatterjee, D.; Bhatt, M.; Butler, D.; De Genst, E.; Dobson, C.M.; Messer, A.; Kordower, J.H. Proteasome-targeted nanobodies alleviate pathology and functional decline in an α-synuclein-based Parkinson’s disease model. npj Park. Dis. 2018, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.C.; Joshi, S.N.; Genst, E.D.; Baghel, A.S.; Dobson, C.M.; Messer, A. Bifunctional anti-non-amyloid component α-synuclein nanobodies are protective in situ. PLoS ONE 2016, 11, e0165964. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, C.; Waal, N.; Offner, T.; Fornasiero, E.F.; Wender, N.; Verbarg, H.; Manzini, I.; Trenkwalder, C.; Mollenhauer, B.; Strohäker, T.; et al. A nanobody-based fluorescent reporter reveals human α-synuclein in the cell cytosol. Nat. Commun. 2020, 11, 2729. [Google Scholar] [CrossRef] [PubMed]
- Iljina, M.; Hong, L.; Horrocks, M.H.; Ludtmann, M.H.; Choi, M.L.; Hughes, C.D.; Ruggeri, F.S.; Guilliams, T.; Buell, A.K.; Lee, J.-E.; et al. Nanobodies raised against monomeric α-synuclein inhibit fibril formation and destabilize toxic oligomeric species. BMC Biol. 2017, 15, 57. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Singh, R.K.; Soliman, A.; Guaitoli, G.; Störmer, E.; von Zweydorf, F.; Dal Maso, T.; Oun, A.; Van Rillaer, L.; Schmidt, S.H.; Chatterjee, D.; et al. Nanobodies as allosteric modulators of Parkinson’s disease-associated LRRK2. Proc. Natl. Acad. Sci. USA 2022, 119, e2112712119. [Google Scholar] [CrossRef]
- Spagnolli, G.; Requena, J.R.; Biasini, E. Understanding prion structure and conversion. Prog. Mol. Biol. Transl. Sci. 2020, 175, 19–30. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhu, J.; Lu, H. Single domain antibody-based vectors in the delivery of biologics across the blood–brain barrier: A review. Drug Deliv. Transl. Res. 2021, 11, 1818–1828. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef]
- Deleault, N.R.; Walsh, D.J.; Piro, J.R.; Wang, F.; Wang, X.; Ma, J.; Rees, J.R.; Supattapone, S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. USA 2012, 109, E1938–E1946. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D.; Pepe, A.; Zurzolo, C. Cell biology of prion protein. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2017; Volume 150, pp. 57–82. ISBN 978-0-12811-226-7. [Google Scholar] [CrossRef]
- White, A.R.; Enever, P.; Tayebi, M.; Mushens, R.; Linehan, J.; Brandner, S.; Anstee, D.; Collinge, J.; Hawke, S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003, 422, 80–83. [Google Scholar] [CrossRef]
- Ma, Y.; Ma, J. Immunotherapy against prion disease. Pathogens 2020, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Song, C.-H.; Furuoka, H.; Kim, C.-L.; Ogino, M.; Suzuki, A.; Hasebe, R.; Horiuchi, M. Effect of intraventricular infusion of anti-prion protein monoclonal antibodies on disease progression in prion-infected mice. J. Gen. Virol. 2008, 89, 1533–1544. [Google Scholar] [CrossRef]
- Dyer, C. British man with CJD gets experimental treatment in world first. BMJ 2018, 363, k4608. [Google Scholar] [CrossRef]
- Mead, S.; Khalili-Shirazi, A.; Potter, C.; Mok, T.; Nihat, A.; Hyare, H.; Canning, S.; Schmidt, C.; Campbell, T.; Darwent, L.; et al. Prion protein monoclonal antibody (PRN100) therapy for Creutzfeldt–Jakob disease: Evaluation of a first-in-human treatment programme. Lancet Neurol. 2022, 21, 342–354. [Google Scholar] [CrossRef]
- David, M.A.; Jones, D.R.; Tayebi, M. Potential candidate camelid antibodies for the treatment of protein-misfolding diseases. J. Neuroimmunol. 2014, 272, 76–85. [Google Scholar] [CrossRef]
- Muruganandam, A.; Tanha, J.; Narang, S.; Stanimirovic, D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB J. 2002, 16, 240–242. [Google Scholar] [CrossRef]
- Haqqani, A.S.; Caram-Salas, N.; Ding, W.; Brunette, E.; Delaney, C.E.; Baumann, E.; Boileau, E.; Stanimirovic, D. Multiplexed evaluation of serum and CSF pharmacokinetics of brain-targeting single-domain antibodies using a nanoLC-SRM-ILIS method. Mol. Pharm. 2013, 10, 1542–1556. [Google Scholar] [CrossRef]
- Stanimirovic, D.; Kemmerich, K.; Haqqani, A.S.; Sulea, T.; Arbabi-Ghahroudi, M.; Massie, B.; Gilbert, R. Insulin-Like Growth Factor 1 Receptor-Specific Antibodies and Uses Thereof. Patent NZ724866A, 29 April 2022. [Google Scholar]
- Li, T.; Bourgeois, J.-P.; Celli, S.; Glacial, F.; Le Sourd, A.-M.; Mecheri, S.; Weksler, B.; Romero, I.; Couraud, P.-O.; Rougeon, F.; et al. Cell-penetrating anti-GFAP VHH and corresponding fluorescent fusion protein VHH-GFP spontaneously cross the blood-brain barrier and specifically recognize astrocytes: Application to brain imaging. FASEB J. 2012, 26, 3969–3979. [Google Scholar] [CrossRef]
- Nabuurs, R.J.A.; Rutgers, K.S.; Welling, M.M.; Metaxas, A.; de Backer, M.E.; Rotman, M.; Bacskai, B.J.; van Buchem, M.A.; van der Maarel, S.M.; van der Weerd, L. In vivo detection of amyloid-β deposits using heavy chain antibody fragments in a transgenic mouse model for Alzheimer’s disease. PLoS ONE 2012, 7, e38284. [Google Scholar] [CrossRef]
- Rutgers, K.S.; Nabuurs, R.J.A.; van den Berg, S.A.A.; Schenk, G.J.; Rotman, M.; Verrips, C.T.; van Duinen, S.G.; Maat-Schieman, M.L.; van Buchem, M.A.; de Boer, A.G.; et al. Transmigration of beta amyloid specific heavy chain antibody fragments across the in vitro blood–brain barrier. Neuroscience 2011, 190, 37–42. [Google Scholar] [CrossRef]
- Meier, S.R.; Sehlin, D.; Syvänen, S. Passive and receptor mediated brain delivery of an anti-GFAP nanobody. Nucl. Med. Biol. 2022, 114–115, 128–134. [Google Scholar] [CrossRef]
- Solár, P.; Zamani, A.; Kubíčková, L.; Dubový, P.; Joukal, M. Choroid plexus and the blood–cerebrospinal fluid barrier in disease. Fluids Barriers CNS 2020, 17, 35. [Google Scholar] [CrossRef]
- Shen, D.D.; Artru, A.A.; Adkison, K.K. Principles and applicability of CSF sampling for the assessment of CNS drug delivery and pharmacodynamics. Adv. Drug Deliv. Rev. 2004, 56, 1825–1857. [Google Scholar] [CrossRef]
- Pardridge, W. Re-engineering therapeutic antibodies for Alzheimer’s disease as blood-brain barrier penetrating bi-specific antibodies. Expert Opin. Bio Ther. 2016, 16, 1455–1468. [Google Scholar] [CrossRef]
- Caljon, G.; Caveliers, V.; Lahoutte, T.; Stijlemans, B.; Ghassabeh, G.; Van Den Abbeele, J.; Smolders, I.; De Baetselier, P.; Michotte, Y.; Muyldermans, S.; et al. Using microdialysis to analyse the passage of monovalent nanobodies through the blood-brain barrier: Brain disposition of monovalent nanobodies. Br. J. Pharmacol. 2012, 165, 2341–2353. [Google Scholar] [CrossRef]
- Custers, M.-L.; Wouters, Y.; Jaspers, T.; De Bundel, D.; Dewilde, M.; Van Eeckhaut, A.; Smolders, I. Applicability of cerebral open flow microperfusion and microdialysis to quantify a brain-penetrating nanobody in mice. Anal. Chim. Acta 2021, 1178, 338803. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-Y.; Morrow, K.; Bonacquisti, E.; Zhang, W.; Shah, D.K. Antibody pharmacokinetics in rat brain determined using microdialysis. mAbs 2018, 10, 843–853. [Google Scholar] [CrossRef]
- Jadhav, S.B.; Khaowroongrueng, V.; Derendorf, H. Microdialysis of large molecules. J. Pharm. Sci. 2016, 105, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Ao, X.; Stenken, J.A. Microdialysis sampling of cytokines. Methods 2006, 38, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Custers, M.-L.; Nestor, L.; De Bundel, D.; Van Eeckhaut, A.; Smolders, I. Current approaches to monitor macromolecules directly from the cerebral interstitial fluid. Pharmaceutics 2022, 14, 1051. [Google Scholar] [CrossRef]
- Lesniak, W.G.; Chu, C.; Jablonska, A.; Azad, B.B.; Zwaenepoel, O.; Zawadzki, M.; Lisok, A.; Pomper, M.; Walczak, P.; Gettemans, J.; et al. PET imaging of distinct brain uptake of a nanobody and similarly-sized PAMAM dendrimers after intra-arterial administration. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1940–1951. [Google Scholar] [CrossRef]
- Farrington, G.K.; Caram-Salas, N.; Haqqani, A.S.; Brunette, E.; Eldredge, J.; Pepinsky, B.; Antognetti, G.; Baumann, E.; Ding, W.; Garber, E.; et al. A novel platform for engineering blood-brain barrier-crossing bispecific biologics. FASEB J. 2014, 28, 4764–4778. [Google Scholar] [CrossRef]
- Webster, C.I.; Caram-Salas, N.; Haqqani, A.S.; Thom, G.; Brown, L.; Rennie, K.; Yogi, A.; Costain, W.; Brunette, E.; Stanimirovic, D.B. Brain penetration, target engagement, and disposition of the blood-brain barrier-crossing bispecific antibody antagonist of metabotropic glutamate receptor type 1. FASEB J. 2016, 30, 1927–1940. [Google Scholar] [CrossRef]
- Lagerström, M.C.; Schiöth, H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 339–357. [Google Scholar] [CrossRef]
- Wouters, Y.; Jaspers, T.; De Strooper, B.; Dewilde, M. Identification and in vivo characterization of a brain-penetrating nanobody. Fluids Barriers CNS 2020, 17, 62. [Google Scholar] [CrossRef]
- Alata, W.; Yogi, A.; Brunette, E.; Delaney, C.E.; van Faassen, H.; Hussack, G.; Iqbal, U.; Kemmerich, K.; Haqqani, A.S.; Moreno, M.J.; et al. Targeting insulin-like growth factor-1 receptor (IGF1R) for brain delivery of biologics. FASEB J. 2022, 36, e22208. [Google Scholar] [CrossRef] [PubMed]
- Stocki, P.; Szary, J.; Rasmussen, C.L.M.; Demydchuk, M.; Northall, L.; Logan, D.B.; Gauhar, A.; Thei, L.; Moos, T.; Walsh, F.S.; et al. Blood-brain barrier transport using a high affinity, brain-selective VNAR antibody targeting transferrin receptor 1. FASEB J. 2021, 35, e21172. [Google Scholar] [CrossRef]

| Antibody | Target | Clinical Status | Dose | Key Findings/Mode of Action | References |
|---|---|---|---|---|---|
| Natalizumab (Tysabri®) Humanized mAb | α4β1 integrin | Approved by FDA and EMA | i.v. infusion of 300 mg every 4 weeks | Blocks binding of α4β1 integrin to VCAM | [26] |
| Rituximab (RituxanTM) Chimeric mAb | CD20 | Phase III | i.v. infusion of 500 or 1000 mg every 6–12 months | Reduced recurrences by lysing circulating B cells and stops MS inflammation | [28,29] |
| Ocrelizumab (OcrevusTM) Humanized mAb | Approved by FDA and EMA | i.v. infusion of 300 mg day 1 and 5 and 600 mg every 6 months | [30] | ||
| Ofatumumab (Kesimpta®) Humanized mAb | Approved by FDA and EMA | s.c. injection 20 mg on weeks 0, 1, and 2, and 20 mg each month | [40] | ||
| Ublituximab (TG-1101) Chimeric IgG1 mAb | Phase III Pending FDA approval | i.v. infusion of 150 mg day 1, 450 mg day 15 and 450 mg every 6 months | [31] | ||
| Alemtuzumab (LemtradaTM) Humanized mAb | CD52 | Approved by FDA and EMA | i.v. infusion 12 mg/day for 5 days and 1 year later 12 mg/day for 3 days | Lysis of T and B lymphocytes | [32] |
| Opicinumab (BIIB033) Human mAb | LINGO-1 | Phase II Development stopped | i.v. infusion 750 mg every 4 weeks for 96 weeks | Inhibition of LINGO-1, differentiation of oligodendrocyte precursor cells into mature oligodendrocytes allowing remyelination | [33] |
| Elezanumab (ABT-555) Human mAb | RMGa | Phase II | i.v. infusion of 1.800 mg monthly or bimonthly | Inhibition of RMGa promoting regeneration | [34] |
| Temelimab (GNbAC1) Humanized IgG4 mAb | MSRV-Env protein | Phase II | i.v. infusion of a single dose of 36, 60, 85 or 110 mg/kg | Expected inhibition of MSRV-Env decreasing proinflammatory and autoimmune cascades | [35] |
| Antibody | Target | Clinical Status | Dose | Key Findings/Mode of Action | References |
|---|---|---|---|---|---|
| Erenumab (Aimovig®) Human mAb | CGRP receptor | Approved by FDA and EMA | s.c. injection of 70 mg once per month | Blocks the CGRP receptor and inhibits the activity of CGRP, preventing the onset of pain | [53] |
| Fremanezumab (AjovyTM) Humanized IgG2 mAb | α and β isoforms of CGRP | s.c. injection of 225 mg once per month or 675 mg every 3 months | Targets the CGRP ligand and blocks its binding to the receptor | [51] | |
| Galcanezumab (Emgality®) Humanized mAb | s.c. injection of 120 mg once per month | [50,54,55] | |||
| Eptinezumab (VyeptyTM) Humanized IgG1 mAb | Infusion of 100 mg every 3 months | [49,56] |
| Antibody | Target | Clinical Status | Dose | Key Findings/Mode of Action | References |
|---|---|---|---|---|---|
| Prasinezumab (PRX002) Humanized IgG1 mAb | Aggregated α-synuclein | Phase II | i.v. infusion of 1500 or 4500 mg every 4 weeks for 52 weeks | Trend toward benefit in motor functions | [105] |
| Cinpanemab (BIIB054) Human-derived mAb | Aggregated α-synuclein | Phase II—discontinued | i.v. infusion of 250, 1250 or 2500 mg every 4 weeks for 52 weeks | No improvement in motor functions | [108] |
| MEDI1341 (TAK-341) mAb | Monomeric and aggregated α-synuclein | Phase I | i.v. infusion of 3 doses given at 4 weeks interval | No results released | / |
| PFFNB2 sdAb | α-synuclein preformed fibrils | Preclinical stages | Intraventricular injection of AAV encoding PFFNB2 in the transgenic mouse model PACTg(SNCAWT) | In vitro dissociation of fibrils. Prevents the spreading of pathological α-synuclein to the cortex | [114] |
| NbSyn2 sdAb | Monomeric and fibrillar α-synuclein | / | Inhibit the formation of fibrils and converts toxic oligomers into less toxic species | [115,116,117,118,119,120] | |
| NbSyn87 sdAb | Injection in the substantia nigra of AAV encoding NbSyn87 into rats |
| Antibody | Target | Clinical Status | Dose | Key Findings/Mode of Action | References |
|---|---|---|---|---|---|
| PRN100 Humanized IgG4κ mAb | PrPSc | No clinical trial—“special exemption” | i.v. infusion of 80–120 mg/kg every 2 weeks until death | Treatment is safe and reached CSF and brain tissue concentrations expected | [133] |
| PrioV3 sdAb | Preclinical stages | i.p. injection of 5 mg/kg in WT mice | Crosses the BBB | [134] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsitokana, M.E.; Lafon, P.-A.; Prézeau, L.; Pin, J.-P.; Rondard, P. Targeting the Brain with Single-Domain Antibodies: Greater Potential Than Stated So Far? Int. J. Mol. Sci. 2023, 24, 2632. https://doi.org/10.3390/ijms24032632
Tsitokana ME, Lafon P-A, Prézeau L, Pin J-P, Rondard P. Targeting the Brain with Single-Domain Antibodies: Greater Potential Than Stated So Far? International Journal of Molecular Sciences. 2023; 24(3):2632. https://doi.org/10.3390/ijms24032632
Chicago/Turabian StyleTsitokana, Mireille Elodie, Pierre-André Lafon, Laurent Prézeau, Jean-Philippe Pin, and Philippe Rondard. 2023. "Targeting the Brain with Single-Domain Antibodies: Greater Potential Than Stated So Far?" International Journal of Molecular Sciences 24, no. 3: 2632. https://doi.org/10.3390/ijms24032632
APA StyleTsitokana, M. E., Lafon, P.-A., Prézeau, L., Pin, J.-P., & Rondard, P. (2023). Targeting the Brain with Single-Domain Antibodies: Greater Potential Than Stated So Far? International Journal of Molecular Sciences, 24(3), 2632. https://doi.org/10.3390/ijms24032632