

Modulating Glycolysis to Improve Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
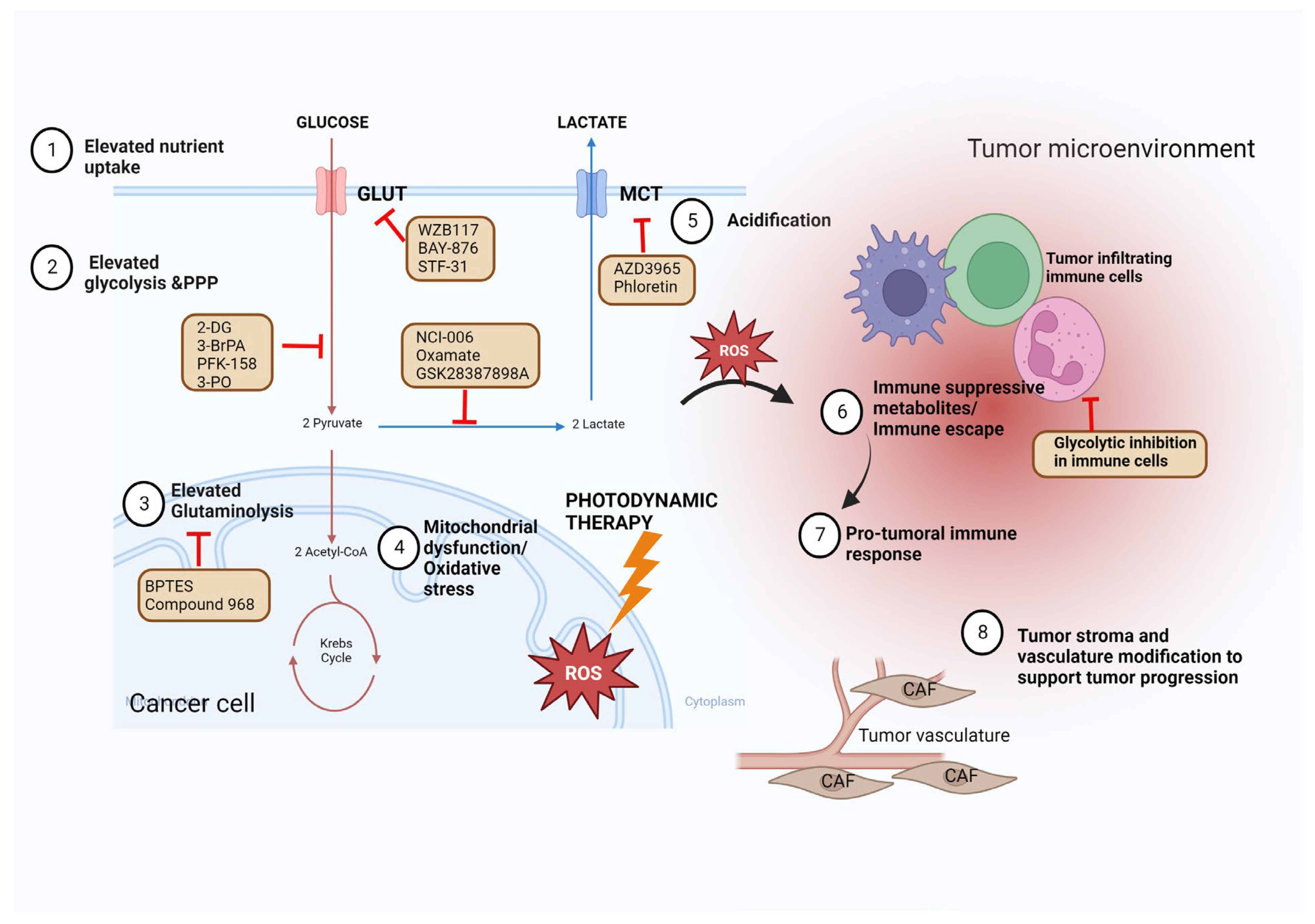
2. Modulating Glycolysis to Improve Chemotherapy and Radiotherapy
2.1. Targeting Glucose Transporters and Glucose Uptake to Improve Chemotherapy
2.2. Targeting Glycolysis Enzymes to Improve Chemotherapy and Radiotherapy
2.3. Modulating Glycolysis to Overcome Drug Resistance
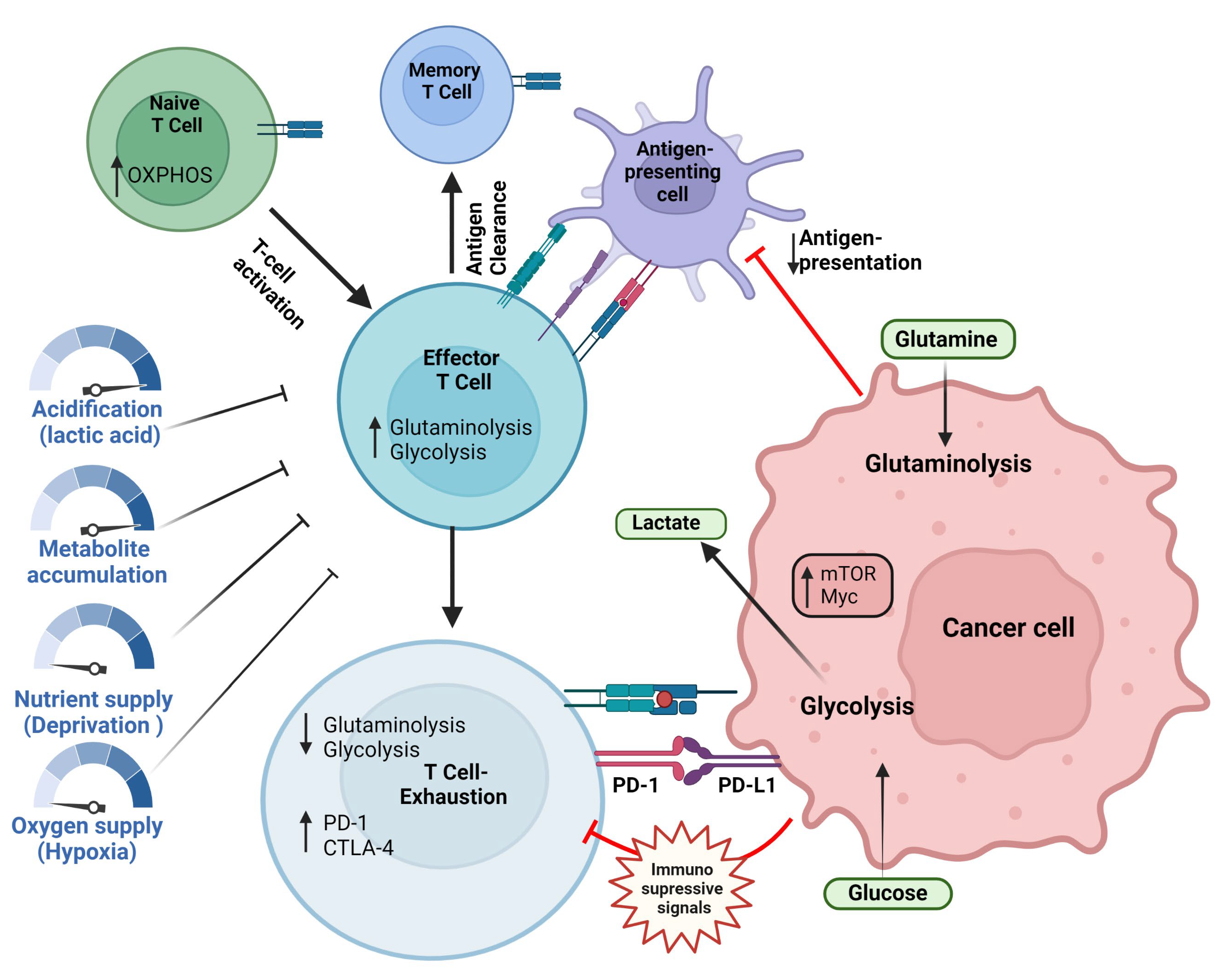
3. Targeting Glycolysis to Enhance Immunotherapy
3.1. Glucose Metabolism in the Immune Cells of the Tumor Microenvironment
3.2. Signaling Mechanisms Regulating Glycolysis
3.3. Targeting Glycolysis to Improve Immunotherapy
3.4. Glycolysis-Targeting Therapies to Improve Immunotherapy Efficacy
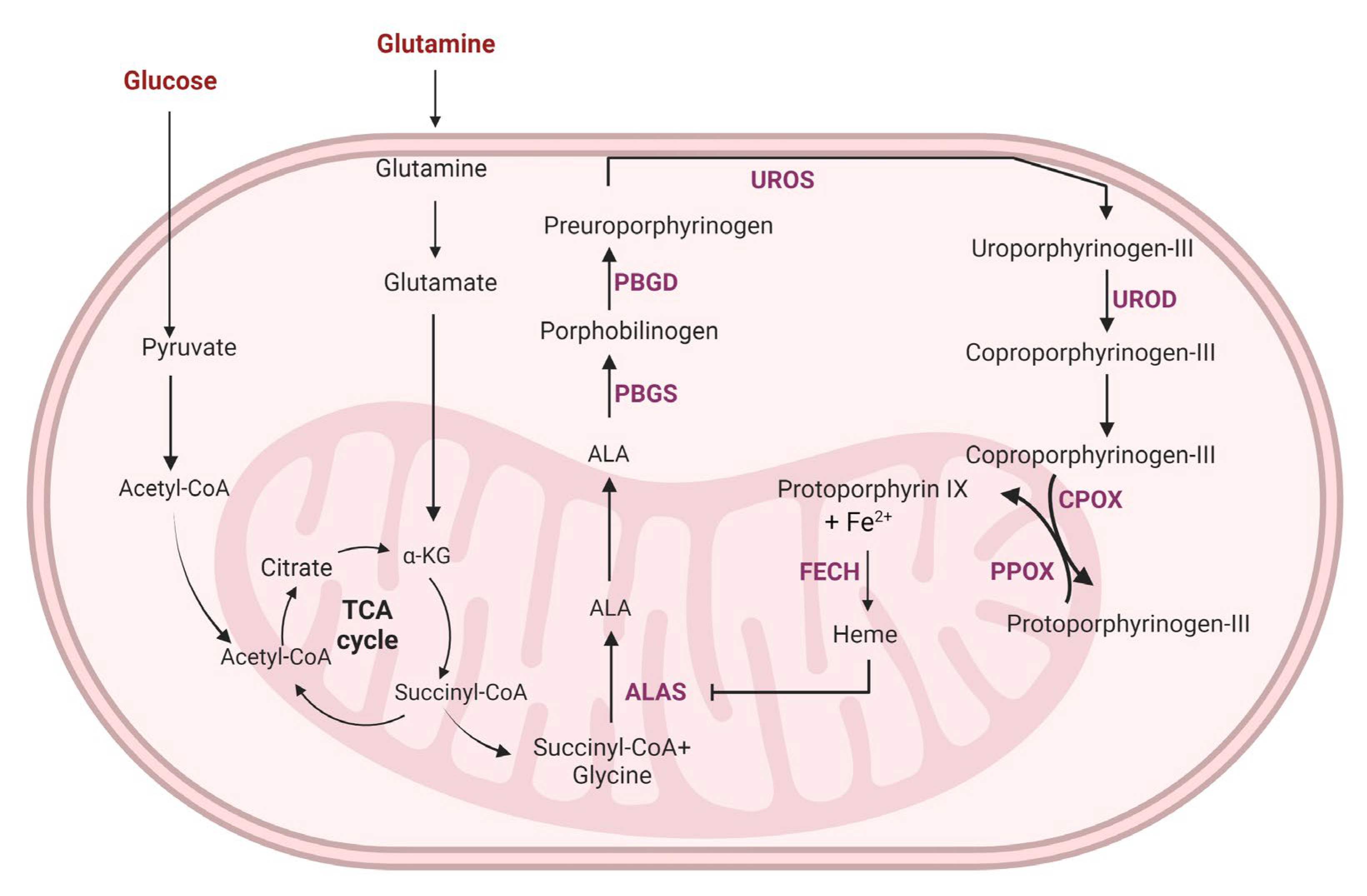
4. Targeting Glycolysis to Enhance Hormonal Therapy
5. Targeting Glycolysis to Improve Photodynamic Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, P.L. Tumor mitochondria and the bioenergetics of cancer cells. Prog. Exp. Tumor Res. 1978, 22, 190–274. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Lemberg, K.M.; Gori, S.S.; Tsukamoto, T.; Rais, R.; Slusher, B.S. Clinical development of metabolic inhibitors for oncology. J. Clin. Investig. 2022, 132, e148550. [Google Scholar] [CrossRef]
- Peng, J.; Cui, Y.; Xu, S.; Wu, X.; Huang, Y.; Zhou, W.; Wang, S.; Fu, Z.; Xie, H. Altered glycolysis results in drug-resistant in clinical tumor therapy. Oncol. Lett. 2021, 21, 369. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Pusapati, R.V.; Daemen, A.; Wilson, C.; Sandoval, W.; Gao, M.; Haley, B.; Baudy, A.R.; Hatzivassiliou, G.; Evangelista, M.; Settleman, J. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell 2016, 29, 548–562. [Google Scholar] [CrossRef]
- Fu, V.; Moroishi, T.; Guan, K.L. Glycoholics Anonymous: Cancer Sobers Up with mTORC1. Cancer Cell 2016, 29, 432–434. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, M.; Ozsvari, B.; Sotgia, F.; Lisanti, M.P. High ATP Production Fuels Cancer Drug Resistance and Metastasis: Implications for Mitochondrial ATP Depletion Therapy. Front. Oncol. 2021, 11, 740720. [Google Scholar] [CrossRef]
- Wang, T.; Ma, F.; Qian, H.L. Defueling the cancer: ATP synthase as an emerging target in cancer therapy. Mol. Ther. Oncolytics 2021, 23, 82–95. [Google Scholar] [CrossRef]
- Vultaggio-Poma, V.; Sarti, A.C.; Di Virgilio, F. Extracellular ATP: A Feasible Target for Cancer Therapy. Cells 2020, 9, 2496. [Google Scholar] [CrossRef]
- Rai, Y.; Yadav, P.; Kumari, N.; Kalra, N.; Bhatt, A.N. Hexokinase II inhibition by 3-bromopyruvate sensitizes myeloid leukemic cells K-562 to anti-leukemic drug, daunorubicin. Biosci. Rep. 2019, 39, BSR20190880. [Google Scholar] [CrossRef] [Green Version]
- Almouhanna, F.; Blagojevic, B.; Can, S.; Ghanem, A.; Wolfl, S. Pharmacological activation of pyruvate kinase M2 reprograms glycolysis leading to TXNIP depletion and AMPK activation in breast cancer cells. Cancer Metab. 2021, 9, 5. [Google Scholar] [CrossRef]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol. Cancer 2010, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, K.C.; Cunha, I.W.; Rocha, R.M.; Ayala, F.R.; Cajaiba, M.M.; Begnami, M.D.; Vilela, R.S.; Paiva, G.R.; Andrade, R.G.; Soares, F.A. GLUT1 expression in malignant tumors and its use as an immunodiagnostic marker. Clinics 2011, 66, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The prognostic value of GLUT1 in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 43356–43367. [Google Scholar] [CrossRef] [Green Version]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Queckborner, S.; Turnbull, A.; Michie, C.O.; Williams, A.R.W.; Rye, T.; Gourley, C.; Langdon, S.P. Expression of glycolytic enzymes in ovarian cancers and evaluation of the glycolytic pathway as a strategy for ovarian cancer treatment. BMC Cancer 2018, 18, 636. [Google Scholar] [CrossRef]
- Shin, E.; Koo, J.S. Glucose Metabolism and Glucose Transporters in Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 728759. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Xu, X.; Luan, H.; Li, L.; Dai, W.; Li, Z.; Bian, J. The progress and development of GLUT1 inhibitors targeting cancer energy metabolism. Future Med. Chem. 2019, 11, 2333–2352. [Google Scholar] [CrossRef] [PubMed]
- Sawayama, H.; Ogata, Y.; Ishimoto, T.; Mima, K.; Hiyoshi, Y.; Iwatsuki, M.; Baba, Y.; Miyamoto, Y.; Yoshida, N.; Baba, H. Glucose transporter 1 regulates the proliferation and cisplatin sensitivity of esophageal cancer. Cancer Sci. 2019, 110, 1705–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, H.C.; Sung, C.J.; Hsu, J.L.; Leu, W.J.; Guh, J.H.; Kung, F.L.; Hsu, L.C. The Combination of a Novel GLUT1 Inhibitor and Cisplatin Synergistically Inhibits Breast Cancer Cell Growth By Enhancing the DNA Damaging Effect and Modulating the Akt/mTOR and MAPK Signaling Pathways. Front. Pharmacol. 2022, 13, 879748. [Google Scholar] [CrossRef] [PubMed]
- Pliszka, M.; Szablewski, L. Glucose Transporters as a Target for Anticancer Therapy. Cancers 2021, 13, 4184. [Google Scholar] [CrossRef]
- Wu, Q.; Ba-Alawi, W.; Deblois, G.; Cruickshank, J.; Duan, S.; Lima-Fernandes, E.; Haight, J.; Tonekaboni, S.A.M.; Fortier, A.M.; Kuasne, H.; et al. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat. Commun. 2020, 11, 4205. [Google Scholar] [CrossRef]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 2015, 6, 651–661. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.L.; Weng, H.C.; Hsu, J.L.; Lin, S.W.; Guh, J.H.; Hsu, L.C. The Combination of MK-2206 and WZB117 Exerts a Synergistic Cytotoxic Effect Against Breast Cancer Cells. Front. Pharmacol. 2019, 10, 1311. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Meng, Y.Q.; Xu, X.F.; Gu, J. Blockade of GLUT1 by WZB117 resensitizes breast cancer cells to adriamycin. Anticancer Drugs 2017, 28, 880–887. [Google Scholar] [CrossRef]
- Zhao, F.; Ming, J.; Zhou, Y.; Fan, L. Inhibition of Glut1 by WZB117 sensitizes radioresistant breast cancer cells to irradiation. Cancer Chemother. Pharmacol. 2016, 77, 963–972. [Google Scholar] [CrossRef]
- Mori, Y.; Yamawaki, K.; Ishiguro, T.; Yoshihara, K.; Ueda, H.; Sato, A.; Ohata, H.; Yoshida, Y.; Minamino, T.; Okamoto, K.; et al. ALDH-Dependent Glycolytic Activation Mediates Stemness and Paclitaxel Resistance in Patient-Derived Spheroid Models of Uterine Endometrial Cancer. Stem Cell Rep. 2019, 13, 730–746. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.K.; Choi, J.S. Effects of silibinin, inhibitor of CYP3A4 and P-glycoprotein in vitro, on the pharmacokinetics of paclitaxel after oral and intravenous administration in rats. Pharmacology 2010, 85, 350–356. [Google Scholar] [CrossRef]
- Pashaei-Asl, F.; Pashaei-Asl, R.; Khodadadi, K.; Akbarzadeh, A.; Ebrahimie, E.; Pashaiasl, M. Enhancement of anticancer activity by silibinin and paclitaxel combination on the ovarian cancer. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1483–1487. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jiang, C.; Wang, Q.; Yang, S.; Cao, Y.; Hao, J.N.; Niu, D.; Chen, Y.; Han, B.; Jia, X.; et al. A “Valve-Closing” Starvation Strategy for Amplification of Tumor-Specific Chemotherapy. Adv. Sci. 2022, 9, e2104671. [Google Scholar] [CrossRef]
- Tilekar, K.; Upadhyay, N.; Iancu, C.V.; Pokrovsky, V.; Choe, J.Y.; Ramaa, C.S. Power of two: Combination of therapeutic approaches involving glucose transporter (GLUT) inhibitors to combat cancer. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188457. [Google Scholar] [CrossRef]
- Icard, P.; Teboul, B.; El Baze, P. A Simple Method to Optimize the Effectiveness of Chemotherapy: Modulation of Glucose Intake During Chemotherapy. Anticancer Res. 2017, 37, 6199–6202. [Google Scholar] [CrossRef]
- Omar, E.M.; Omran, G.A.; Mustafa, M.F.; El-Khodary, N.M. Intermittent fasting during adjuvant chemotherapy may promote differential stress resistance in breast cancer patients. J. Egypt. Natl. Cancer Inst. 2022, 34, 38. [Google Scholar] [CrossRef]
- Vaziri-Gohar, A.; Hue, J.J.; Graor, H.G.; Abbas, A.; Zarei, M.; Hajihassani, O.; Titomihelakis, G.; Feczko, J.; Rathore, M.; Wang, R.; et al. Increased glucose availability sensitizes pancreatic cancer to chemotherapy. bioRxiv 2022. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Mao, X.; Qi, Q.; Zhu, M.; Zhang, C.; Pan, X.; Ling, Y. Palliative treatment efficacy of glucose inhibition combined with chemotherapy for non-small cell lung cancer with widespread bone and brain metastases: A case report. Biomed. Rep. 2017, 7, 553–557. [Google Scholar] [CrossRef]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef]
- Ahn, K.J.; Hwang, H.S.; Park, J.H.; Bang, S.H.; Kang, W.J.; Yun, M.; Lee, J.D. Evaluation of the role of hexokinase type II in cellular proliferation and apoptosis using human hepatocellular carcinoma cell lines. J. Nucl. Med. 2009, 50, 1525–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato-Tadano, A.; Suzuki, T.; Amari, M.; Takagi, K.; Miki, Y.; Tamaki, K.; Watanabe, M.; Ishida, T.; Sasano, H.; Ohuchi, N. Hexokinase II in breast carcinoma: A potent prognostic factor associated with hypoxia-inducible factor-1alpha and Ki-67. Cancer Sci. 2013, 104, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zajicek, G. The normal and the pathological. Harefuah 1994, 127, 24–25. [Google Scholar] [PubMed]
- Zhang, X.D.; Deslandes, E.; Villedieu, M.; Poulain, L.; Duval, M.; Gauduchon, P.; Schwartz, L.; Icard, P. Effect of 2-deoxy-D-glucose on various malignant cell lines in vitro. Anticancer Res. 2006, 26, 3561–3566. [Google Scholar]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; Ledoux, S.P.; Wilson, G.L.; Voellmy, R.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011, 71, 4585–4597. [Google Scholar] [CrossRef] [Green Version]
- Maschek, G.; Savaraj, N.; Priebe, W.; Braunschweiger, P.; Hamilton, K.; Tidmarsh, G.F.; De Young, L.R.; Lampidis, T.J. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004, 64, 31–34. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Fan, T.; Sun, G.; Zhou, Y.; Huang, Y.; Zhang, N.; Zhao, L.; Zhong, R.; Peng, Y. 2-Deoxy-D-glucose increases the sensitivity of glioblastoma cells to BCNU through the regulation of glycolysis, ROS and ERS pathways: In vitro and in vivo validation. Biochem. Pharmacol. 2022, 199, 115029. [Google Scholar] [CrossRef]
- Zhou, N.; Liu, Q.; Wang, X.; He, L.; Zhang, T.; Zhou, H.; Zhu, X.; Zhou, T.; Deng, G.; Qiu, C. The combination of hydroxychloroquine and 2-deoxyglucose enhances apoptosis in breast cancer cells by blocking protective autophagy and sustaining endoplasmic reticulum stress. Cell Death Discov. 2022, 8, 286. [Google Scholar] [CrossRef]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef]
- Geng, C.; Li, J.; Ding, F.; Wu, G.; Yang, Q.; Sun, Y.; Zhang, Z.; Dong, T.; Tian, X. Curcumin suppresses 4-hydroxytamoxifen resistance in breast cancer cells by targeting SLUG/Hexokinase 2 pathway. Biochem. Biophys. Res. Commun. 2016, 473, 147–153. [Google Scholar] [CrossRef]
- Pedersen, P.L. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J. Bioenerg. Biomembr. 2007, 39, 211–222. [Google Scholar] [CrossRef]
- Pedersen, P.L.; Mathupala, S.; Rempel, A.; Geschwind, J.F.; Ko, Y.H. Mitochondrial bound type II hexokinase: A key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim. Biophys. Acta 2002, 1555, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K. 3-Bromopyruvate kills cancer cells in animals. Lancet Oncol. 2002, 3, 524. [Google Scholar] [CrossRef]
- Fan, T.; Sun, G.; Sun, X.; Zhao, L.; Zhong, R.; Peng, Y. Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers 2019, 11, 317. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Pan, J.; Lubet, R.A.; Komas, S.M.; Kalyanaraman, B.; Wang, Y.; You, M. Enhanced antitumor activity of 3-bromopyruvate in combination with rapamycin in vivo and in vitro. Cancer Prev. Res. 2015, 8, 318–326. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Ren, Y.; Lu, J.; Ma, J.; Shen, X.; Zhuang, Z. Synergistic Effect of 3-Bromopyruvate in Combination with Rapamycin Impacted Neuroblastoma Metabolism by Inhibiting Autophagy. Onco Targets Ther. 2020, 13, 11125–11137. [Google Scholar] [CrossRef]
- Li, J.; Pan, J.; Liu, Y.; Luo, X.; Yang, C.; Xiao, W.; Li, Q.; Yang, L.; Zhang, X. 3-Bromopyruvic acid regulates glucose metabolism by targeting the c-Myc/TXNIP axis and induces mitochondria-mediated apoptosis in TNBC cells. Exp. Ther. Med. 2022, 24, 520. [Google Scholar] [CrossRef]
- Attia, Y.M.; El-Abhar, H.S.; Al Marzabani, M.M.; Shouman, S.A. Targeting glycolysis by 3-bromopyruvate improves tamoxifen cytotoxicity of breast cancer cell lines. BMC Cancer 2015, 15, 838. [Google Scholar] [CrossRef] [Green Version]
- Wicks, R.T.; Azadi, J.; Mangraviti, A.; Zhang, I.; Hwang, L.; Joshi, A.; Bow, H.; Hutt-Cabezas, M.; Martin, K.L.; Rudek, M.A.; et al. Local delivery of cancer-cell glycolytic inhibitors in high-grade glioma. Neuro-Oncol. 2015, 17, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Na, J.; Youn, H.; Yu, J.S.; Kang, K.W.; Chung, J.-K. Enhanced therapeutic efficacy of combined Sorafenib and 3-bromopyruvate in murine orthotopic liver cancer assessed by bioluminescence image. J. Nucl. Med. 2016, 57, 1331. [Google Scholar]
- Roy, S.K.; Dukic, T.; Bhandary, B.; Acharya, A.; Tu, K.J.; Ko, Y.H.; Shukla, H.D. 3-Bromopyruvate in combination with radiation inhibits pancreatic cancer growth by dismantling mitochondria and ATP generation in a preclinical mouse model. Cancer Res. 2022, 82, 5243–5243. [Google Scholar] [CrossRef]
- Skaripa-Koukelli, I.; Hauton, D.; Walsby-Tickle, J.; Thomas, E.; Owen, J.; Lakshminarayanan, A.; Able, S.; McCullagh, J.; Carlisle, R.C.; Vallis, K.A. 3-Bromopyruvate-mediated MCT1-dependent metabolic perturbation sensitizes triple negative breast cancer cells to ionizing radiation. Cancer Metab. 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e1287. [Google Scholar] [CrossRef] [Green Version]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [Green Version]
- Involvement of physicians in capital punishment. N. Y. State J. Med. 1991, 91, 271–272.
- Morais-Santos, F.; Granja, S.; Miranda-Goncalves, V.; Moreira, A.H.; Queiros, S.; Vilaca, J.L.; Schmitt, F.C.; Longatto-Filho, A.; Paredes, J.; Baltazar, F.; et al. Targeting lactate transport suppresses in vivo breast tumour growth. Oncotarget 2015, 6, 19177–19189. [Google Scholar] [CrossRef] [Green Version]
- Saulle, E.; Spinello, I.; Quaranta, M.T.; Pasquini, L.; Pelosi, E.; Iorio, E.; Castelli, G.; Chirico, M.; Pisanu, M.E.; Ottone, T.; et al. Targeting Lactate Metabolism by Inhibiting MCT1 or MCT4 Impairs Leukemic Cell Proliferation, Induces Two Different Related Death-Pathways and Increases Chemotherapeutic Sensitivity of Acute Myeloid Leukemia Cells. Front. Oncol. 2020, 10, 621458. [Google Scholar] [CrossRef]
- Halford, S.E.; Walter, H.; McKay, P.; Townsend, W.; Linton, K.; Heinzmann, K.; Dragoni, I.; Brotherton, L.; Veal, G.; Siskos, A.; et al. Phase I expansion study of the first-in-class monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in patients with diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL). J. Clin. Oncol. 2021, 39, 3115. [Google Scholar] [CrossRef]
- Halford, S.E.R.; Jones, P.; Wedge, S.; Hirschberg, S.; Katugampola, S.; Veal, G.; Payne, G.; Bacon, C.; Potter, S.; Griffin, M.; et al. A first-in-human first-in-class (FIC) trial of the monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in patients with advanced solid tumours. J. Clin. Oncol. 2017, 35 (Suppl. 15), 2516. [Google Scholar] [CrossRef]
- Claps, G.; Faouzi, S.; Quidville, V.; Chehade, F.; Shen, S.; Vagner, S.; Robert, C. The multiple roles of LDH in cancer. Nat. Rev. Clin. Oncol. 2022, 19, 749–762. [Google Scholar] [CrossRef]
- Flores, A.; Sandoval-Gonzalez, S.; Takahashi, R.; Krall, A.; Sathe, L.; Wei, L.; Radu, C.; Joly, J.H.; Graham, N.A.; Christofk, H.R.; et al. Increased lactate dehydrogenase activity is dispensable in squamous carcinoma cells of origin. Nat. Commun. 2019, 10, 91. [Google Scholar] [CrossRef] [Green Version]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef]
- Lu, R.; Jiang, M.; Chen, Z.; Xu, X.; Hu, H.; Zhao, X.; Gao, X.; Guo, L. Lactate dehydrogenase 5 expression in Non-Hodgkin lymphoma is associated with the induced hypoxia regulated protein and poor prognosis. PLoS ONE 2013, 8, e74853. [Google Scholar] [CrossRef]
- Zha, X.; Wang, F.; Wang, Y.; He, S.; Jing, Y.; Wu, X.; Zhang, H. Lactate dehydrogenase B is critical for hyperactive mTOR-mediated tumorigenesis. Cancer Res. 2011, 71, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [Green Version]
- He, T.L.; Zhang, Y.J.; Jiang, H.; Li, X.H.; Zhu, H.; Zheng, K.L. The c-Myc-LDHA axis positively regulates aerobic glycolysis and promotes tumor progression in pancreatic cancer. Med. Oncol. 2015, 32, 187. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wu, Q.; Xu, X.; Yang, C.; You, J.; Chen, F.; Zeng, Y. Cancer/testis antigen LDHC promotes proliferation and metastasis by activating the PI3K/Akt/GSK-3beta-signaling pathway and the in lung adenocarcinoma. Exp. Cell Res. 2021, 398, 112414. [Google Scholar] [CrossRef]
- Jin, L.; Chun, J.; Pan, C.; Alesi, G.N.; Li, D.; Magliocca, K.R.; Kang, Y.; Chen, Z.G.; Shin, D.M.; Khuri, F.R.; et al. Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis. Oncogene 2017, 36, 3797–3806. [Google Scholar] [CrossRef] [Green Version]
- Ferraris, A.M.; Giuntini, P.; Gaetani, G.F. Serum lactic dehydrogenase as a prognostic tool for non-Hodgkin lymphomas. Blood 1979, 54, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, G.H.; Olde Damink, S.W.; Malago, M.; Dhar, D.K.; Pereira, S.P. Pyruvate Kinase M2 and Lactate Dehydrogenase A Are Overexpressed in Pancreatic Cancer and Correlate with Poor Outcome. PLoS ONE 2016, 11, e0151635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrelli, F.; Cabiddu, M.; Coinu, A.; Borgonovo, K.; Ghilardi, M.; Lonati, V.; Barni, S. Prognostic role of lactate dehydrogenase in solid tumors: A systematic review and meta-analysis of 76 studies. Acta Oncol. 2015, 54, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Hanai, J.; Ren, J.G.; Kats, L.; Burgess, K.; Bhargava, P.; Signoretti, S.; Billiard, J.; Duffy, K.J.; Grant, A.; et al. Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014, 19, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [Green Version]
- Altinoz, M.A.; Ozpinar, A. Oxamate targeting aggressive cancers with special emphasis to brain tumors. Biomed. Pharmacother. 2022, 147, 112686. [Google Scholar] [CrossRef]
- Zhao, Z.; Han, F.; Yang, S.; Wu, J.; Zhan, W. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: Involvement of the Akt-mTOR signaling pathway. Cancer Lett. 2015, 358, 17–26. [Google Scholar] [CrossRef]
- Miskimins, W.K.; Ahn, H.J.; Kim, J.Y.; Ryu, S.; Jung, Y.S.; Choi, J.Y. Synergistic anti-cancer effect of phenformin and oxamate. PLoS ONE 2014, 9, e85576. [Google Scholar] [CrossRef] [Green Version]
- Xing, B.C.; Wang, C.; Ji, F.J.; Zhang, X.B. Synergistically suppressive effects on colorectal cancer cells by combination of mTOR inhibitor and glycolysis inhibitor, Oxamate. Int. J. Clin. Exp. Pathol. 2018, 11, 4439–4445. [Google Scholar]
- Almaguel, F.A.; Sanchez, T.W.; Ortiz-Hernandez, G.L.; Casiano, C.A. Alpha-Enolase: Emerging Tumor-Associated Antigen, Cancer Biomarker, and Oncotherapeutic Target. Front. Genet. 2020, 11, 614726. [Google Scholar] [CrossRef]
- He, P.; Naka, T.; Serada, S.; Fujimoto, M.; Tanaka, T.; Hashimoto, S.; Shima, Y.; Yamadori, T.; Suzuki, H.; Hirashima, T.; et al. Proteomics-based identification of alpha-enolase as a tumor antigen in non-small lung cancer. Cancer Sci. 2007, 98, 1234–1240. [Google Scholar] [CrossRef]
- Hsiao, K.C.; Shih, N.Y.; Chu, P.Y.; Hung, Y.M.; Liao, J.Y.; Chou, S.W.; Yang, Y.Y.; Chang, G.C.; Liu, K.J. Anti-alpha-enolase is a prognostic marker in postoperative lung cancer patients. Oncotarget 2015, 6, 35073–35086. [Google Scholar] [CrossRef]
- Fu, Q.F.; Liu, Y.; Fan, Y.; Hua, S.N.; Qu, H.Y.; Dong, S.W.; Li, R.L.; Zhao, M.Y.; Zhen, Y.; Yu, X.L.; et al. Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. J. Hematol. Oncol. 2015, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Zhan, P.; Zhao, S.; Yan, H.; Yin, C.; Xiao, Y.; Wang, Y.; Ni, R.; Chen, W.; Wei, G.; Zhang, P. alpha-enolase promotes tumorigenesis and metastasis via regulating AMPK/mTOR pathway in colorectal cancer. Mol. Carcinog. 2017, 56, 1427–1437. [Google Scholar] [CrossRef]
- Capello, M.; Ferri-Borgogno, S.; Riganti, C.; Chattaragada, M.S.; Principe, M.; Roux, C.; Zhou, W.; Petricoin, E.F.; Cappello, P.; Novelli, F. Targeting the Warburg effect in cancer cells through ENO1 knockdown rescues oxidative phosphorylation and induces growth arrest. Oncotarget 2016, 7, 5598–5612. [Google Scholar] [CrossRef]
- Song, K.; Rajasekaran, N.; Chelakkot, C.; Lee, H.S.; Paek, S.M.; Yang, H.; Jia, L.; Park, H.G.; Son, W.S.; Kim, Y.J.; et al. Macrosphelide A Exhibits a Specific Anti-Cancer Effect by Simultaneously Inactivating ENO1, ALDOA, and FH. Pharmaceuticals 2021, 14, 1060. [Google Scholar] [CrossRef]
- Qian, X.; Xu, W.; Xu, J.; Shi, Q.; Li, J.; Weng, Y.; Jiang, Z.; Feng, L.; Wang, X.; Zhou, J.; et al. Enolase 1 stimulates glycolysis to promote chemoresistance in gastric cancer. Oncotarget 2017, 8, 47691–47708. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.K.; Sun, Y.; Lv, L.; Ping, Y. ENO1 and Cancer. Mol. Ther. Oncolytics 2022, 24, 288–298. [Google Scholar] [CrossRef]
- Lin, Y.H.; Satani, N.; Hammoudi, N.; Yan, V.C.; Barekatain, Y.; Khadka, S.; Ackroyd, J.J.; Georgiou, D.K.; Pham, C.D.; Arthur, K.; et al. An enolase inhibitor for the targeted treatment of ENO1-deleted cancers. Nat. Metab. 2020, 2, 1413–1426. [Google Scholar] [CrossRef]
- Jones, B.C.; Pohlmann, P.R.; Clarke, R.; Sengupta, S. Treatment against glucose-dependent cancers through metabolic PFKFB3 targeting of glycolytic flux. Cancer Metastasis Rev. 2022, 41, 447–458. [Google Scholar] [CrossRef]
- Zhu, W.; Ye, L.; Zhang, J.; Yu, P.; Wang, H.; Ye, Z.; Tian, J. PFK15, a Small Molecule Inhibitor of PFKFB3, Induces Cell Cycle Arrest, Apoptosis and Inhibits Invasion in Gastric Cancer. PLoS ONE 2016, 11, e0163768. [Google Scholar] [CrossRef]
- Telang, S.; O’Neal, J.; Tapolsky, G.; Clem, B.; Kerr, A.; Imbert-Ferndandez, Y.; Chesney, J. Discovery of a PFKFB3 inhibitor for phase I trial testing that synergizes with the B-Raf inhibitor vemurafenib. Cancer Metab. 2014, 2, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Phase 1 Safety Study of ACT-PFK-158, 2HCl in Patients with Advanced Solid Malignancies_NCT02044861. Available online: https://clinicaltrials.gov/ct2/show/NCT02044861 (accessed on 30 September 2015).
- Redman, R.A.; Pohlmann, P.R.; Kurman, M.R.; Tapolsky, G.; Chesney, J. A phase I, dose-escalation, multicenter study of ACT-PFK-158, 2HCl in patients with advanced solid malignancies explores a first-in-human inhibitor of glycolysis. J. Clin. Oncol. 2015, 33, TPS494. [Google Scholar] [CrossRef]
- Yan, S.; Zhou, N.; Zhang, D.; Zhang, K.; Zheng, W.; Bao, Y.; Yang, W. PFKFB3 Inhibition Attenuates Oxaliplatin-Induced Autophagy and Enhances Its Cytotoxicity in Colon Cancer Cells. Int. J. Mol. Sci. 2019, 20, 5415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- Emini Veseli, B.; Van Wielendaele, P.; Delibegovic, M.; Martinet, W.; De Meyer, G.R.Y. The PFKFB3 Inhibitor AZ67 Inhibits Angiogenesis Independently of Glycolysis Inhibition. Int. J. Mol. Sci. 2021, 22, 5970. [Google Scholar] [CrossRef]
- Zhang, J.; Xue, W.; Xu, K.; Yi, L.; Guo, Y.; Xie, T.; Tong, H.; Zhou, B.; Wang, S.; Li, Q.; et al. Dual inhibition of PFKFB3 and VEGF normalizes tumor vasculature, reduces lactate production, and improves chemotherapy in glioblastoma: Insights from protein expression profiling and MRI. Theranostics 2020, 10, 7245–7259. [Google Scholar] [CrossRef]
- Hsu, M.C.; Hung, W.C. Pyruvate kinase M2 fuels multiple aspects of cancer cells: From cellular metabolism, transcriptional regulation to extracellular signaling. Mol. Cancer 2018, 17, 35. [Google Scholar] [CrossRef] [Green Version]
- Allen, A.E.; Locasale, J.W. Glucose Metabolism in Cancer: The Saga of Pyruvate Kinase Continues. Cancer Cell 2018, 33, 337–339. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Azoitei, N.; Becher, A.; Steinestel, K.; Rouhi, A.; Diepold, K.; Genze, F.; Simmet, T.; Seufferlein, T. PKM2 promotes tumor angiogenesis by regulating HIF-1alpha through NF-kappaB activation. Mol. Cancer 2016, 15, 3. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Hu, L.; Chen, M.; Cao, W.; Chen, H.; He, T. Pyruvate kinase M2 overexpression and poor prognosis in solid tumors of digestive system: Evidence from 16 cohort studies. Onco Targets Ther. 2016, 9, 4277–4288. [Google Scholar] [CrossRef]
- Zhu, H.; Luo, H.; Zhu, X.; Hu, X.; Zheng, L.; Zhu, X. Pyruvate kinase M2 (PKM2) expression correlates with prognosis in solid cancers: A meta-analysis. Oncotarget 2017, 8, 1628–1640. [Google Scholar] [CrossRef] [Green Version]
- Tennant, D.A.; Duran, R.V.; Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar] [CrossRef]
- Shi, H.S.; Li, D.; Zhang, J.; Wang, Y.S.; Yang, L.; Zhang, H.L.; Wang, X.H.; Mu, B.; Wang, W.; Ma, Y.; et al. Silencing of pkm2 increases the efficacy of docetaxel in human lung cancer xenografts in mice. Cancer Sci. 2010, 101, 1447–1453. [Google Scholar] [CrossRef]
- Yuan, S.; Qiao, T.; Zhuang, X.; Chen, W.; Xing, N.; Zhang, Q. Knockdown of the M2 Isoform of Pyruvate Kinase (PKM2) with shRNA Enhances the Effect of Docetaxel in Human NSCLC Cell Lines In Vitro. Yonsei Med. J. 2016, 57, 1312–1323. [Google Scholar] [CrossRef]
- Papadaki, C.; Sfakianaki, M.; Lagoudaki, E.; Giagkas, G.; Ioannidis, G.; Trypaki, M.; Tsakalaki, E.; Voutsina, A.; Koutsopoulos, A.; Mavroudis, D.; et al. PKM2 as a biomarker for chemosensitivity to front-line platinum-based chemotherapy in patients with metastatic non-small-cell lung cancer. Br. J. Cancer 2014, 111, 1757–1764. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Zhang, Y.; Chen, T.; Wang, Y.; Xue, J.; Zhang, Y.; Xiao, W.; Mo, X.; Lu, Y. Efficacy of RNAi targeting of pyruvate kinase M2 combined with cisplatin in a lung cancer model. J. Cancer Res. Clin. Oncol. 2011, 137, 65–72. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Kunjithapatham, R.; Geschwind, J.F. Glyceraldehyde-3-phosphate dehydrogenase: A promising target for molecular therapy in hepatocellular carcinoma. Oncotarget 2012, 3, 940–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornalley, P.J.; Rabbani, N. Glyoxalase in tumourigenesis and multidrug resistance. Semin. Cell Dev. Biol. 2011, 22, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Vali, M.; Kunjithapatham, R.; Buijs, M.; Syed, L.H.; Rao, P.P.; Ota, S.; Kwak, B.K.; Loffroy, R.; Geschwind, J.F. 3-bromopyruvate: A new targeted antiglycolytic agent and a promise for cancer therapy. Curr. Pharm. Biotechnol. 2010, 11, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Kunjithapatham, R.; Geschwind, J.F. Anticancer efficacy of the metabolic blocker 3-bromopyruvate: Specific molecular targeting. Anticancer Res. 2013, 33, 13–20. [Google Scholar]
- Gao, Y.; Zhang, T.; Terai, H.; Ficarro, S.B.; Kwiatkowski, N.; Hao, M.F.; Sharma, B.; Christensen, C.L.; Chipumuro, E.; Wong, K.K.; et al. Overcoming Resistance to the THZ Series of Covalent Transcriptional CDK Inhibitors. Cell Chem. Biol. 2018, 25, 135–142.e5. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C. Glycolysis-induced drug resistance in tumors-A response to danger signals? Neoplasia 2021, 23, 234–245.e5. [Google Scholar] [CrossRef]
- Sun, H.; Wang, H.; Wang, X.; Aoki, Y.; Wang, X.; Yang, Y.; Cheng, X.; Wang, Z.; Wang, X. Aurora-A/SOX8/FOXK1 signaling axis promotes chemoresistance via suppression of cell senescence and induction of glucose metabolism in ovarian cancer organoids and cells. Theranostics 2020, 10, 6928–6945. [Google Scholar] [CrossRef]
- Efimova, E.V.; Takahashi, S.; Shamsi, N.A.; Wu, D.; Labay, E.; Ulanovskaya, O.A.; Weichselbaum, R.R.; Kozmin, S.A.; Kron, S.J. Linking Cancer Metabolism to DNA Repair and Accelerated Senescence. Mol. Cancer Res. 2016, 14, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Kohnken, R.; Kodigepalli, K.M.; Wu, L. Regulation of deoxynucleotide metabolism in cancer: Novel mechanisms and therapeutic implications. Mol. Cancer 2015, 14, 176. [Google Scholar] [CrossRef] [Green Version]
- Le, T.M.; Poddar, S.; Capri, J.R.; Abt, E.R.; Kim, W.; Wei, L.; Uong, N.T.; Cheng, C.M.; Braas, D.; Nikanjam, M.; et al. ATR inhibition facilitates targeting of leukemia dependence on convergent nucleotide biosynthetic pathways. Nat. Commun. 2017, 8, 241. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.; Kim, M.; Jung, H.S.; Kim, Y.; Jeoung, D. Targeting Autophagy for Overcoming Resistance to Anti-EGFR Treatments. Cancers 2019, 11, 1374. [Google Scholar] [CrossRef] [Green Version]
- Lamoureux, F.; Zoubeidi, A. Dual inhibition of autophagy and the AKT pathway in prostate cancer. Autophagy 2013, 9, 1119–1120. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Castellanos, G.; Abdel Hadi, N.; Carrier, A. Autophagy Contributes to Metabolic Reprogramming and Therapeutic Resistance in Pancreatic Tumors. Cells 2022, 11, 1374. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [Green Version]
- Cerezo, M.; Rocchi, S. Cancer cell metabolic reprogramming: A keystone for the response to immunotherapy. Cell Death Dis. 2020, 11, 964. [Google Scholar] [CrossRef]
- Santos, J.C.; Lima, N.D.S.; Sarian, L.O.; Matheu, A.; Ribeiro, M.L.; Derchain, S.F.M. Exosome-mediated breast cancer chemoresistance via miR-155 transfer. Sci. Rep. 2018, 8, 829. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, G.J.; Saya, H. Therapeutic strategies targeting cancer stem cells. Cancer Sci. 2016, 107, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.T.; Chien, I.H.; Shen, W.H.; Kuo, Y.Z.; Jin, Y.T.; Wong, T.Y.; Hsiao, J.R.; Wang, H.P.; Shih, N.Y.; Wu, L.W. ENO1, a potential prognostic head and neck cancer marker, promotes transformation partly via chemokine CCL20 induction. Eur. J. Cancer 2010, 46, 1712–1723. [Google Scholar] [CrossRef]
- Song, Y.; Luo, Q.; Long, H.; Hu, Z.; Que, T.; Zhang, X.; Li, Z.; Wang, G.; Yi, L.; Liu, Z.; et al. Alpha-enolase as a potential cancer prognostic marker promotes cell growth, migration, and invasion in glioma. Mol. Cancer 2014, 13, 65. [Google Scholar] [CrossRef] [Green Version]
- Mahammedi, H.; Planchat, E.; Pouget, M.; Durando, X.; Cure, H.; Guy, L.; Van-Praagh, I.; Savareux, L.; Atger, M.; Bayet-Robert, M.; et al. The New Combination Docetaxel, Prednisone and Curcumin in Patients with Castration-Resistant Prostate Cancer: A Pilot Phase II Study. Oncology 2016, 90, 69–78. [Google Scholar] [CrossRef]
- Huang, K.; Liang, Q.; Zhou, Y.; Jiang, L.L.; Gu, W.M.; Luo, M.Y.; Tang, Y.B.; Wang, Y.; Lu, W.; Huang, M.; et al. A Novel Allosteric Inhibitor of Phosphoglycerate Mutase 1 Suppresses Growth and Metastasis of Non-Small-Cell Lung Cancer. Cell Metab. 2019, 30, 1107–1119.e8. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.H.; Chang, C.C.; Chen, C.S.; Tam, K.W.; Wang, Y.J.; Lee, C.H.; Lin, H.W.; Cheng, T.C.; Huang, C.S.; Chu, J.S.; et al. Increased expression of enolase alpha in human breast cancer confers tamoxifen resistance in human breast cancer cells. Breast Cancer Res. Treat. 2010, 121, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Principe, M.; Borgoni, S.; Cascione, M.; Chattaragada, M.S.; Ferri-Borgogno, S.; Capello, M.; Bulfamante, S.; Chapelle, J.; Di Modugno, F.; Defilippi, P.; et al. Alpha-enolase (ENO1) controls alpha v/beta 3 integrin expression and regulates pancreatic cancer adhesion, invasion, and metastasis. J. Hematol. Oncol. 2017, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Qu, M.; Huang, S.; Fu, Y.; Yang, L.; He, S.; Li, L.; Zhang, Z.; Lin, Q.; Zhang, L. A novel alpha-enolase-targeted drug delivery system for high efficacy prostate cancer therapy. Nanoscale 2018, 10, 13673–13683. [Google Scholar] [CrossRef]
- Santana-Rivera, Y.; Rabelo-Fernandez, R.J.; Quinones-Diaz, B.I.; Grafals-Ruiz, N.; Santiago-Sanchez, G.; Lozada-Delgado, E.L.; Echevarria-Vargas, I.M.; Apiz, J.; Soto, D.; Rosado, A.; et al. Reduced expression of enolase-1 correlates with high intracellular glucose levels and increased senescence in cisplatin-resistant ovarian cancer cells. Am. J. Transl. Res. 2020, 12, 1275–1292. [Google Scholar]
- Jiang, Y.X.; Siu, M.K.Y.; Wang, J.J.; Leung, T.H.Y.; Chan, D.W.; Cheung, A.N.Y.; Ngan, H.Y.S.; Chan, K.K.L. PFKFB3 Regulates Chemoresistance, Metastasis and Stemness via IAP Proteins and the NF-kappaB Signaling Pathway in Ovarian Cancer. Front. Oncol. 2022, 12, 748403. [Google Scholar] [CrossRef]
- Mondal, S.; Roy, D.; Sarkar Bhattacharya, S.; Jin, L.; Jung, D.; Zhang, S.; Kalogera, E.; Staub, J.; Wang, Y.; Xuyang, W.; et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int. J. Cancer 2019, 144, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Meng, M.B.; Wang, H.H.; Guo, W.H.; Wu, Z.Q.; Zeng, X.L.; Zaorsky, N.G.; Shi, H.S.; Qian, D.; Niu, Z.M.; Jiang, B.; et al. Targeting pyruvate kinase M2 contributes to radiosensitivity of non-small cell lung cancer cells in vitro and in vivo. Cancer Lett. 2015, 356, 985–993. [Google Scholar] [CrossRef]
- Sizemore, S.T.; Zhang, M.; Cho, J.H.; Sizemore, G.M.; Hurwitz, B.; Kaur, B.; Lehman, N.L.; Ostrowski, M.C.; Robe, P.A.; Miao, W.; et al. Pyruvate kinase M2 regulates homologous recombination-mediated DNA double-strand break repair. Cell Res. 2018, 28, 1090–1102. [Google Scholar] [CrossRef] [Green Version]
- Sfakianaki, M.; Papadaki, C.; Tzardi, M.; Trypaki, M.; Manolakou, S.; Messaritakis, I.; Saridaki, Z.; Athanasakis, E.; Mavroudis, D.; Tsiaoussis, J.; et al. PKM2 Expression as Biomarker for Resistance to Oxaliplatin-Based Chemotherapy in Colorectal Cancer. Cancers 2020, 12, 2058. [Google Scholar] [CrossRef]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wu, J.; Zhang, W.; Luo, H.; Shen, Z.; Cheng, H.; Zhu, X. PKM2 enhances chemosensitivity to cisplatin through interaction with the mTOR pathway in cervical cancer. Sci. Rep. 2016, 6, 30788. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Lv, F.; Liu, F.; Guo, X.; Fan, Y.; Gu, F.; Gu, J.; Fu, L. High Expression of Pyruvate Kinase M2 is Associated with Chemosensitivity to Epirubicin and 5-Fluorouracil in Breast Cancer. J. Cancer 2015, 6, 1130–1139. [Google Scholar] [CrossRef] [Green Version]
- Yoo, B.C.; Ku, J.L.; Hong, S.H.; Shin, Y.K.; Park, S.Y.; Kim, H.K.; Park, J.G. Decreased pyruvate kinase M2 activity linked to cisplatin resistance in human gastric carcinoma cell lines. Int. J. Cancer 2004, 108, 532–539. [Google Scholar] [CrossRef]
- Martinez-Balibrea, E.; Plasencia, C.; Gines, A.; Martinez-Cardus, A.; Musulen, E.; Aguilera, R.; Manzano, J.L.; Neamati, N.; Abad, A. A proteomic approach links decreased pyruvate kinase M2 expression to oxaliplatin resistance in patients with colorectal cancer and in human cell lines. Mol. Cancer Ther. 2009, 8, 771–778. [Google Scholar] [CrossRef]
- Al-Sawaf, O.; Bazeos, A.; Robrecht, S.; Bahlo, J.; Gower, C.; Fink, A.M.; Tresckow, J.; Cramer, P.; Langerbeins, P.; Kutsch, N.; et al. Mode of progression after first line treatment correlates with outcome of chronic lymphocytic leukemia (CLL). Am. J. Hematol. 2019, 94, 1002–1006. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Bell, R.B.; Bifulco, C.B.; Burtness, B.; Gillison, M.L.; Harrington, K.J.; Le, Q.T.; Lee, N.Y.; Leidner, R.; Lewis, R.L.; et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J. Immunother. Cancer 2019, 7, 184. [Google Scholar] [CrossRef] [Green Version]
- Reck, M.; Remon, J.; Hellmann, M.D. First-Line Immunotherapy for Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 586–597. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePeaux, K.; Delgoffe, G.M. Metabolic barriers to cancer immunotherapy. Nat. Rev. Immunol. 2021, 21, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.J.; Vignali, P.D.A.; Mullett, S.J.; Overacre-Delgoffe, A.E.; Peralta, R.M.; Grebinoski, S.; Menk, A.V.; Rittenhouse, N.L.; DePeaux, K.; Whetstone, R.D.; et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 2021, 591, 645–651. [Google Scholar] [CrossRef]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.A.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K.; et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef]
- Jancewicz, I.; Szarkowska, J.; Konopinski, R.; Stachowiak, M.; Swiatek, M.; Blachnio, K.; Kubala, S.; Oksinska, P.; Cwiek, P.; Rusetska, N.; et al. PD-L1 Overexpression, SWI/SNF Complex Deregulation, and Profound Transcriptomic Changes Characterize Cancer-Dependent Exhaustion of Persistently Activated CD4(+) T Cells. Cancers 2021, 13, 4148. [Google Scholar] [CrossRef]
- Martins, C.P.; New, L.A.; O’Connor, E.C.; Previte, D.M.; Cargill, K.R.; Tse, I.L.; Sims-Lucas, S.; Piganelli, J.D. Glycolysis Inhibition Induces Functional and Metabolic Exhaustion of CD4(+) T Cells in Type 1 Diabetes. Front. Immunol. 2021, 12, 669456. [Google Scholar] [CrossRef]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Netea-Maier, R.T.; Smit, J.W.A.; Netea, M.G. Metabolic changes in tumor cells and tumor-associated macrophages: A mutual relationship. Cancer Lett. 2018, 413, 102–109. [Google Scholar] [CrossRef]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Dai, Y.; Tang, X.; Yin, T.; Wang, C.; Wang, T.; Dong, L.; Shi, M.; Qin, J.; et al. Single-cell RNA-seq analysis reveals BHLHE40-driven pro-tumour neutrophils with hyperactivated glycolysis in pancreatic tumour microenvironment. Gut 2022, 1–14. [Google Scholar] [CrossRef]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103.e6. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lu, Y.; Hang, J.; Zhang, J.; Zhang, T.; Huo, Y.; Liu, J.; Lai, S.; Luo, D.; Wang, L.; et al. Lactate-Modulated Immunosuppression of Myeloid-Derived Suppressor Cells Contributes to the Radioresistance of Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 1440–1451. [Google Scholar] [CrossRef]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987.e974. [Google Scholar] [CrossRef]
- Song, B.S.; Moon, J.S.; Tian, J.; Lee, H.Y.; Sim, B.C.; Kim, S.H.; Kang, S.G.; Kim, J.T.; Nga, H.T.; Benfeitas, R.; et al. Mitoribosomal defects aggravate liver cancer via aberrant glycolytic flux and T cell exhaustion. J. Immunother. Cancer 2022, 10, e004337. [Google Scholar] [CrossRef]
- Yu, Y.R.; Imrichova, H.; Wang, H.; Chao, T.; Xiao, Z.; Gao, M.; Rincon-Restrepo, M.; Franco, F.; Genolet, R.; Cheng, W.C.; et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat. Immunol. 2020, 21, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- Kooshki, L.; Mahdavi, P.; Fakhri, S.; Akkol, E.K.; Khan, H. Targeting lactate metabolism and glycolytic pathways in the tumor microenvironment by natural products: A promising strategy in combating cancer. Biofactors 2022, 48, 359–383. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Lewis, K.D.; Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Biomarkers of treatment benefit with atezolizumab plus vemurafenib plus cobimetinib in BRAF(V600) mutation-positive melanoma. Ann. Oncol. 2022, 33, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [Green Version]
- Mendler, A.N.; Hu, B.; Prinz, P.U.; Kreutz, M.; Gottfried, E.; Noessner, E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int. J. Cancer 2012, 131, 633–640. [Google Scholar] [CrossRef]
- Shime, H.; Yabu, M.; Akazawa, T.; Kodama, K.; Matsumoto, M.; Seya, T.; Inoue, N. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J. Immunol. 2008, 180, 7175–7183. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Zhao, Y.; Lee, W.C.; Ong, L.T.; Lee, P.L.; Jiang, Z.; Oguz, G.; Niu, Z.; Liu, M.; Goh, J.Y.; et al. Hypoxia induces HIF1alpha-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-PD-1 immunotherapy. Nat. Commun. 2022, 13, 4118. [Google Scholar] [CrossRef]
- Ford, B.R.; Vignali, P.D.A.; Rittenhouse, N.L.; Scharping, N.E.; Peralta, R.; Lontos, K.; Frisch, A.T.; Delgoffe, G.M.; Poholek, A.C. Tumor microenvironmental signals reshape chromatin landscapes to limit the functional potential of exhausted T cells. Sci. Immunol. 2022, 7, eabj9123. [Google Scholar] [CrossRef]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Li, J.; Sarde, A.; Lines, J.L.; Lee, Y.C.; Qian, D.C.; Pechenick, D.A.; Manivanh, R.; Le Mercier, I.; Lowrey, C.H.; et al. Hypoxia-Induced VISTA Promotes the Suppressive Function of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Cancer Immunol. Res. 2019, 7, 1079–1090. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.B.; Wong, C.W.; Liu, J.; Luo, X.J.; Zhou, W.Y.; Chen, Y.X.; Luo, H.Y.; Zeng, Z.L.; Ren, C.; Xie, X.M.; et al. Glucose metabolism inhibitor PFK-015 combined with immune checkpoint inhibitor is an effective treatment regimen in cancer. Oncoimmunology 2022, 11, 2079182. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Chow, E.; Yang, A.; Chung, C.H.L.; Chan, J.C.N. A Clinical Perspective of the Multifaceted Mechanism of Metformin in Diabetes, Infections, Cognitive Dysfunction, and Cancer. Pharmaceuticals 2022, 15, 442. [Google Scholar] [CrossRef]
- Rubiño, M.E.; Carrillo, E.; Alcalá, G.; Domínguez-Martín, A.; Marchal, J.A.; Boulaiz, H. Phenformin as an Anticancer Agent: Challenges and Prospects. Int. J. Mol. Sci. 2019, 20, 3316. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- MacIver, N.J.; Blagih, J.; Saucillo, D.C.; Tonelli, L.; Griss, T.; Rathmell, J.C.; Jones, R.G. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J. Immunol. 2011, 187, 4187–4198. [Google Scholar] [CrossRef] [Green Version]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Icard, P.; Simula, L.; Fournel, L.; Leroy, K.; Lupo, A.; Damotte, D.; Charpentier, M.C.; Durdux, C.; Loi, M.; Schussler, O.; et al. The strategic roles of four enzymes in the interconnection between metabolism and oncogene activation in non-small cell lung cancer: Therapeutic implications. Drug Resist. Updat. 2022, 63, 100852. [Google Scholar] [CrossRef]
- Xu, K.; Yin, N.; Peng, M.; Stamatiades, E.G.; Shyu, A.; Li, P.; Zhang, X.; Do, M.H.; Wang, Z.; Capistrano, K.J.; et al. Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Science 2021, 371, 405–410. [Google Scholar] [CrossRef]
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008, 15, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell 2016, 30, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.R.; Li, Q.; Odunsi, K.; Shrikant, P.A. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 2010, 32, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Madden, M.Z.; Rathmell, J.C. The Complex Integration of T-cell Metabolism and Immunotherapy. Cancer Discov. 2021, 11, 1636–1643. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Huang, J.; Huang, X.; Huang, J. CAR-T cell therapy for hematological malignancies: Limitations and optimization strategies. Front. Immunol. 2022, 13, 1019115. [Google Scholar] [CrossRef]
- Qin, X.; Wu, F.; Chen, C.; Li, Q. Recent advances in CAR-T cells therapy for colorectal cancer. Front. Immunol. 2022, 13, 904137. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D., Jr. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Cohen, S.; Guy, C.; Shrestha, S.; Neale, G.; Brown, S.A.; Cloer, C.; Kishton, R.J.; Gao, X.; Youngblood, B.; et al. mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity 2016, 45, 540–554. [Google Scholar] [CrossRef] [Green Version]
- Sabharwal, S.S.; Rosen, D.B.; Grein, J.; Tedesco, D.; Joyce-Shaikh, B.; Ueda, R.; Semana, M.; Bauer, M.; Bang, K.; Stevenson, C.; et al. GITR Agonism Enhances Cellular Metabolism to Support CD8(+) T-cell Proliferation and Effector Cytokine Production in a Mouse Tumor Model. Cancer Immunol. Res. 2018, 6, 1199–1211. [Google Scholar] [CrossRef] [Green Version]
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555. [Google Scholar] [CrossRef] [Green Version]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Shen, L.; Xiao, Y.; Zhang, C.; Li, S.; Teng, X.; Cui, L.; Liu, T.; Wu, N.; Lu, Z. Metabolic reprogramming by ex vivo glutamine inhibition endows CAR-T cells with less-differentiated phenotype and persistent antitumor activity. Cancer Lett. 2022, 538, 215710. [Google Scholar] [CrossRef]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Crompton, J.G.; Leonardi, A.J.; Yamamoto, T.N.; Chandran, S.S.; Eil, R.L.; Sukumar, M.; Vodnala, S.K.; Hu, J.; Ji, Y.; et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight 2017, 2, e95103. [Google Scholar] [CrossRef] [Green Version]
- Klein Geltink, R.I.; Edwards-Hicks, J.; Apostolova, P.; O’Sullivan, D.; Sanin, D.E.; Patterson, A.E.; Puleston, D.J.; Ligthart, N.A.M.; Buescher, J.M.; Grzes, K.M.; et al. Metabolic conditioning of CD8(+) effector T cells for adoptive cell therapy. Nat. Metab. 2020, 2, 703–716. [Google Scholar] [CrossRef]
- Gemta, L.F.; Siska, P.J.; Nelson, M.E.; Gao, X.; Liu, X.; Locasale, J.W.; Yagita, H.; Slingluff, C.L., Jr.; Hoehn, K.L.; Rathmell, J.C.; et al. Impaired enolase 1 glycolytic activity restrains effector functions of tumor-infiltrating CD8(+) T cells. Sci. Immunol. 2019, 4, eaap9520. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siska, P.J.; Beckermann, K.E.; Mason, F.M.; Andrejeva, G.; Greenplate, A.R.; Sendor, A.B.; Chiang, Y.J.; Corona, A.L.; Gemta, L.F.; Vincent, B.G.; et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2017, 2, e93411. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef]
- Lee, M.J.; Yun, S.J.; Lee, B.; Jeong, E.; Yoon, G.; Kim, K.; Park, S. Association of TIM-3 expression with glucose metabolism in Jurkat T cells. BMC Immunol. 2020, 21, 48. [Google Scholar] [CrossRef]
- Tsurutani, N.; Mittal, P.; St Rose, M.C.; Ngoi, S.M.; Svedova, J.; Menoret, A.; Treadway, F.B.; Laubenbacher, R.; Suarez-Ramirez, J.E.; Cauley, L.S.; et al. Costimulation Endows Immunotherapeutic CD8 T Cells with IL-36 Responsiveness during Aerobic Glycolysis. J. Immunol. 2016, 196, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Previte, D.M.; Martins, C.P.; O’Connor, E.C.; Marre, M.L.; Coudriet, G.M.; Beck, N.W.; Menk, A.V.; Wright, R.H.; Tse, H.M.; Delgoffe, G.M.; et al. Lymphocyte Activation Gene-3 Maintains Mitochondrial and Metabolic Quiescence in Naive CD4(+) T Cells. Cell Rep. 2019, 27, 129–141.e4. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Res 2016, 5, 2078. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.; Luo, S.; Guo, H.; Kong, D. Development of PI3Kalpha inhibitors for tumor therapy. J. Biomol. Struct. Dyn. 2022, 1–18. [Google Scholar] [CrossRef]
- Kopf, H.; de la Rosa, G.M.; Howard, O.M.; Chen, X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int. Immunopharmacol. 2007, 7, 1819–1824. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Siska, P.J.; van der Windt, G.J.; Kishton, R.J.; Cohen, S.; Eisner, W.; MacIver, N.J.; Kater, A.P.; Weinberg, J.B.; Rathmell, J.C. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J. Immunol. 2016, 197, 2532–2540. [Google Scholar] [CrossRef] [Green Version]
- Chapman, N.M.; Zeng, H.; Nguyen, T.M.; Wang, Y.; Vogel, P.; Dhungana, Y.; Liu, X.; Neale, G.; Locasale, J.W.; Chi, H. mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat. Commun. 2018, 9, 2095. [Google Scholar] [CrossRef]
- Nozhat, Z.; Mohammadi-Yeganeh, S.; Azizi, F.; Zarkesh, M.; Hedayati, M. Effects of metformin on the PI3K/AKT/FOXO1 pathway in anaplastic thyroid Cancer cell lines. Daru 2018, 26, 93–103. [Google Scholar] [CrossRef]
- Shao, S.; Zhao, L.; An, G.; Zhang, L.; Jing, X.; Luo, M.; Li, W.; Meng, D.; Ning, Q.; Zhao, X.; et al. Metformin suppresses HIF-1alpha expression in cancer-associated fibroblasts to prevent tumor-stromal cross talk in breast cancer. FASEB J. 2020, 34, 10860–10870. [Google Scholar] [CrossRef]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef] [Green Version]
- Chao, R.; Nishida, M.; Yamashita, N.; Tokumasu, M.; Zhao, W.; Kudo, I.; Udono, H. Nutrient Condition in the Microenvironment Determines Essential Metabolisms of CD8(+) T Cells for Enhanced IFNgamma Production by Metformin. Front. Immunol. 2022, 13, 864225. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, X.; Yang, X.; Li, W.; Li, S.; Hu, Z.; Ling, C.; Shi, R.; Liu, J.; Chen, G.; et al. Dual Blockade of Lactate/GPR81 and PD-1/PD-L1 Pathways Enhances the Anti-Tumor Effects of Metformin. Biomolecules 2021, 11, 1373. [Google Scholar] [CrossRef]
- Chung, Y.M.; Khan, P.P.; Wang, H.; Tsai, W.B.; Qiao, Y.; Yu, B.; Larrick, J.W.; Hu, M.C. Sensitizing tumors to anti-PD-1 therapy by promoting NK and CD8+ T cells via pharmacological activation of FOXO3. J. Immunother. Cancer 2021, 9, e002772. [Google Scholar] [CrossRef] [PubMed]
- Song, C.W.; Kim, H.; Cho, H.; Kim, M.S.; Paek, S.H.; Park, H.J.; Griffin, R.J.; Terezakis, S.; Cho, L.C. HIF-1alpha Inhibition Improves Anti-Tumor Immunity and Promotes the Efficacy of Stereotactic Ablative Radiotherapy (SABR). Cancers 2022, 14, 3273. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, S.; Zalpoor, H.; Hassan, Z.M. The promising therapeutic effects of metformin on metabolic reprogramming of cancer-associated fibroblasts in solid tumors. Cell. Mol. Biol. Lett. 2022, 27, 58. [Google Scholar] [CrossRef] [PubMed]
- Pouyssegur, J.; Marchiq, I.; Parks, S.K.; Durivault, J.; Zdralevic, M.; Vucetic, M. ‘Warburg effect’ controls tumor growth, bacterial, viral infections and immunity—Genetic deconstruction and therapeutic perspectives. Semin. Cancer Biol. 2022, 86, 334–346. [Google Scholar] [CrossRef]
- Luo, Y.; Li, Y.; Huang, Z.; Li, X.; Wang, Y.; Hou, J.; Zhou, S. A Nanounit Strategy Disrupts Energy Metabolism and Alleviates Immunosuppression for Cancer Therapy. Nano Lett. 2022, 22, 6418–6427. [Google Scholar] [CrossRef]
- Xu, R.; Wu, M.; Liu, S.; Shang, W.; Li, R.; Xu, J.; Huang, L.; Wang, F. Glucose metabolism characteristics and TLR8-mediated metabolic control of CD4(+) Treg cells in ovarian cancer cells microenvironment. Cell Death Dis. 2021, 12, 22. [Google Scholar] [CrossRef]
- Guo, D.; Tong, Y.; Jiang, X.; Meng, Y.; Jiang, H.; Du, L.; Wu, Q.; Li, S.; Luo, S.; Li, M.; et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2-mediated phosphorylation of IkappaBalpha. Cell Metab. 2022, 34, 1312–1324.e6. [Google Scholar] [CrossRef]
- Wu, L.; Hollinshead, K.E.R.; Hao, Y.; Au, C.; Kroehling, L.; Ng, C.; Lin, W.Y.; Li, D.; Silva, H.M.; Shin, J.; et al. Niche-Selective Inhibition of Pathogenic Th17 Cells by Targeting Metabolic Redundancy. Cell 2020, 182, 641–654.e20. [Google Scholar] [CrossRef]
- Lei, J.; Yang, Y.; Lu, Z.; Pan, H.; Fang, J.; Jing, B.; Chen, Y.; Yin, L. Taming metabolic competition via glycolysis inhibition for safe and potent tumor immunotherapy. Biochem. Pharmacol. 2022, 202, 115153. [Google Scholar] [CrossRef]
- Shan, Y.; Ni, Q.; Zhang, Q.; Zhang, M.; Wei, B.; Cheng, L.; Zhong, C.; Wang, X.; Wang, Q.; Liu, J.; et al. Targeting tumor endothelial hyperglycolysis enhances immunotherapy through remodeling tumor microenvironment. Acta Pharm. Sin. B 2022, 12, 1825–1839. [Google Scholar] [CrossRef]
- Chen, D.P.; Ning, W.R.; Jiang, Z.Z.; Peng, Z.P.; Zhu, L.Y.; Zhuang, S.M.; Kuang, D.M.; Zheng, L.; Wu, Y. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3-PD-L1 axis in human hepatocellular carcinoma. J. Hepatol. 2019, 71, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, H.; Gu, H.; Zhang, E.; He, J.; Cao, W.; Qu, J.; Xu, R.; Cao, L.; He, D.; et al. Myeloma-derived IL-32gamma induced PD-L1 expression in macrophages facilitates immune escape via the PFKFB3-JAK1 axis. Oncoimmunology 2022, 11, 2057837. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liang, Y.; Li, D.; Wang, L.; Liang, Z.; Chen, Y.; Ma, G.; Wu, H.; Jiao, W.; Niu, H. Glucose metabolism involved in PD-L1-mediated immune escape in the malignant kidney tumour microenvironment. Cell Death Discov. 2021, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; Dyck, L.; Zaslona, Z.; Menon, D.; McGettrick, A.F.; Mills, K.H.G.; O’Neill, L.A. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front. Immunol. 2017, 8, 1300. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Jia, J.; Hu, C.; Lu, J.; Li, J.; Xu, H.; Fang, J.; Feng, D.; Wang, L.; Chen, Y. Tumor-associated macrophages promote PD-L1 expression in tumor cells by regulating PKM2 nuclear translocation in pancreatic ductal adenocarcinoma. Oncogene 2022, 41, 865–877. [Google Scholar] [CrossRef]
- Xu, W.; Yang, W.; Wu, C.; Ma, X.; Li, H.; Zheng, J. Enolase 1 Correlated with Cancer Progression and Immune-Infiltrating in Multiple Cancer Types: A Pan-Cancer Analysis. Front. Oncol. 2020, 10, 593706. [Google Scholar] [CrossRef]
- Cappello, P.; Tomaino, B.; Chiarle, R.; Ceruti, P.; Novarino, A.; Castagnoli, C.; Migliorini, P.; Perconti, G.; Giallongo, A.; Milella, M.; et al. An integrated humoral and cellular response is elicited in pancreatic cancer by alpha-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int. J. Cancer 2009, 125, 639–648. [Google Scholar] [CrossRef]
- Cappello, P.; Rolla, S.; Chiarle, R.; Principe, M.; Cavallo, F.; Perconti, G.; Feo, S.; Giovarelli, M.; Novelli, F. Vaccination with ENO1 DNA prolongs survival of genetically engineered mice with pancreatic cancer. Gastroenterology 2013, 144, 1098–1106. [Google Scholar] [CrossRef] [Green Version]
- Cappello, P.; Principe, M.; Bulfamante, S.; Novelli, F. Alpha-Enolase (ENO1), a potential target in novel immunotherapies. Front. Biosci. (Landmark Ed.) 2017, 22, 944–959. [Google Scholar] [CrossRef]
- Mandili, G.; Curcio, C.; Bulfamante, S.; Follia, L.; Ferrero, G.; Mazza, E.; Principe, M.; Cordero, F.; Satolli, M.A.; Spadi, R.; et al. In pancreatic cancer, chemotherapy increases antitumor responses to tumor-associated antigens and potentiates DNA vaccination. J. Immunother. Cancer 2020, 8, e001071. [Google Scholar] [CrossRef]
- Curcio, C.; Brugiapaglia, S.; Bulfamante, S.; Follia, L.; Cappello, P.; Novelli, F. The Glycolytic Pathway as a Target for Novel Onco-Immunology Therapies in Pancreatic Cancer. Molecules 2021, 26, 1642. [Google Scholar] [CrossRef]
- Chen, M.L.; Yuan, T.T.; Chuang, C.F.; Huang, Y.T.; Chung, I.C.; Huang, W.C. A Novel Enolase-1 Antibody Targets Multiple Interacting Players in the Tumor Microenvironment of Advanced Prostate Cancer. Mol. Cancer Ther. 2022, 21, 1337–1347. [Google Scholar] [CrossRef]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Reeves, G.K.; Beral, V.; Green, J.; Gathani, T.; Bull, D.; Million Women Study, C. Hormonal therapy for menopause and breast-cancer risk by histological type: A cohort study and meta-analysis. Lancet Oncol. 2006, 7, 910–918. [Google Scholar] [CrossRef]
- Rugo, H.S.; Rumble, R.B.; Macrae, E.; Barton, D.L.; Connolly, H.K.; Dickler, M.N.; Fallowfield, L.; Fowble, B.; Ingle, J.N.; Jahanzeb, M.; et al. Endocrine Therapy for Hormone Receptor-Positive Metastatic Breast Cancer: American Society of Clinical Oncology Guideline. J. Clin. Oncol. 2016, 34, 3069–3103. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- Fiorillo, M.; Sanchez-Alvarez, R.; Sotgia, F.; Lisanti, M.P. The ER-alpha mutation Y537S confers Tamoxifen-resistance via enhanced mitochondrial metabolism, glycolysis and Rho-GDI/PTEN signaling: Implicating TIGAR in somatic resistance to endocrine therapy. Aging 2018, 10, 4000–4023. [Google Scholar] [CrossRef]
- Kulkoyluoglu-Cotul, E.; Arca, A.; Madak-Erdogan, Z. Crosstalk between Estrogen Signaling and Breast Cancer Metabolism. Trends Endocrinol. Metab. 2019, 30, 25–38. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosio, M.R.; D’Esposito, V.; Costa, V.; Liguoro, D.; Collina, F.; Cantile, M.; Prevete, N.; Passaro, C.; Mosca, G.; De Laurentiis, M.; et al. Glucose impairs tamoxifen responsiveness modulating connective tissue growth factor in breast cancer cells. Oncotarget 2017, 8, 109000–109017. [Google Scholar] [CrossRef] [PubMed]
- Rivenzon-Segal, D.; Boldin-Adamsky, S.; Seger, D.; Seger, R.; Degani, H. Glycolysis and glucose transporter 1 as markers of response to hormonal therapy in breast cancer. Int. J. Cancer 2003, 107, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yang, Y.; Qian, K.; Li, L.; Zhang, C.; Fu, X.; Zhang, X.; Chen, H.; Liu, Q.; Cao, S.; et al. A novel tumor suppressor ZBTB1 regulates tamoxifen resistance and aerobic glycolysis through suppressing HER2 expression in breast cancer. J. Biol. Chem. 2020, 295, 14140–14152. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Yang, S.; Zhu, Y.; Zhai, X.; Li, S.; Tao, X.; Dong, D. Relationship between metabolic reprogramming and drug resistance in breast cancer. Front. Oncol. 2022, 12, 942064. [Google Scholar] [CrossRef]
- Woo, Y.M.; Shin, Y.; Lee, E.J.; Lee, S.; Jeong, S.H.; Kong, H.K.; Park, E.Y.; Kim, H.K.; Han, J.; Chang, M.; et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1alpha Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLoS ONE 2015, 10, e0132285. [Google Scholar] [CrossRef]
- Steifensand, F.; Gallwas, J.; Bauerschmitz, G.; Grundker, C. Inhibition of Metabolism as a Therapeutic Option for Tamoxifen-Resistant Breast Cancer Cells. Cells 2021, 10, 2398. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Y.; Warden, C.; Chen, S. Cross-talk between ER and HER2 regulates c-MYC-mediated glutamine metabolism in aromatase inhibitor resistant breast cancer cells. J. Steroid Biochem. Mol. Biol. 2015, 149, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Sevigny, C.M.; Liu, X.; Jin, L.; Pohlmann, P.R.; Clarke, R. Targeting glycolysis enzyme, PFKFB3, in endocrine therapy resistant breast cancers. Cancer Res. 2018, 78, 907. [Google Scholar] [CrossRef]
- Jones, B.C.; Sengupta, S.; Sevigny, C.M.; Jin, L.; Pohlmann, P.R.; Shajahan-Haq, A.; Clarke, R. Pfkfb3 inhibition significantly decreases endocrine-resistant breast cancer growth and induces necroptotic cell death. In Proceedings of the San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 7–10 December 2021. [Google Scholar]
- Bacci, M.; Giannoni, E.; Fearns, A.; Ribas, R.; Gao, Q.; Taddei, M.L.; Pintus, G.; Dowsett, M.; Isacke, C.M.; Martin, L.A.; et al. miR-155 Drives Metabolic Reprogramming of ER+ Breast Cancer Cells Following Long-Term Estrogen Deprivation and Predicts Clinical Response to Aromatase Inhibitors. Cancer Res. 2016, 76, 1615–1626. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Q.; Mogol, A.N.; Liu, Y.J.; Santaliz Casiano, A.; Chien, C.; Drnevich, J.; Imir, O.B.; Kulkoyluoglu-Cotul, E.; Park, N.H.; Shapiro, D.J.; et al. Targeting Metabolic Adaptations in the Breast Cancer-Liver Metastatic Niche Using Dietary Approaches to Improve Endocrine Therapy Efficacy. Mol. Cancer Res. 2022, 20, 923–937. [Google Scholar] [CrossRef]
- Blundon, M.A.; Dasgupta, S. Metabolic Dysregulation Controls Endocrine Therapy-Resistant Cancer Recurrence and Metastasis. Endocrinology 2019, 160, 1811–1820. [Google Scholar] [CrossRef]
- Pertega-Gomes, N.; Felisbino, S.; Massie, C.E.; Vizcaino, J.R.; Coelho, R.; Sandi, C.; Simoes-Sousa, S.; Jurmeister, S.; Ramos-Montoya, A.; Asim, M.; et al. A glycolytic phenotype is associated with prostate cancer progression and aggressiveness: A role for monocarboxylate transporters as metabolic targets for therapy. J. Pathol. 2015, 236, 517–530. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xu, W.; Wang, B.; Lin, G.; Wei, Y.; Abudurexiti, M.; Zhu, W.; Liu, C.; Qin, X.; Dai, B.; et al. GLUT1 is an AR target contributing to tumor growth and glycolysis in castration-resistant and enzalutamide-resistant prostate cancers. Cancer Lett. 2020, 485, 45–55. [Google Scholar] [CrossRef]
- Gonzalez-Menendez, P.; Hevia, D.; Alonso-Arias, R.; Alvarez-Artime, A.; Rodriguez-Garcia, A.; Kinet, S.; Gonzalez-Pola, I.; Taylor, N.; Mayo, J.C.; Sainz, R.M. GLUT1 protects prostate cancer cells from glucose deprivation-induced oxidative stress. Redox Biol. 2018, 17, 112–127. [Google Scholar] [CrossRef]
- Gonzalez-Menendez, P.; Hevia, D.; Mayo, J.C.; Sainz, R.M. The dark side of glucose transporters in prostate cancer: Are they a new feature to characterize carcinomas? Int. J. Cancer 2018, 142, 2414–2424. [Google Scholar] [CrossRef]
- Cui, Y.; Nadiminty, N.; Liu, C.; Lou, W.; Schwartz, C.T.; Gao, A.C. Upregulation of glucose metabolism by NF-kappaB2/p52 mediates enzalutamide resistance in castration-resistant prostate cancer cells. Endocr. Relat. Cancer 2014, 21, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Efstathiou, E.; Titus, M.; Wen, S.; Hoang, A.; Karlou, M.; Ashe, R.; Tu, S.M.; Aparicio, A.; Troncoso, P.; Mohler, J.; et al. Molecular characterization of enzalutamide-treated bone metastatic castration-resistant prostate cancer. Eur. Urol. 2015, 67, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Atif, F.; Yousuf, S.; Stein, D.G. Anti-tumor effects of progesterone in human glioblastoma multiforme: Role of PI3K/Akt/mTOR signaling. J. Steroid Biochem. Mol. Biol. 2015, 146, 62–73. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Cengel, K.A.; Simone, C.B., 2nd; Glatstein, E. PDT: What’s Past Is Prologue. Cancer Res. 2016, 76, 2497–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblin, M.R. Photodynamic Therapy for Cancer: What’s Past is Prologue. Photochem. Photobiol. 2020, 96, 506–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Ogura, S.; Maruyama, K.; Hagiya, Y.; Sugiyama, Y.; Tsuchiya, K.; Takahashi, K.; Abe, F.; Tabata, K.; Okura, I.; Nakajima, M.; et al. The effect of 5-aminolevulinic acid on cytochrome c oxidase activity in mouse liver. BMC Res. Notes 2011, 4, 66. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Koda, A.; Nozawa, N.; Ota, U.; Kondo, H.; Nakagawa, H.; Kamiya, A.; Miyashita, K.; Itoh, H.; Nakajima, M.; et al. Combination of 5-aminolevulinic acid and ferrous ion reduces plasma glucose and hemoglobin A1c levels in Zucker diabetic fatty rats. FEBS Open Bio 2016, 6, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Grigalavicius, M.; Ezzatpanah, S.; Papakyriakou, A.; Raabe, T.T.H.; Yannakopoulou, K.; Theodossiou, T.A. 5-ALA Is a Potent Lactate Dehydrogenase Inhibitor but Not a Substrate: Implications for Cell Glycolysis and New Avenues in 5-ALA-Mediated Anticancer Action. Cancers 2022, 14, 4003. [Google Scholar] [CrossRef]
- Kaur, P.; Nagar, S.; Bhagwat, M.; Uddin, M.; Zhu, Y.; Vancurova, I.; Vancura, A. Activated heme synthesis regulates glycolysis and oxidative metabolism in breast and ovarian cancer cells. PLoS ONE 2021, 16, e0260400. [Google Scholar] [CrossRef]
- Simcox, J.A.; Mitchell, T.C.; Gao, Y.; Just, S.F.; Cooksey, R.; Cox, J.; Ajioka, R.; Jones, D.; Lee, S.H.; King, D.; et al. Dietary iron controls circadian hepatic glucose metabolism through heme synthesis. Diabetes 2015, 64, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Yasuzawa, T.; Uesaka, A.; Izumi, Y.; Kamiya, A.; Tsuchiya, K.; Kobayashi, Y.; Kuwahata, M.; Kido, Y. Type 2 diabetic conditions in Otsuka Long-Evans Tokushima Fatty rats are ameliorated by 5-aminolevulinic acid. Nutr. Res. 2014, 34, 544–551. [Google Scholar] [CrossRef]
- Handschin, C.; Lin, J.; Rhee, J.; Peyer, A.K.; Chin, S.; Wu, P.H.; Meyer, U.A.; Spiegelman, B.M. Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell 2005, 122, 505–515. [Google Scholar] [CrossRef] [Green Version]
- McNicholas, K.; MacGregor, M.N.; Gleadle, J.M. In order for the light to shine so brightly, the darkness must be present-why do cancers fluoresce with 5-aminolaevulinic acid? Br. J. Cancer 2019, 121, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Golding, J.P.; Wardhaugh, T.; Patrick, L.; Turner, M.; Phillips, J.B.; Bruce, J.I.; Kimani, S.G. Targeting tumour energy metabolism potentiates the cytotoxicity of 5-aminolevulinic acid photodynamic therapy. Br. J. Cancer 2013, 109, 976–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, A.; Tsuji, D.; Miki, H.; Cui, Q.; El Sayed, S.M.; Ikegame, A.; Oda, A.; Amou, H.; Nakamura, S.; Harada, T.; et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS ONE 2011, 6, e27222. [Google Scholar] [CrossRef] [PubMed]
- Hagiya, Y.; Fukuhara, H.; Matsumoto, K.; Endo, Y.; Nakajima, M.; Tanaka, T.; Okura, I.; Kurabayashi, A.; Furihata, M.; Inoue, K.; et al. Expression levels of PEPT1 and ABCG2 play key roles in 5-aminolevulinic acid (ALA)-induced tumor-specific protoporphyrin IX (PpIX) accumulation in bladder cancer. Photodiagnosis Photodyn. Ther. 2013, 10, 288–295. [Google Scholar] [CrossRef]
- Kobuchi, H.; Moriya, K.; Ogino, T.; Fujita, H.; Inoue, K.; Shuin, T.; Yasuda, T.; Utsumi, K.; Utsumi, T. Mitochondrial localization of ABC transporter ABCG2 and its function in 5-aminolevulinic acid-mediated protoporphyrin IX accumulation. PLoS ONE 2012, 7, e50082. [Google Scholar] [CrossRef] [Green Version]
- Alkarakooly, Z.; Al-Anbaky, Q.A.; Kannan, K.; Ali, N. Metabolic reprogramming by Dichloroacetic acid potentiates photodynamic therapy of human breast adenocarcinoma MCF-7 cells. PLoS ONE 2018, 13, e0206182. [Google Scholar] [CrossRef]
- Kiesslich, T.; Plaetzer, K.; Oberdanner, C.B.; Berlanda, J.; Obermair, F.J.; Krammer, B. Differential effects of glucose deprivation on the cellular sensitivity towards photodynamic treatment-based production of reactive oxygen species and apoptosis-induction. FEBS Lett. 2005, 579, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Lilge, L.; Belcuig, M.; Singh, G.; Grenier, J.; Li, Y.; Chopp, M. Photodynamic therapy using Photofrin in combination with buthionine sulfoximine (BSO) to treat 9L gliosarcoma in rat brain. Lasers Surg. Med. 1998, 23, 161–166. [Google Scholar] [CrossRef]
- Perotti, C.; Casas, A.; Del, C.B.A.M. Scavengers protection of cells against ALA-based photodynamic therapy-induced damage. Lasers Med. Sci. 2002, 17, 222–229. [Google Scholar] [CrossRef]
- Mascaraque-Checa, M.; Gallego-Rentero, M.; Nicolas-Morala, J.; Portillo-Esnaola, M.; Cuezva, J.M.; Gonzalez, S.; Gilaberte, Y.; Juarranz, A. Metformin overcomes metabolic reprogramming-induced resistance of skin squamous cell carcinoma to photodynamic therapy. Mol. Metab. 2022, 60, 101496. [Google Scholar] [CrossRef]
- Xie, R.; Xu, T.; Zhu, J.; Wei, X.; Zhu, W.; Li, L.; Wang, Y.; Han, Y.; Zhou, J.; Bai, Y. The Combination of Glycolytic Inhibitor 2-Deoxyglucose and Microbubbles Increases the Effect of 5-Aminolevulinic Acid-Sonodynamic Therapy in Liver Cancer Cells. Ultrasound Med. Biol. 2017, 43, 2640–2650. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, Y.; Wang, P.; Liu, Q.; Wang, X. Energy metabolism targeted drugs synergize with photodynamic therapy to potentiate breast cancer cell death. Photochem. Photobiol. Sci. 2014, 13, 1793–1803. [Google Scholar] [CrossRef]
- Yang, X.; Palasuberniam, P.; Myers, K.A.; Wang, C.; Chen, B. Her2 oncogene transformation enhances 5-aminolevulinic acid-mediated protoporphyrin IX production and photodynamic therapy response. Oncotarget 2016, 7, 57798–57810. [Google Scholar] [CrossRef] [Green Version]
- Zhong, G.; Li, Q.; Luo, Y.; Liu, Y.; Liu, D.; Li, B.; Wang, T. FECH Expression Correlates with the Prognosis and Tumor Immune Microenvironment in Clear Cell Renal Cell Carcinoma. J. Oncol. 2022, 2022, 8943643. [Google Scholar] [CrossRef]
- Chelakkot, V.S.; Liu, K.; Yoshioka, E.; Saha, S.; Xu, D.; Licursi, M.; Dorward, A.; Hirasawa, K. MEK reduces cancer-specific PpIX accumulation through the RSK-ABCB1 and HIF-1alpha-FECH axes. Sci. Rep. 2020, 10, 22124. [Google Scholar] [CrossRef]
- Yoshioka, E.; Chelakkot, V.S.; Licursi, M.; Rutihinda, S.G.; Som, J.; Derwish, L.; King, J.J.; Pongnopparat, T.; Mearow, K.; Larijani, M.; et al. Enhancement of Cancer-Specific Protoporphyrin IX Fluorescence by Targeting Oncogenic Ras/MEK Pathway. Theranostics 2018, 8, 2134–2146. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Franklin, D.A.; Sharick, J.T.; Ericsson-Gonzalez, P.I.; Sanchez, V.; Dean, P.T.; Opalenik, S.R.; Cairo, S.; Judde, J.G.; Lewis, M.T.; Chang, J.C.; et al. MEK activation modulates glycolysis and supports suppressive myeloid cells in TNBC. JCI Insight 2020, 5, e134290. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Yang, M.; Soga, T.; Pollard, P.J. Oncometabolites: Linking altered metabolism with cancer. J. Clin. Investig. 2013, 123, 3652–3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groves, A.M.; Win, T.; Haim, S.B.; Ell, P.J. Non-[18F] FDG PET in clinical oncology. Lancet Oncol. 2007, 8, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Q.; Huang, Z.; Li, B.; Nice, E.C.; Huang, C.; Wei, L.; Zou, B. Targeting Glucose Metabolism Enzymes in Cancer Treatment: Current and Emerging Strategies. Cancers 2022, 14, 4568. [Google Scholar] [CrossRef]
- Zdralevic, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.M.; et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Cassim, S.; Vucetic, M.; Zdralevic, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef]
- Noble, R.A.; Thomas, H.; Zhao, Y.; Herendi, L.; Howarth, R.; Dragoni, I.; Keun, H.C.; Vellano, C.P.; Marszalek, J.R.; Wedge, S.R. Simultaneous targeting of glycolysis and oxidative phosphorylation as a therapeutic strategy to treat diffuse large B-cell lymphoma. Br. J. Cancer 2022, 127, 937–947. [Google Scholar] [CrossRef]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Yan, X.; Chen, J.; Zhan, Q.; Hua, Y.; Xu, S.; Li, Z.; Wang, Z.; Dong, Y.; Zuo, D.; et al. Hexokinase 2 discerns a novel circulating tumor cell population associated with poor prognosis in lung cancer patients. Proc. Natl. Acad. Sci. USA 2021, 118, e2012228118. [Google Scholar] [CrossRef]
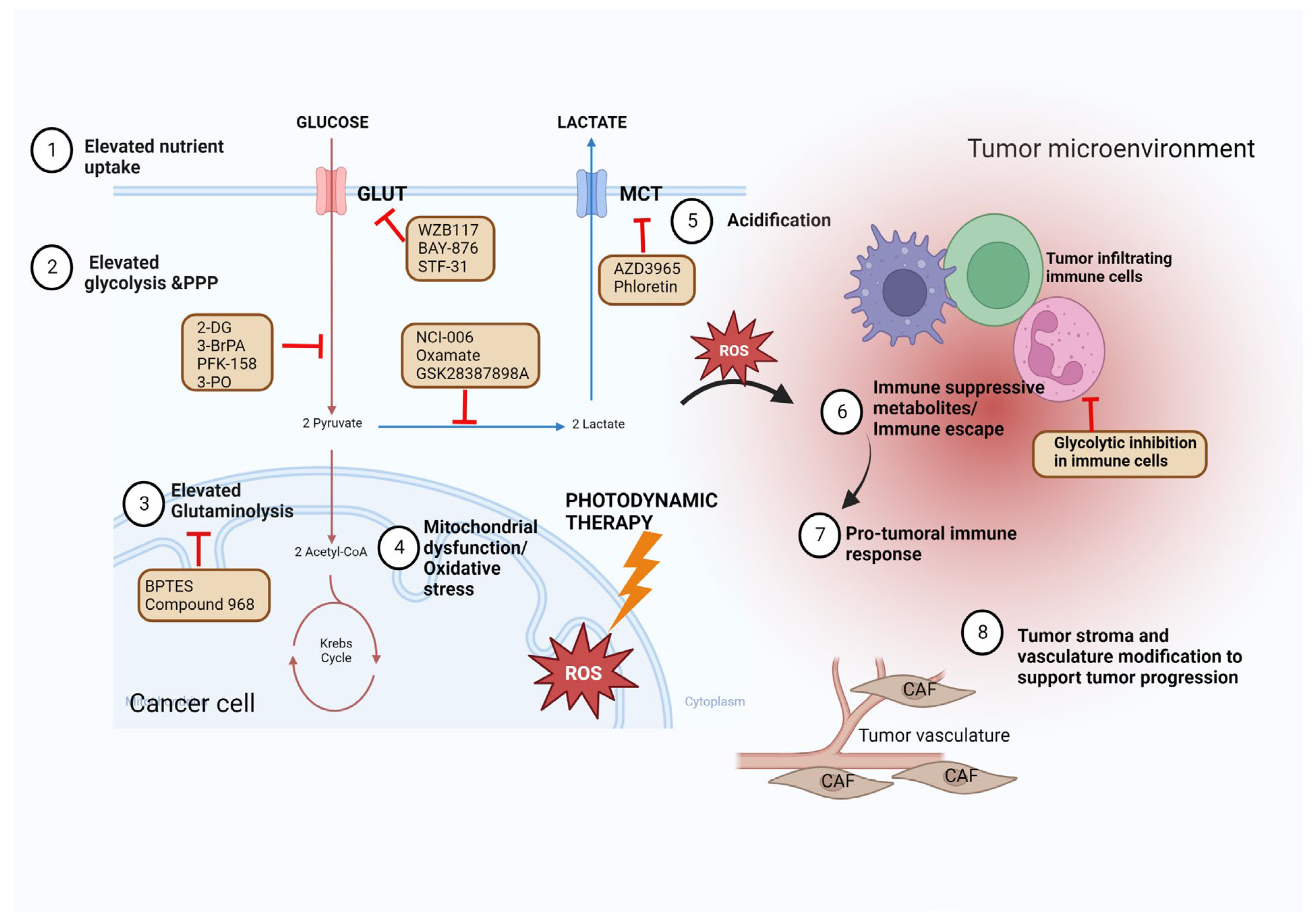
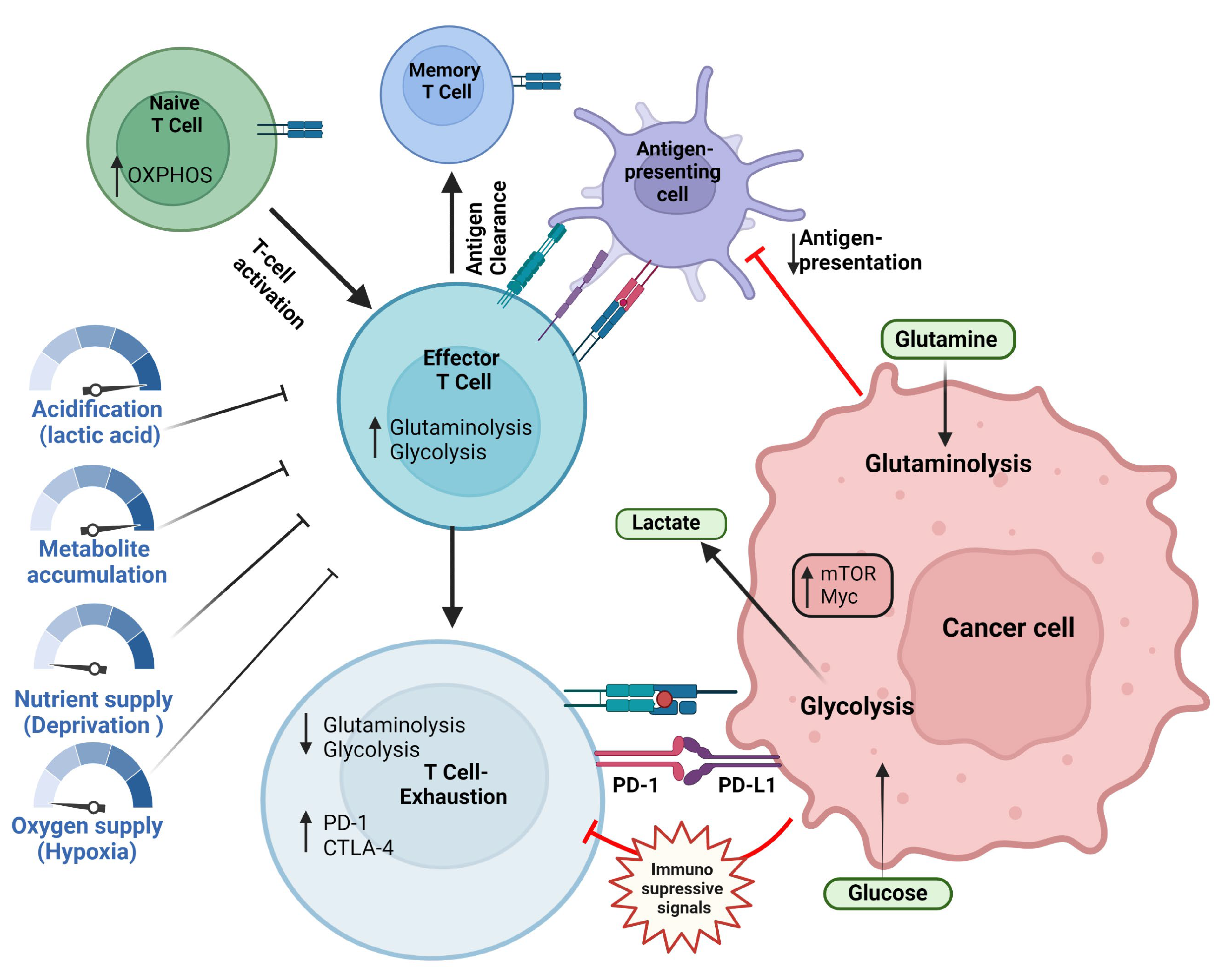
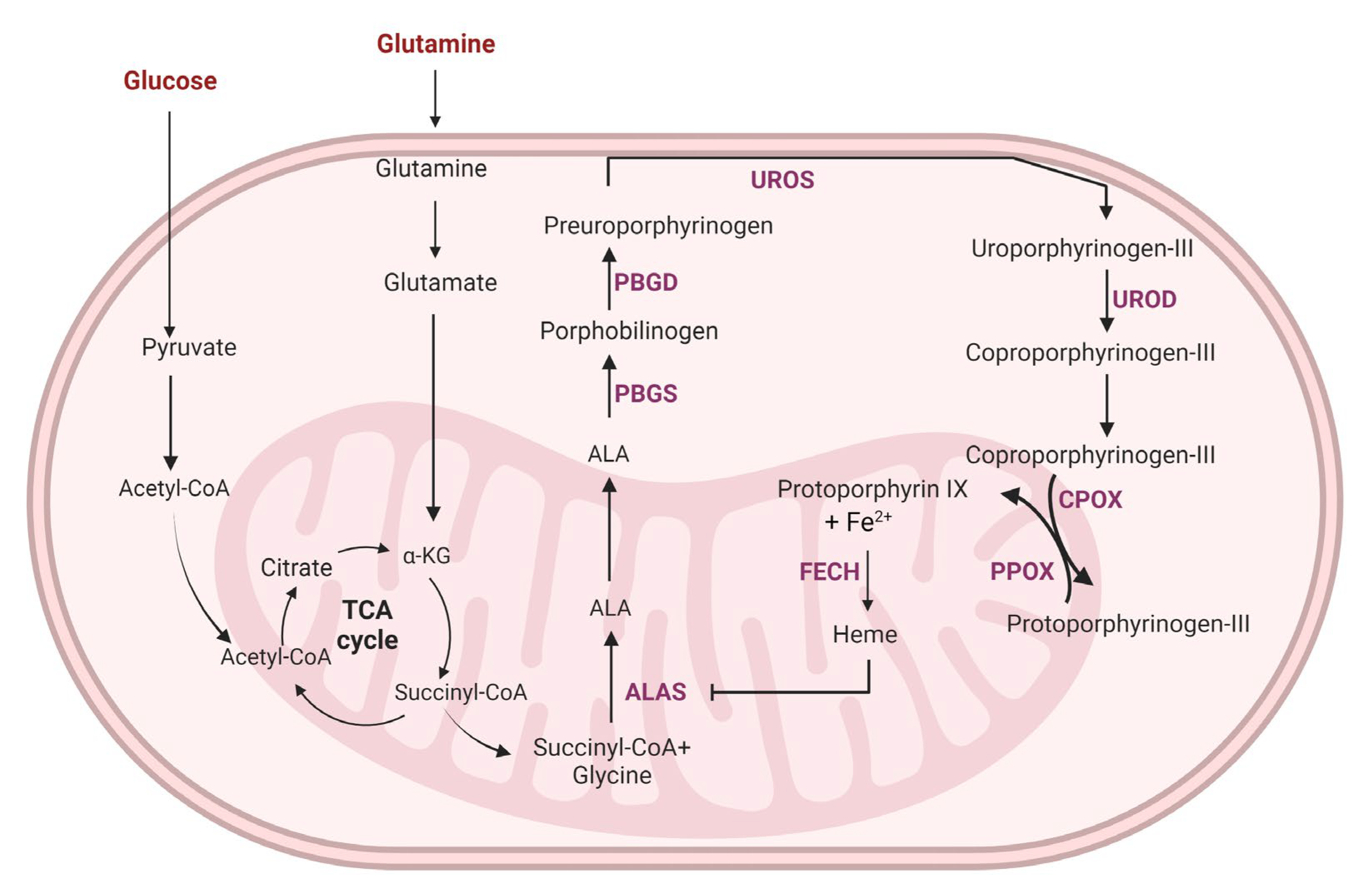
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chelakkot, C.; Chelakkot, V.S.; Shin, Y.; Song, K. Modulating Glycolysis to Improve Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 2606. https://doi.org/10.3390/ijms24032606
Chelakkot C, Chelakkot VS, Shin Y, Song K. Modulating Glycolysis to Improve Cancer Therapy. International Journal of Molecular Sciences. 2023; 24(3):2606. https://doi.org/10.3390/ijms24032606
Chicago/Turabian StyleChelakkot, Chaithanya, Vipin Shankar Chelakkot, Youngkee Shin, and Kyoung Song. 2023. "Modulating Glycolysis to Improve Cancer Therapy" International Journal of Molecular Sciences 24, no. 3: 2606. https://doi.org/10.3390/ijms24032606
APA StyleChelakkot, C., Chelakkot, V. S., Shin, Y., & Song, K. (2023). Modulating Glycolysis to Improve Cancer Therapy. International Journal of Molecular Sciences, 24(3), 2606. https://doi.org/10.3390/ijms24032606