
MetaSEM: Gene Regulatory Network Inference from Single-Cell RNA Data by Meta-Learning



Abstract
1. Introduction
2. Results and Discussion
2.1. Comparison with Existing Methods
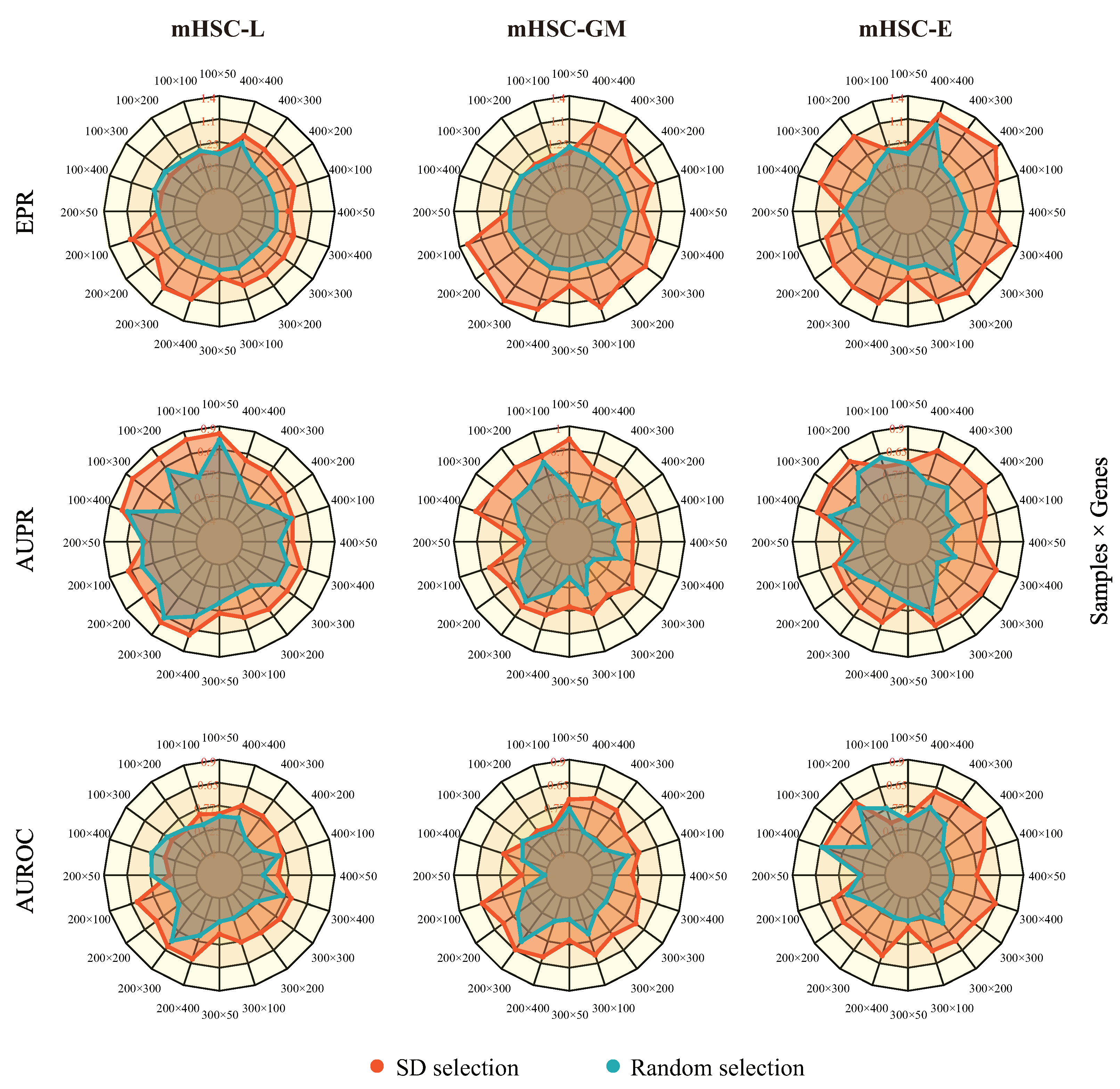
2.2. MetaSEM Can Adapt to High Dimensions and Sparse Characteristics
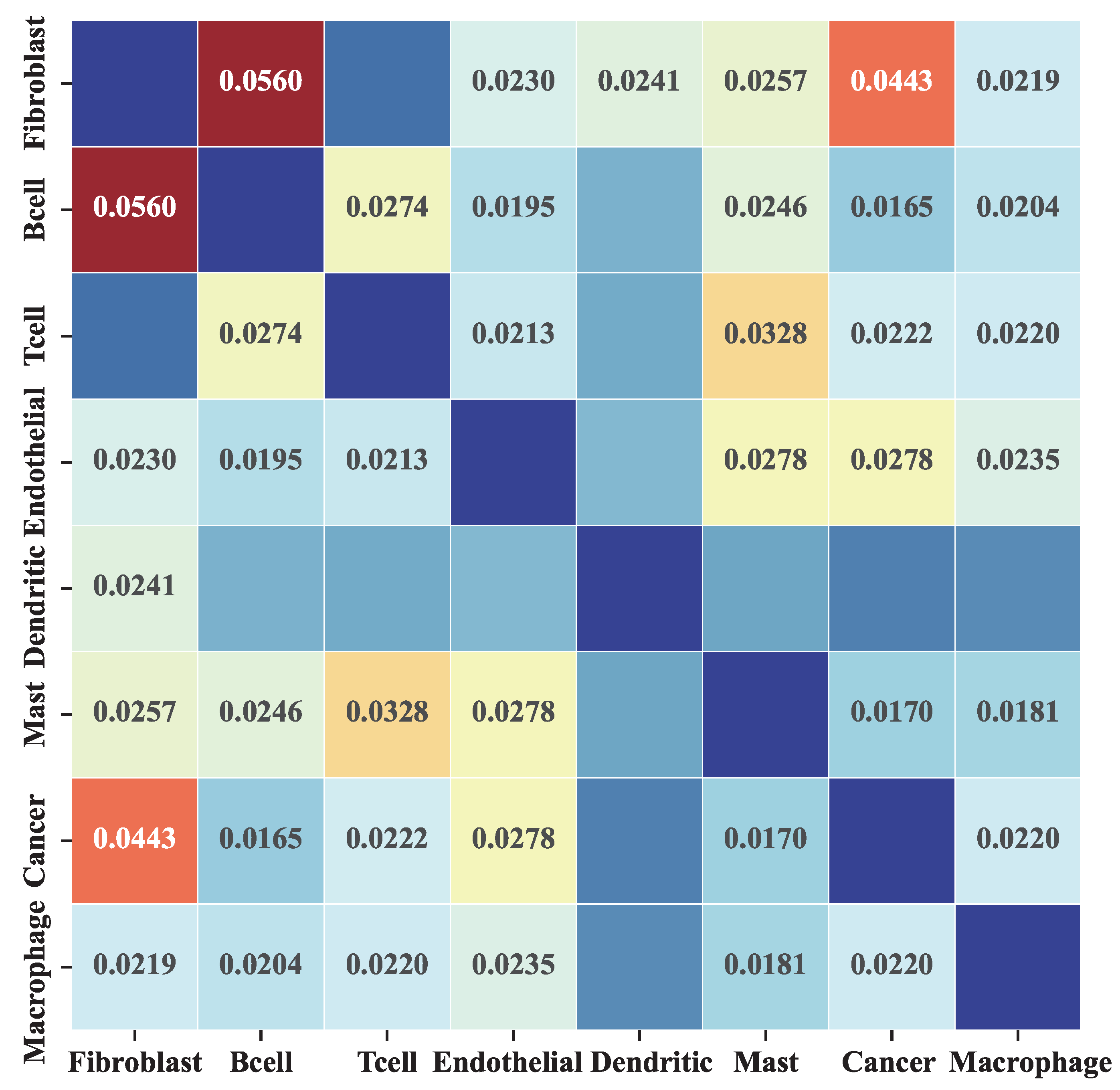
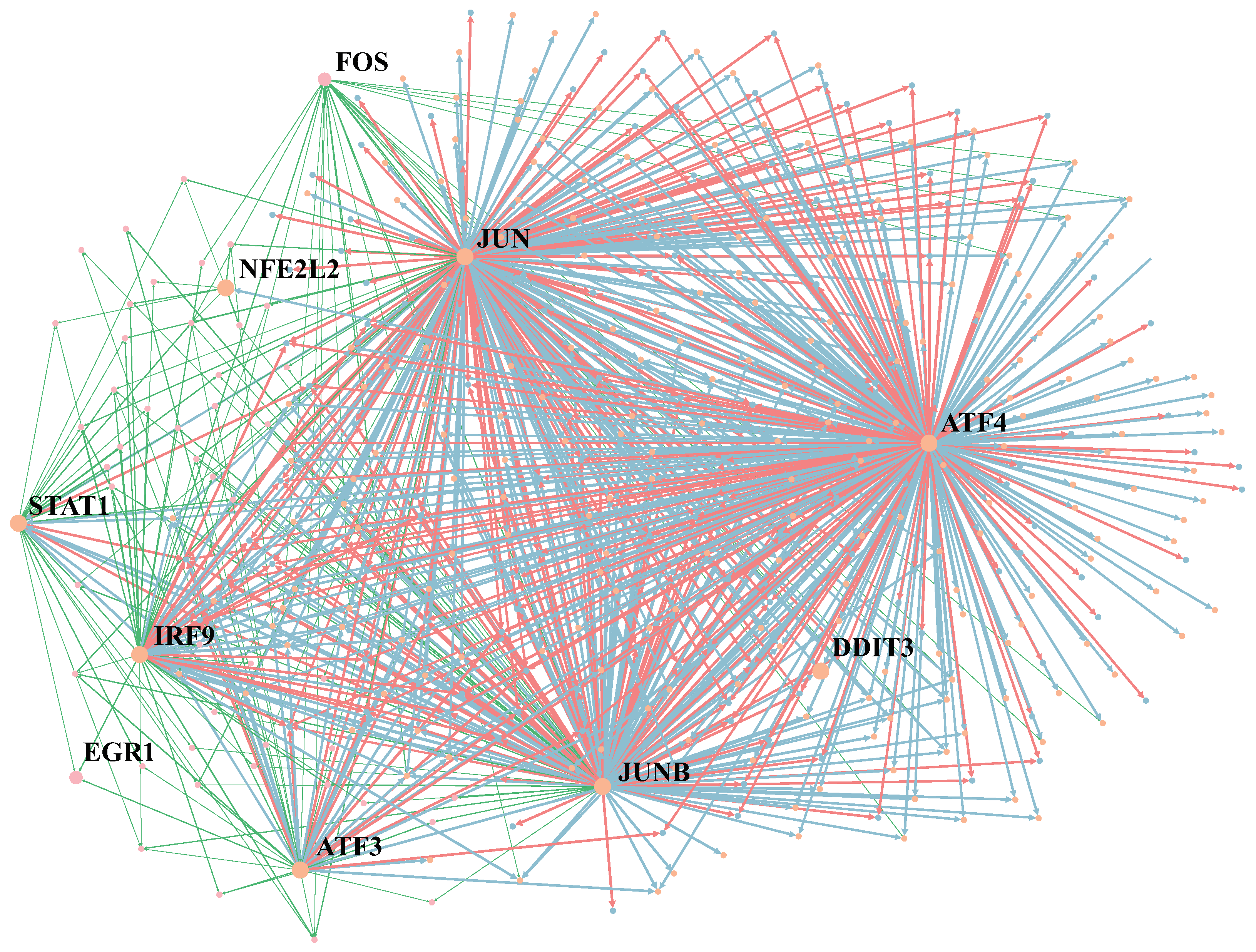
2.3. MetaSEM Reveals That GRN Specificity Is Related to Gene Expression
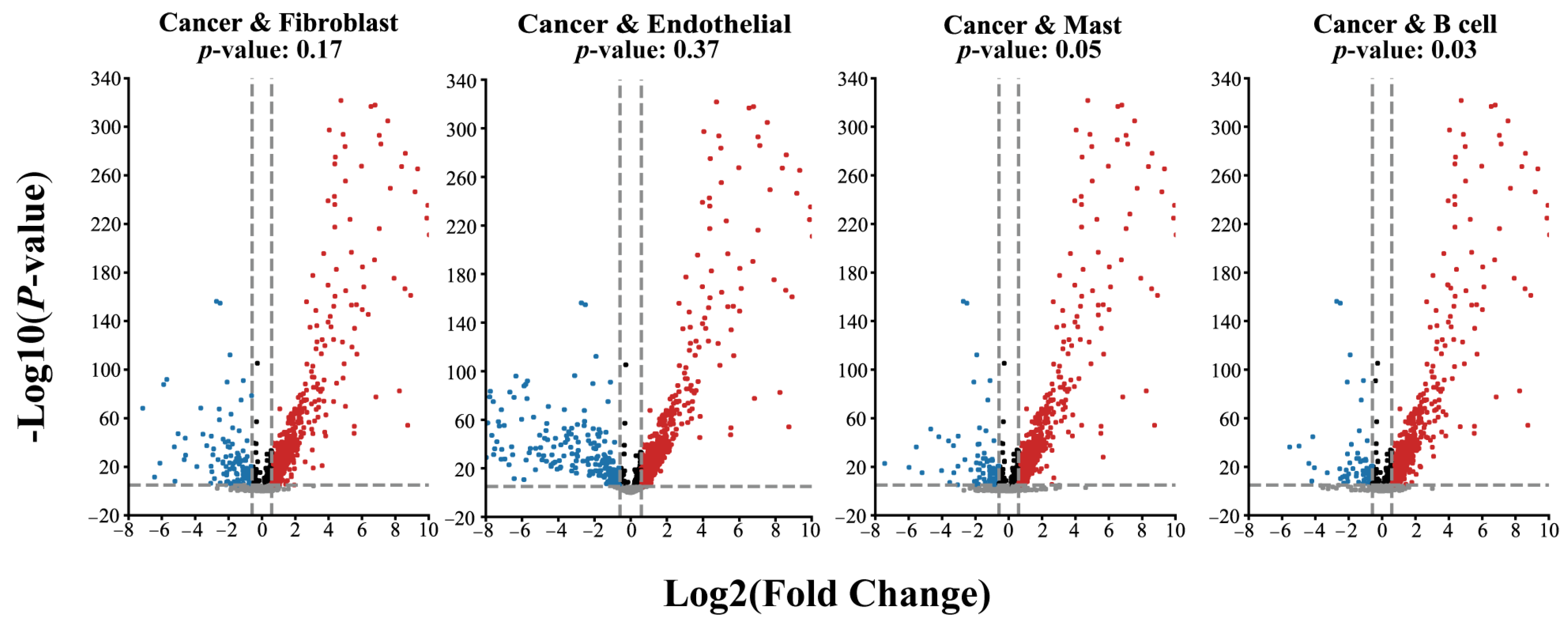
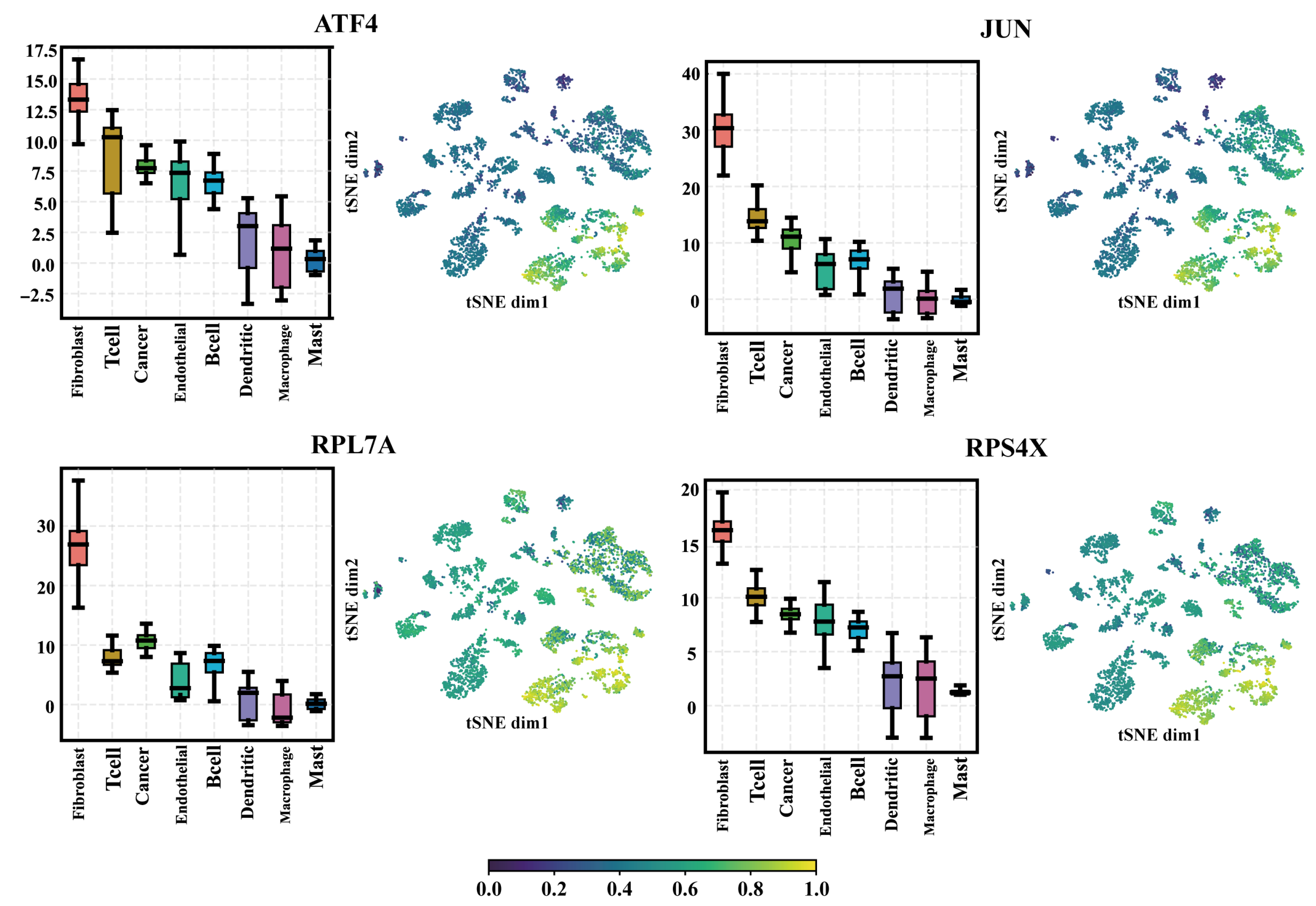
2.4. The Selected Regulators from the SEM Model Have a Higher Expression Level
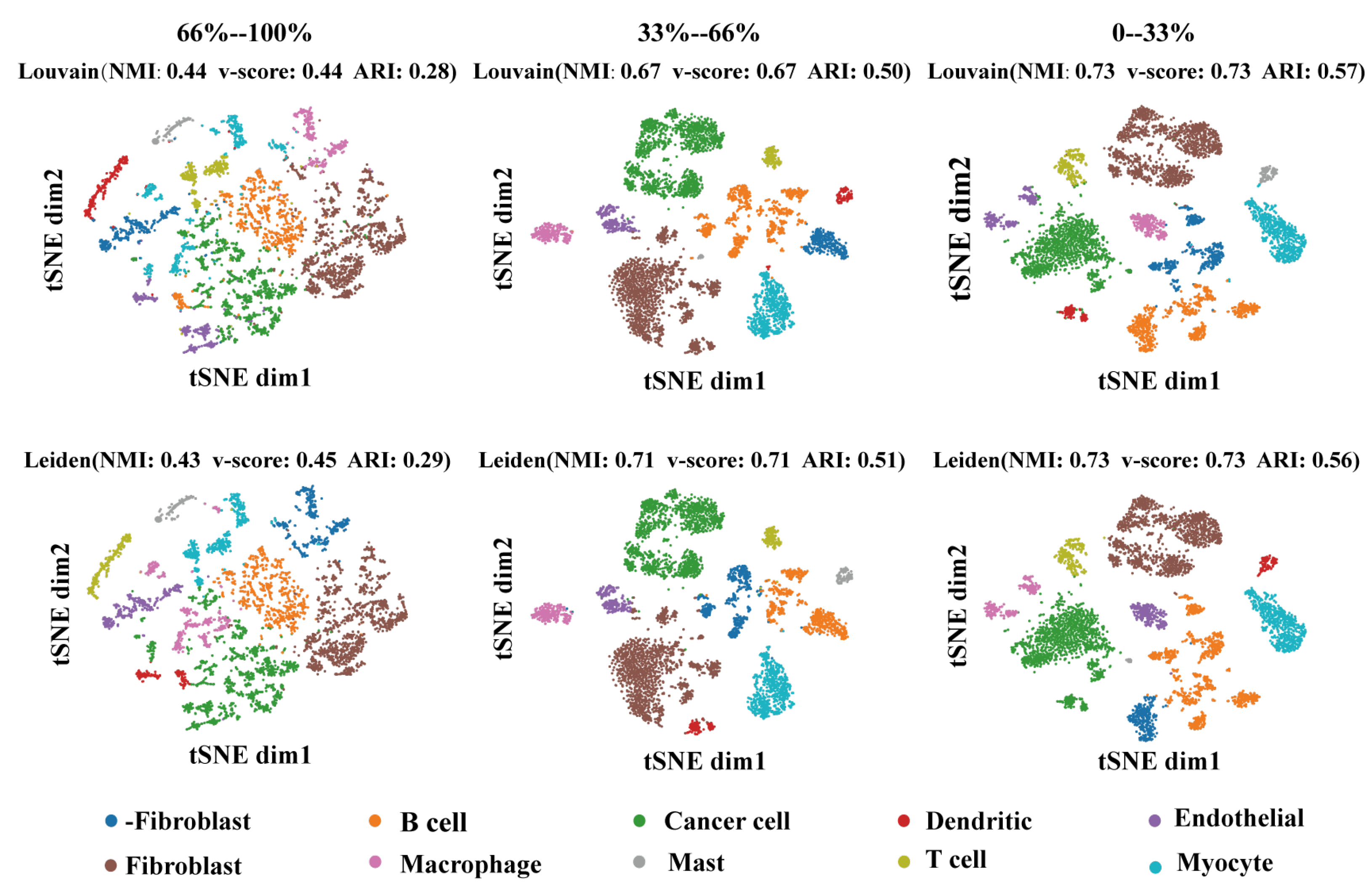
2.5. The Selected Regulators Are the Main Factors of Cell-Type Identification
3. Materials and Methods
3.1. Data Preparation
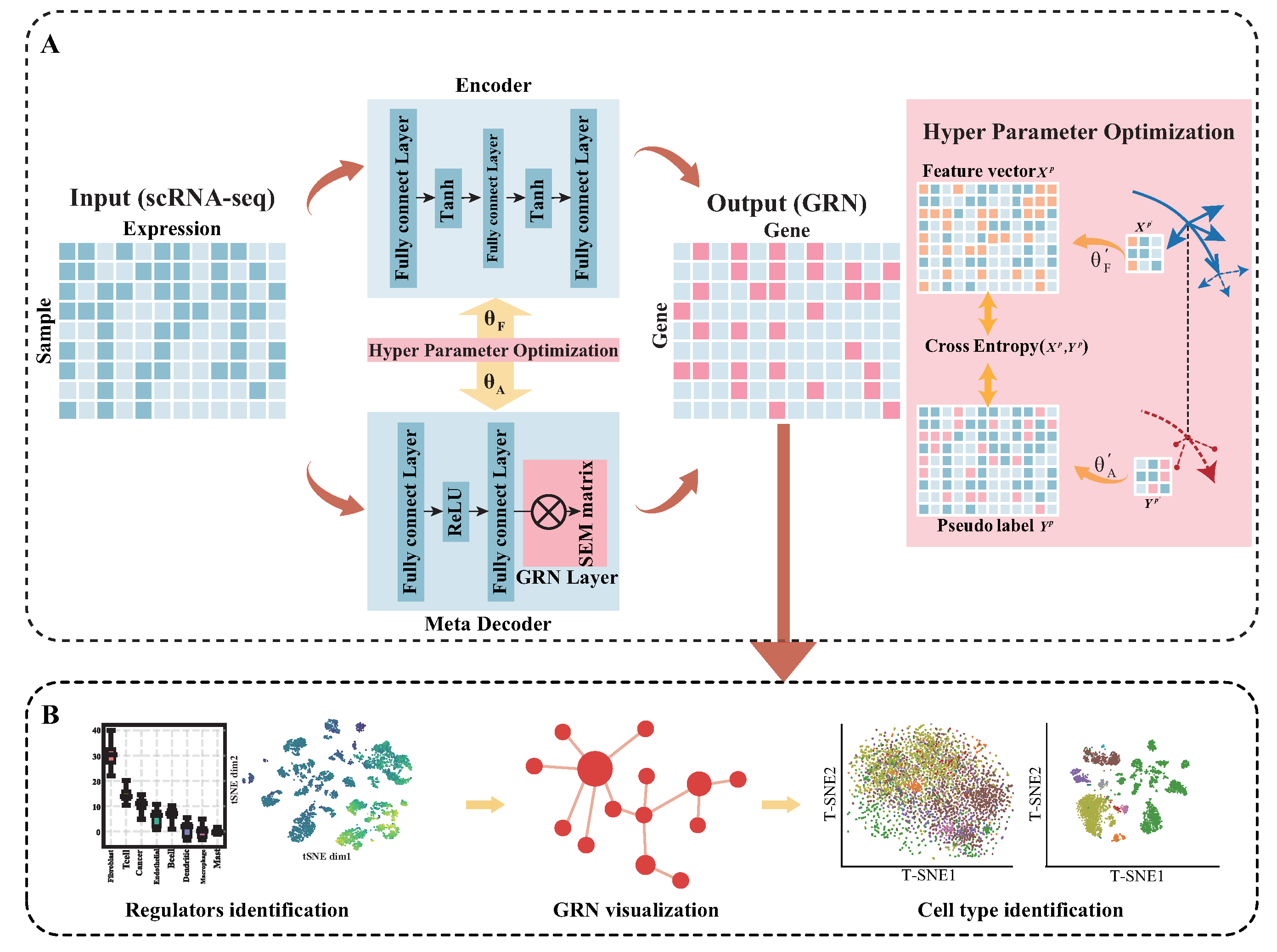
3.2. Model Description
3.2.1. GRN Layer
3.2.2. Encoder
3.2.3. Meta-Decoder
3.2.4. Hyperparameter Optimization
3.3. Implementation
3.4. Evaluation Metrics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wouters, J.; Atak, Z.K.; Aerts, S. Decoding transcriptional states in cancer. Curr. Opin. Genet. Dev. 2017, 43, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.E.; Stumpf, M.P.; Babtie, A.C. Gene regulatory network inference from single-cell data using multivariate information measures. Cell Syst. 2017, 5, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Fiers, M.W.; Minnoye, L.; Aibar, S.; Bravo González-Blas, C.; Kalender Atak, Z.; Aerts, S. Mapping gene regulatory networks from single-cell omics data. Briefings Funct. Genom. 2018, 17, 246–254. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, K. Tools for the analysis of high-dimensional single-cell RNA sequencing data. Nat. Rev. Nephrol. 2020, 16, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ning, B.; Shi, T. Single-cell RNA-seq technologies and related computational data analysis. Front. Genet. 2019, 317. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.E.; Loh, S.K.; Low, S.T.; Mohamad, M.S.; Deris, S.; Zakaria, Z. A review on the computational approaches for gene regulatory network construction. Comput. Biol. Med. 2014, 48, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Ding, W.; Zhang, L.; Ziemek, D.; Zarringhalam, K. A biological network-based regularized artificial neural network model for robust phenotype prediction from gene expression data. BMC Bioinform. 2017, 18, 565. [Google Scholar] [CrossRef]
- Biswas, S.; Acharyya, S. Neural model of gene regulatory network: A survey on supportive meta-heuristics. Theory Biosci. 2016, 135, 1–19. [Google Scholar] [CrossRef]
- Van de Sande, B.; Flerin, C.; Davie, K.; De Waegeneer, M.; Hulselmans, G.; Aibar, S.; Seurinck, R.; Saelens, W.; Cannoodt, R.; Rouchon, Q.; et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020, 15, 2247–2276. [Google Scholar] [CrossRef]
- Zhao, M.; He, W.; Tang, J.; Zou, Q.; Guo, F. A hybrid deep learning framework for gene regulatory network inference from single-cell transcriptomic data. Briefings Bioinform. 2022, 23, bbab568. [Google Scholar] [CrossRef]
- Chen, J.; Cheong, C.; Lan, L.; Zhou, X.; Liu, J.; Lyu, A.; Cheung, W.K.; Zhang, L. DeepDRIM: A deep neural network to reconstruct cell-type-specific gene regulatory network using single-cell RNA-seq data. Briefings Bioinform. 2021, 22, bbab325. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, A.; Ma, Q.; Xu, D.; Joshi, T.J.C.; Journal, S.B. Inductive inference of gene regulatory network using supervised and semi-supervised graph neural networks. Comput. Struct. Biotechnol. J. 2020, 18, 3335–3343. [Google Scholar] [CrossRef] [PubMed]
- Karaaslanli, A.; Saha, S.; Aviyente, S.; Maiti, T. scSGL: Kernelized signed graph learning for single-cell gene regulatory network inference. Bioinformatics 2022, 38, 3011–3019. [Google Scholar] [CrossRef]
- Seninge, L.; Anastopoulos, I.; Ding, H.; Stuart, J. VEGA is an interpretable generative model for inferring biological network activity in single-cell transcriptomics. Nat. Commun. 2021, 12, 5684. [Google Scholar] [CrossRef]
- Shu, H.; Zhou, J.; Lian, Q.; Li, H.; Zhao, D.; Zeng, J.; Ma, J. Modeling gene regulatory networks using neural network architectures. Nat. Comput. Sci. 2021, 1, 491–501. [Google Scholar] [CrossRef]
- Matsumoto, H.; Kiryu, H.; Furusawa, C.; Ko, M.S.; Ko, S.B.; Gouda, N.; Hayashi, T.; Nikaido, I.J.B. SCODE: An efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 2017, 33, 2314–2321. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Geurts, P. dynGENIE3: Dynamical GENIE3 for the inference of gene networks from time-series expression data. Sci. Rep. 2018, 8, 3384. [Google Scholar] [CrossRef] [PubMed]
- Moerman, T.; Aibar Santos, S.; Bravo González-Blas, C.; Simm, J.; Moreau, Y.; Aerts, J.; Aerts, S. GRNBoost2 and Arboreto: Efficient and scalable inference of gene regulatory networks. Bioinformatics 2019, 35, 2159–2161. [Google Scholar] [CrossRef]
- Vilalta, R.; Drissi, Y.J. A perspective view and survey of meta-learning. Artif. Intell. Rev. 2002, 18, 77–95. [Google Scholar] [CrossRef]
- Fu, K.; Zhang, T.; Zhang, Y.; Wang, Z.; Sun, X. Few-shot SAR target classification via meta-learning. IEEE Trans. Geosci. Remote Sens. 2021, 60, 1–14. [Google Scholar]
- Chowdhury, A.; Chaudhari, D.; Chaudhuri, S.; Jermaine, C. Meta-Meta Classification for One-Shot Learning. In Proceedings of the IEEE/CVF Winter Conference on Applications of Computer Vision, Waikoloa, HI, USA, 3–8 January 2022; pp. 177–186. [Google Scholar]
- Chen, Z.; Fu, Y.; Wang, Y.X.; Ma, L.; Liu, W.; Hebert, M. Image deformation meta-networks for one-shot learning. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Long Beach, CA, USA, 15–20 June 2019; pp. 8680–8689. [Google Scholar]
- Yu, Y.; Chen, J.; Gao, T.; Yu, M. DAG-GNN: DAG structure learning with graph neural networks. In Proceedings of the International Conference on Machine Learning, PMLR, Long Beach, CA, USA, 9–15 June 2019; pp. 7154–7163. [Google Scholar]
- Zheng, S.; Zou, Y.; Tang, Y.; Yang, A.; Liang, J.Y.; Wu, L.; Tian, W.; Xiao, W.; Xie, X.; Yang, L.; et al. Landscape of cancer-associated fibroblasts identifies the secreted biglycan as a protumor and immunosuppressive factor in triple-negative breast cancer. Oncoimmunology 2022, 11, 2020984. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed]
- Kaviany, S.; Bartkowiak, T.; Dulek, D.E.; Khan, Y.W.; Hayes, M.J.; Schaefer, S.G.; Ye, X.; Dahunsi, D.O.; Connelly, J.A.; Irish, J.M.J.I. Systems Immunology Analyses of STAT1 Gain-of-Function Immune Phenotypes Reveal Heterogeneous Response to IL-6 and Broad Immunometabolic Roles for STAT1. ImmunoHorizons 2022, 6, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, M.; Fukasawa, M.; Nishihara, A.; Takada, L.; Asano, M. JunB can enhance the transcription of IL-8 in oral squamous cell carcinoma. J. Cell. Physiol. 2021, 236, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Pratapa, A.; Jalihal, A.P.; Law, J.N.; Bharadwaj, A.; Murali, T.J. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods 2020, 17, 147–154. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. Współczesna Onkol. 2015, 2015, 68–77. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Zeng, Y.; Zhou, J.; Zou, Q. High-resolution transcription factor binding sites prediction improved performance and interpretability by the deep learning method. Briefings Bioinform. 2021, 22, bbab273. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Zeng, Y.; Liu, Y.; Xiong, S.; Wang, M.; Zhou, J.; Zou, Q. A novel convolution attention model for predicting transcription factor binding sites by combining sequence and shape. Briefings Bioinform. 2022, 23, bbab525. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiao, S.; Zeng, Y.; Gao, D.; Han, N.; Zhou, J. CAE-CNN: Predicting transcription factor binding site with convolutional autoencoder and convolutional neural network. Expert Syst. Appl. 2021, 183, 115404. [Google Scholar] [CrossRef]
- Liu, R.; Gao, J.; Zhang, J.; Meng, D.; Lin, Z. Investigating Bi-Level Optimization for Learning and Vision from a Unified Perspective: A Survey and Beyond. IEEE Trans. Pattern Anal. Mach. Intell. 2021. [Google Scholar] [CrossRef]
- Li, X.; Wang, K.; Lyu, Y.; Pan, H.; Zhang, J.; Stambolian, D.; Susztak, K.; Reilly, M.P.; Hu, G.; Li, M. Deep learning enables accurate clustering with batch effect removal in single-cell RNA-seq analysis. Nat. Commun. 2020, 11, 2338. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EPR | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Methods | 1000 Gene Datasets | 500 Gene Datasets | ||||||||
| mHSC-L | mHSC-GM | mHSC-E | hESC | mESC | hHep | mDC | mHSC-L | mHSC-GM | mHSC-E | |
| DeepSEM | 1.09 | 1.14 | 1.24 | 1.43 | 1.06 | 1.14 | 2.55 | 1.07 | 1.06 | 1.30 |
| DGRN | - | - | - | - | - | - | - | - | - | - |
| GENIE3 | <1 | 1.03 | 1.01 | 1.00 | 1.06 | 1.12 | 1.01 | 1.00 | 1.06 | 1.03 |
| PIDC | <1 | <1 | <1 | <1 | 1.01 | 1.03 | <1 | <1 | <1 | <1 |
| MetaSEM | 1.36 | 1.41 | 1.24 | 2.29 | 1.24 | 1.53 | 3.36 | 1.13 | 1.20 | 1.69 |
| AUPR | ||||||||||
| Methods | 1000 Gene Datasets | 500 Gene Datasets | ||||||||
| mHSC-L | mHSC-GM | mHSC-E | hESC | mESC | hHep | mDC | mHSC-L | mHSC-GM | mHSC-E | |
| DeepSEM | 0.63 | 0.56 | 0.42 | 0.30 | 0.38 | 0.48 | 0.43 | 0.66 | 0.63 | 0.58 |
| DGRN | 0.15 | 0.16 | 0.25 | - | - | - | - | 0.15 | 0.27 | 0.25 |
| GENIE3 | 0.09 | 0.12 | 0.09 | - | - | - | - | 0.14 | 0.15 | 0.14 |
| PIDC | 0.07 | 0.12 | 0.10 | - | - | - | - | 0.16 | 0.12 | 0.19 |
| MetaSEM | 0.70 | 0.73 | 0.66 | 0.48 | 0.43 | 0.70 | 0.33 | 0.75 | 0.77 | 0.84 |
| AUROC | ||||||||||
| Methods | 1000 Gene Datasets | 500 Gene Datasets | ||||||||
| mHSC-L | mHSC-GM | mHSC-E | hESC | mESC | hHep | mDC | mHSC-L | mHSC-GM | mHSC-E | |
| DeepSEM | 0.51 | 0.57 | 0.63 | 0.52 | 0.51 | 0.54 | 0.77 | 0.52 | 0.52 | 0.67 |
| DGRN | 0.63 | 0.67 | 0.75 | - | - | - | - | 0.63 | 0.71 | 0.77 |
| GENIE3 | 0.63 | 0.63 | 0.59 | - | - | - | - | 0.52 | 0.52 | 0.55 |
| PIDC | 0.57 | 0.61 | 0.60 | - | - | - | - | 0.47 | 0.47 | 0.59 |
| MetaSEM | 0.75 | 0.77 | 0.76 | 0.81 | 0.61 | 0.77 | 0.71 | 0.67 | 0.72 | 0.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, M.; Wang, Z.; Liu, Y.; Xiong, S.; Zou, Q. MetaSEM: Gene Regulatory Network Inference from Single-Cell RNA Data by Meta-Learning. Int. J. Mol. Sci. 2023, 24, 2595. https://doi.org/10.3390/ijms24032595
Zhang Y, Wang M, Wang Z, Liu Y, Xiong S, Zou Q. MetaSEM: Gene Regulatory Network Inference from Single-Cell RNA Data by Meta-Learning. International Journal of Molecular Sciences. 2023; 24(3):2595. https://doi.org/10.3390/ijms24032595
Chicago/Turabian StyleZhang, Yongqing, Maocheng Wang, Zixuan Wang, Yuhang Liu, Shuwen Xiong, and Quan Zou. 2023. "MetaSEM: Gene Regulatory Network Inference from Single-Cell RNA Data by Meta-Learning" International Journal of Molecular Sciences 24, no. 3: 2595. https://doi.org/10.3390/ijms24032595
APA StyleZhang, Y., Wang, M., Wang, Z., Liu, Y., Xiong, S., & Zou, Q. (2023). MetaSEM: Gene Regulatory Network Inference from Single-Cell RNA Data by Meta-Learning. International Journal of Molecular Sciences, 24(3), 2595. https://doi.org/10.3390/ijms24032595