



Alpha-Thalassemia in Southern Italy: Characterization of Five New Deletions Removing the Alpha-Globin Gene Cluster


Abstract
1. Introduction
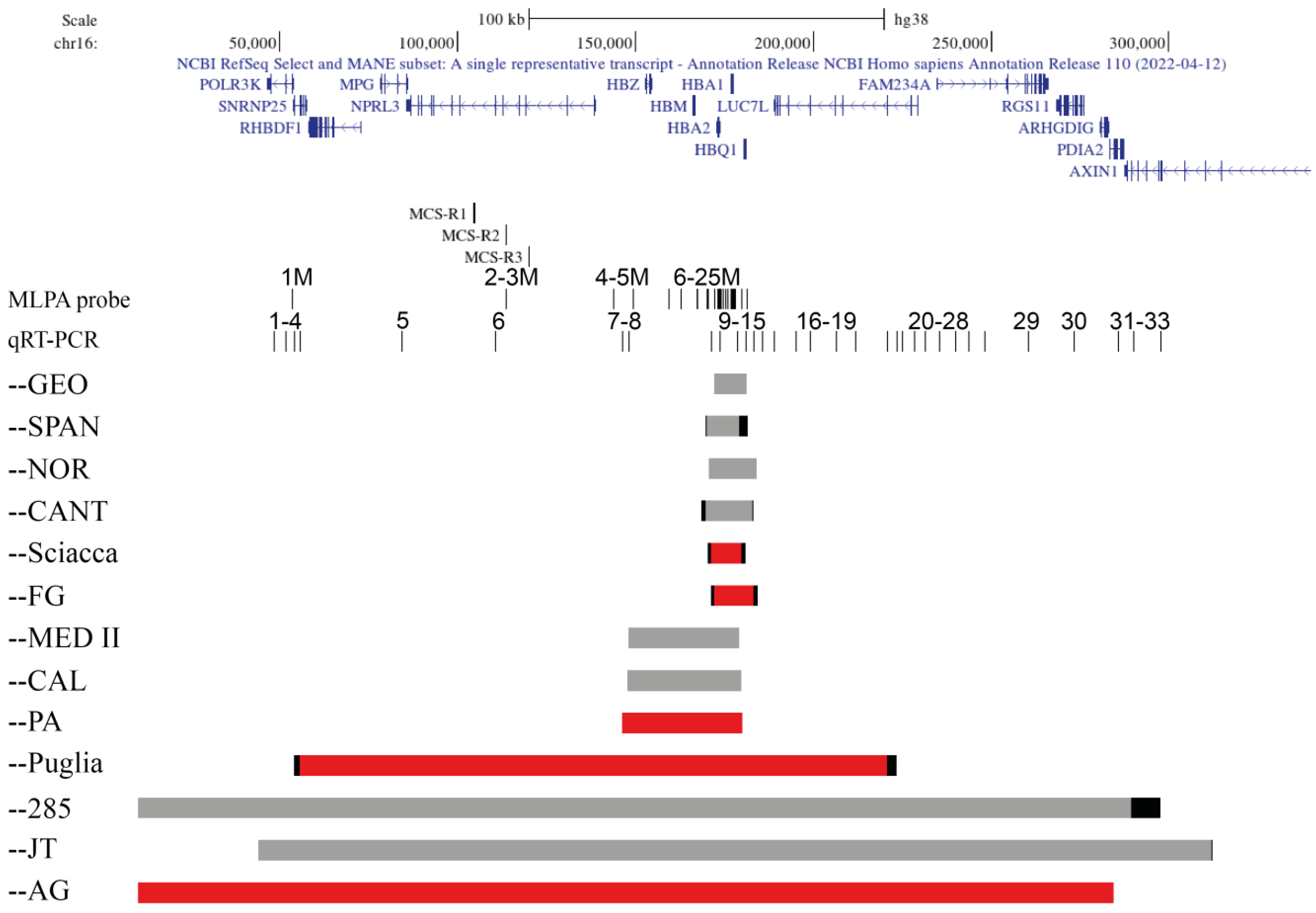
2. Results
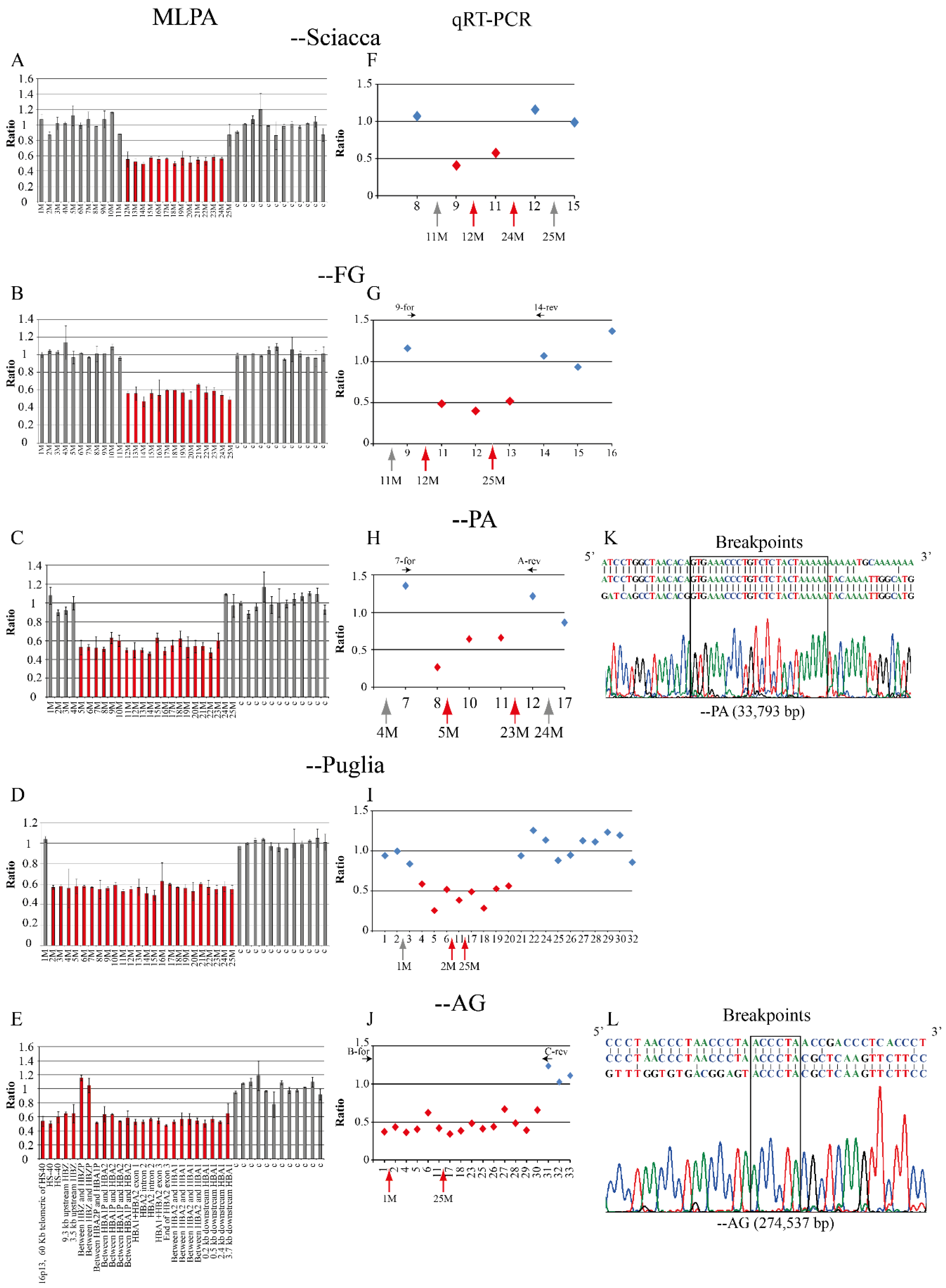
2.1. Family 1, --Sciacca
2.2. Family 2, --FG
2.3. Family 3 and 4, --PA
2.4. Family 5, --Puglia
2.5. Family 6, --AG
3. Discussion
3.1. Deletion Removing the α-Globin Gene Cluster
3.2. Deletions Removing the Regulative Elements and the α-Globin Cluster
4. Materials and Methods
4.1. Patients and Hematological Data
4.2. DNA Analysis
4.3. MLPA Analysis
4.4. qRT-PCR
4.5. Breakpoint Characterization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Waye, J.S.; Eng, B. Diagnostic testing for α-globin gene disorders in a heterogeneous North American population. Int. J. Lab. Hematol. 2013, 35, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Harteveld, C.L.; Higgs, D.R. Alpha-thalassaemia. Orphanet J. Rare Dis. 2010, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Brancaleoni, V.; Di Pierro, E.; Motta, I.; Cappellini, M.D. Laboratory diagnosis of thalassemia. Int. J. Lab. Hematol. 2016, 38 (Suppl. 1), 32–40. [Google Scholar] [CrossRef]
- Higgs, D.; Wood, W. Long-range regulation of alpha globin gene expression during erythropoiesis. Curr. Opin. Hematol. 2008, 15, 176–183. [Google Scholar] [CrossRef]
- Harteveld, C.L.; Voskamp, A.; Phylipsen, M.; Akkermans, N.; den Dunnen, J.T.; White, S.J.; Giordano, P.C. Nine unknown rearrangements in 16p13.3 and 11p15.4 causing alpha- and beta-thalassaemia characterised by high resolution multiplex ligation-dependent probe amplification. J. Med. Genet. 2005, 42, 922–931. [Google Scholar] [CrossRef]
- Colosimo, A.; Gatta, V.; Guida, V.; Leodori, E.; Foglietta, E.; Rinaldi, S.; Cappabianca, M.; Amato, A.; Stuppia, L.; Dallapiccola, B. Application of MLPA assay to characterize unsolved alpha-globin gene rearrangements. Blood Cells Mol. Dis. 2011, 46, 139–144. [Google Scholar] [CrossRef]
- Higgs, D.R. The molecular basis of α-thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011718. [Google Scholar] [CrossRef] [PubMed]
- Cardiero, G.; Musollino, G.; Prezioso, R.; Lacerra, G. mRNA Analysis of Frameshift Mutations with Stop Codon in the Last Exon: The Case of Hemoglobins Campania [α1 cod95 (-C)] and Sciacca [α1 cod109 (-C)]. Biomedicines 2021, 9, 1390. [Google Scholar] [CrossRef] [PubMed]
- Hardison, R.C.; Chui, D.H.; Giardine, B.; Riemer, C.; Patrinos, G.P.; Anagnou, N.; Miller, W.; Wajcman, H. HbVar: A relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Hum. Mutat. 2002, 19, 225–233. [Google Scholar] [CrossRef]
- Lacerra, G.; Fioretti, G.; De Angioletti, M.; Pagano, L.; Guarino, E.; de Bonis, C.; Viola, A.; Maglione, G.; Scarallo, A.; De Rosa, L. (Alpha)alpha 5.3: A novel alpha(+)-thalassemia deletion with the breakpoints in the alpha 2-globin gene and in close proximity to an Alu family repeat between the psi alpha 2- and psi alpha 1-globin genes. Blood 1991, 78, 2740–2746. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; Puzone, S.; Ammirabile, M.; Piscopo, C.; Pagano, L.; Colucci, S.; Izzo, P.; Grosso, M. Identification and molecular characterization of the --CAMPANIA deletion, a novel alpha (0) -thalassemic defect, in two unrelated Italian families [corrected]. Am. J. Hematol. 2010, 85, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Villegas, A.; Ropero, P.; González, F.A.; Anguita, E.; Espinós, D. The thalassemia syndromes: Molecular characterization in the Spanish population. Hemoglobin 2001, 25, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Villegas, A.; Sanchez, J.; Ricard, P.; Gonzalez, F.A.; Del Potro, E.; Armada, B.; Carreno, D.L.; Espinos, D. Characterization of a new alpha-thalassemia-1 mutation in a Spanish family. Hemoglobin 1994, 18, 29–37. [Google Scholar] [CrossRef]
- Villegas, A.; Calero, F.; Vickers, M.A.; Ayyub, H.; Higgs, D.R. Alpha thalassaemia in two Spanish families. Eur. J. Haematol. 1990, 44, 109–115. [Google Scholar]
- Fei, Y.J.; Liu, J.C.; Walker, E.L.; Huisman, T.H. A new gene deletion involving the alpha 2-, alpha 1-, and theta 1-globin genes in a black family with Hb H disease. Am. J. Hematol. 1992, 39, 299–300. [Google Scholar] [CrossRef] [PubMed]
- Grimholt, R.M.; Fjeld, B.; Klingenberg, O. Hemoglobinopathy gone astray-three novel forms of α-thalassemia in Norwegian patients characterized by quantitative real-time PCR and DNA sequencing. Scand. J. Clin. Lab. Investig. 2021, 81, 670–678. [Google Scholar] [CrossRef]
- Harteveld, K.L.; Losekoot, M.; Fodde, R.; Giordano, P.C.; Bernini, L.F. The involvement of Alu repeats in recombination events at the alpha-globin gene cluster: Characterization of two alphazero-thalassaemia deletion breakpoints. Hum. Genet. 1997, 99, 528–534. [Google Scholar] [CrossRef]
- Brieghel, C.; Birgens, H.; Frederiksen, H.; Hertz, J.M.; Steenhof, M.; Petersen, J. Novel 31.2 kb α0 Deletion in a Palestinian Family with α-Thalassemia. Hemoglobin 2015, 39, 346–349. [Google Scholar] [CrossRef]
- Villegas, A.; Sanchez, J.; Gonzalez, F.A.; Carreño, D.L.; Ropero, P. Alpha-thalassemia-1 (--CAL mutation) in a Spanish family. Am. J. Hematol. 1994, 46, 367–368. [Google Scholar] [CrossRef]
- Harteveld, C.L.; Kriek, M.; Bijlsma, E.K.; Erjavec, Z.; Balak, D.; Phylipsen, M.; Voskamp, A.; di Capua, E.; White, S.J.; Giordano, P.C. Refinement of the genetic cause of ATR-16. Hum. Genet. 2007, 122, 283–292. [Google Scholar] [CrossRef]
- Horsley, S.W.; Daniels, R.J.; Anguita, E.; Raynham, H.A.; Peden, J.F.; Villegas, A.; Vickers, M.A.; Green, S.; Waye, J.S.; Chui, D.H.; et al. Monosomy for the most telomeric, gene-rich region of the short arm of human chromosome 16 causes minimal phenotypic effects. Eur. J. Hum. Genet. 2001, 9, 217–225. [Google Scholar] [CrossRef]
- Joly, P.; Lacan, P.; Labalme, A.; Bonhomme, E.; Sanlaville, D.; Francina, A. A novel telomeric (approximately 285 kb) α-thalassemia deletion leading to a phenotypically unusual HbH disease. Haematologica 2010, 95, 850–851. [Google Scholar] [CrossRef]
- Digilio, F.A.; Lanati, A.; Bongiovanni, A.; Mascia, A.; Di Carlo, M.; Barra, A.; Cirafici, A.M.; Colotti, G.; Kisslinger, A.; Lacerra, G.; et al. Quality-based model for Life Sciences research guidelines. Accredit. Qual. Assur. 2016, 21, 221–230. [Google Scholar] [CrossRef]
- Bongiovanni, A.; Colotti, G.; Liguori, G.L.; Di Carlo, M.; Digilio, F.A.; Lacerra, G.; Mascia, A.; Cirafici, A.M.; Barra, A.; Lanati, A.; et al. Applying Quality and Project Management methodologies in biomedical research laboratories: A public research network’s case study. Accredit. Qual. Assur. 2015, 20, 203–213. [Google Scholar] [CrossRef]
- Cardiero, G.; Musollino, G.; Friscia, M.G.; Testa, R.; Virruso, L.; Di Girgenti, C.; Caldora, M.; Bisogno, R.C.; Gaudiano, C.; Manco, G.; et al. Effect of Mutations on mRNA and Globin Stability: The Cases of Hb Bernalda/Groene Hart and Hb Southern Italy. Genes 2020, 11, 870. [Google Scholar] [CrossRef] [PubMed]
- Bisconte, M.G.; Caldora, M.; Musollino, G.; Cardiero, G.; Flagiello, A.; La Porta, G.; Lagona, L.; Prezioso, R.; Qualtieri, G.; Gaudiano, C.; et al. Alpha-Thalassemia Associated with Hb Instability: A Tale of Two Features. The Case of Hb Rogliano or alpha 1 Cod 108(G15)Thr → Asn and Hb Policoro or alpha 2 Cod 124(H7)Ser → Pro. PLoS ONE 2015, 10, e0115738. [Google Scholar] [CrossRef]
- Cardiero, G.; Prezioso, R.; Dembech, S.; Blanco, F.D.V.; Scarano, C.; Lacerra, G. Identification and molecular characterization of a novel 163 kb deletion: The Italian (epsilon gamma delta beta) degrees-thalassemia. Hematology 2016, 21, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Lacerra, G.; Prezioso, R.; Musollino, G.; Piluso, G.; Mastrullo, L.; De Angioletti, M. Identification and molecular characterization of a novel 55-kb deletion recurrent in southern Italy: The Italian (G) γ((A) γδβ)-thalassemia. Eur. J. Haematol. 2013, 90, 214–219. [Google Scholar] [CrossRef]
- Carusone, T.M.; Cardiero, G.; Cerreta, M.; Mandrich, L.; Moran, O.; Porzio, E.; Catara, G.; Lacerra, G.; Manco, G. WTAP and BIRC3 are involved in the posttranscriptional mechanisms that impact on the expression and activity of the human lactonase PON2. Cell Death Dis. 2020, 11, 324. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Raney, B.J.; Dreszer, T.R.; Barber, G.P.; Clawson, H.; Fujita, P.A.; Wang, T.; Nguyen, N.; Paten, B.; Zweig, A.S.; Karolchik, D.; et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics 2014, 30, 1003–1005. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Families (no./Relationship) | Sex | RBC (×1012/L) | Hb (g/dL) | Ht (%) | MCV (fL) | MCH (pg) | MCHC (L/L) | Serum Iron (mg/dL) | Ferritin (ng/mL) | Bilirubin Tot (mg/dL) | Bilirubin Ind (mg/dL) | Hb A2 (%) | Hb F (%) | Hb H (%) | α Genotype | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | I.1 | M | 5.55 | 12.3 | 39.3 | 70.8 | 22.2 | 31.3 | = | = | = | = | 2.5 | 1.9 | = | --Sciacca/αα |
| 2 | I.1 | M | 5.68 | 15.6 | 45.9 | 80.8 | 27.5 | 34.0 | 57 | 168 | 0.68 | 0.53 | 2.2 | = | = | -α3.7/αα |
| I.2 | F | 6.15 | 13.0 | 41.0 | 66.6 | 21.1 | 31.6 | 31 | 12 | = | = | 2.3 | 0.7 | = | --FG/αα | |
| II.1 | M | 6.18 | 11.6 | 39.4 | 63.7 | 18.8 | 29.5 | 67 | 154 | = | = | 1.1 | 0.5 | 6.7 | -α3.7/--FG | |
| II.2 | M | 6.02 | 11.4 | 38.7 | 64.2 | 19.0 | 29.5 | 111 | 172 | = | = | 1.2 | = | 9.9 | -α3.7/--FG | |
| II.3 | F | 4.98 | 9.1 | 30.1 | 60.5 | 18.4 | 30.4 | 66 | 132 | = | = | 1.2 | 0.6 | 7.6 | -α3.7/--FG | |
| 3 | I.1 | F | 4.01 | 12.6 | 37.7 | 94.0 | 31.6 | 33.6 | = | 142 | 0.44 | 0.31 | 2.5 | 0.5 | = | αα/αα |
| II.1 | M | 6.78 | 14.1 | 45.4 | 67.0 | 20.9 | 31.1 | = | 555 | 0.72 | 0.62 | 2.4 | 0.4 | = | --PA/αα | |
| III.1 | F | 5.99 | 12.0 | 37.7 | 63.0 | 20.1 | 31.8 | = | 19 | 0.46 | 0.36 | 2.5 | 0.7 | = | --PA/αα | |
| III.2 | M | 5.39 | 10.9 | 34.4 | 63.8 | 20.3 | 31.8 | = | 49 | 0.68 | 0.54 | 2.6 | 0.4 | = | --PA/αα | |
| 4 | I.1 | M | 6.28 | 15.7 | 43.9 | 70.0 | 25.0 | 35.7 | = | 56 | 0.49 | 0.34 | 2.6 | = | = | --PA/αα |
| II.1 * | M | 5.00 | 14.8 | 35.2 | 70.0 | 29.5 | 41.9 | = | 121 | 0.64 | 0.49 | 6.3 | = | = | --PA/αα | |
| II.2 | M | 3.88 | 16.4 | 35.5 | 91.0 | 42.3 | 46.2 | = | 186 | 0.87 | 0.66 | 3.0 | = | = | αα/αα | |
| 5 | I.1 | F | 6.09 | 13.9 | 44.6 | 73.1 | 22.8 | 31.2 | 78 | 134 | 0.36 | 0.27 | 2.7 | 0.3 | = | --Puglia/αα |
| 6 | I.1 | M | 6.00 | 13.6 | 42.5 | 70.8 | 22.7 | 32.0 | = | 69 | = | = | 2.8 | = | = | --AG/αα |
| I.2 | F | 4.10 | 12.5 | 38.2 | 93.2 | 30.5 | 32.7 | = | 20 | = | = | 3 | = | = | αα/αα | |
| II.1 | F | 4.97 | 10.5 | 33.8 | 68.0 | 21.1 | 31.1 | = | 22 | = | = | 2.5 | = | = | --AG/αα | |
| II.2 | M | 6.71 | 14.1 | 44.4 | 66.2 | 21.0 | 31.8 | = | 43 | = | = | 2.5 | = | = | --AG/αα | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardiero, G.; Musollino, G.; Prezioso, R.; Nigro, V.; Lacerra, G. Alpha-Thalassemia in Southern Italy: Characterization of Five New Deletions Removing the Alpha-Globin Gene Cluster. Int. J. Mol. Sci. 2023, 24, 2577. https://doi.org/10.3390/ijms24032577
Cardiero G, Musollino G, Prezioso R, Nigro V, Lacerra G. Alpha-Thalassemia in Southern Italy: Characterization of Five New Deletions Removing the Alpha-Globin Gene Cluster. International Journal of Molecular Sciences. 2023; 24(3):2577. https://doi.org/10.3390/ijms24032577
Chicago/Turabian StyleCardiero, Giovanna, Gennaro Musollino, Romeo Prezioso, Vincenzo Nigro, and Giuseppina Lacerra. 2023. "Alpha-Thalassemia in Southern Italy: Characterization of Five New Deletions Removing the Alpha-Globin Gene Cluster" International Journal of Molecular Sciences 24, no. 3: 2577. https://doi.org/10.3390/ijms24032577
APA StyleCardiero, G., Musollino, G., Prezioso, R., Nigro, V., & Lacerra, G. (2023). Alpha-Thalassemia in Southern Italy: Characterization of Five New Deletions Removing the Alpha-Globin Gene Cluster. International Journal of Molecular Sciences, 24(3), 2577. https://doi.org/10.3390/ijms24032577