
A Mini Review of Novel Topoisomerase II Inhibitors as Future Anticancer Agents



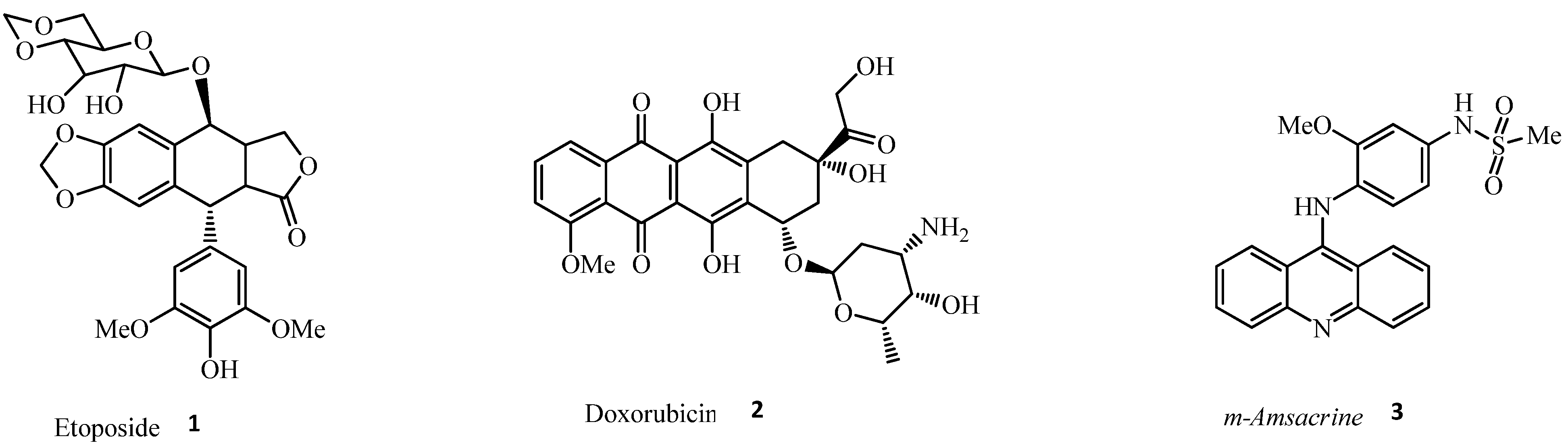{kind=link}
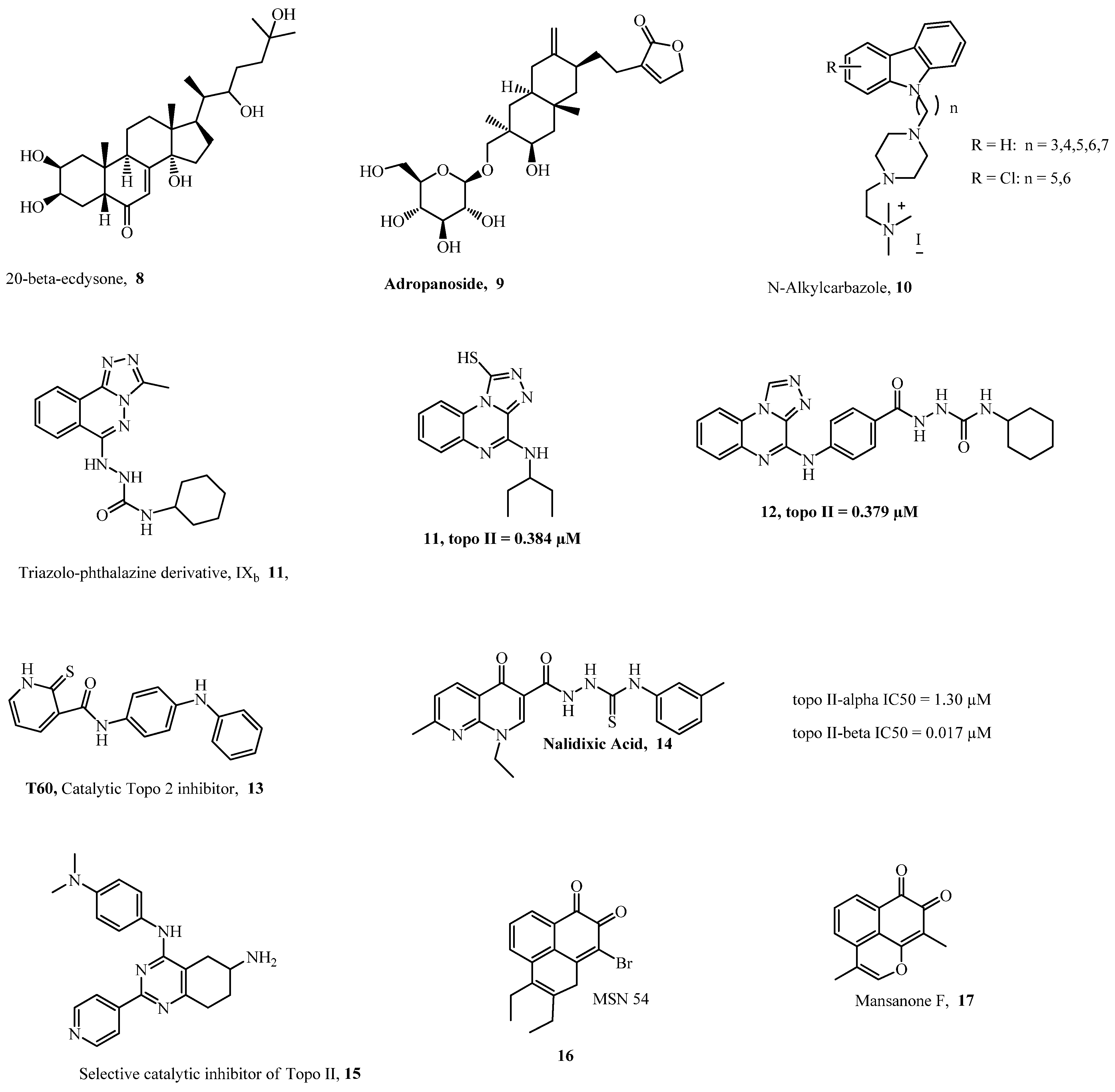{kind=link}
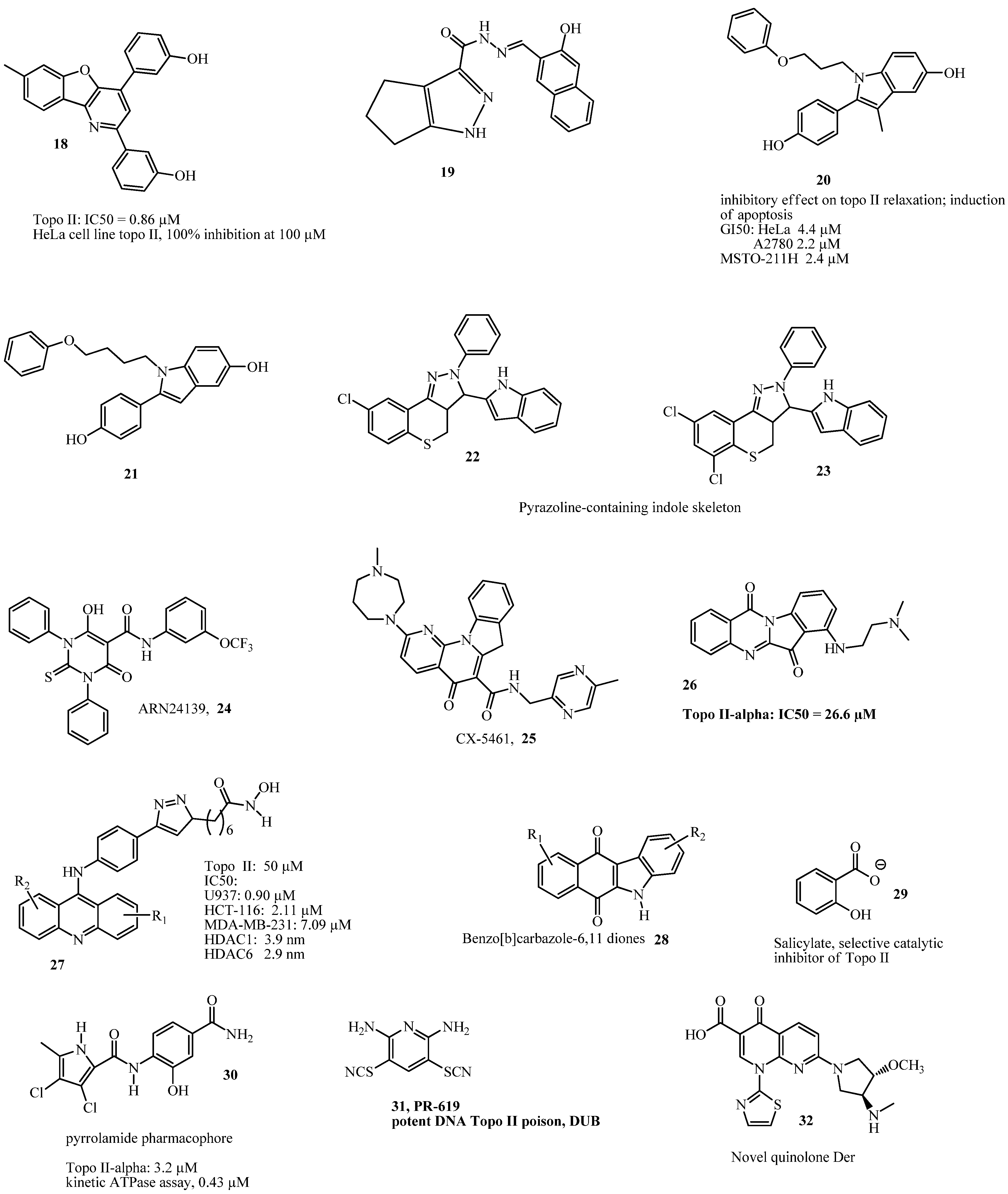{kind=link}
Abstract
1. Introduction
2. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- Leppard, J.B.; Champoux, J.J. Human DNA topoisomerase I: Relaxation, roles, and damage control. Chromosoma 2005, 114, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Pogorelcnik, B.; Perdih, A.; Solmajer, T. Recent advances in the development of catalytic inhibitors of human DNA topoiso-merase IIα as novel anticancer agents. Curr. Med. Chem. 2013, 20, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Pogorelcnik, B.; Perdih, A.; Solmajer, T. Recent Developments of DNA Poisons-Human DNA Topoisomerase IIα Inhibitors-as Anticancer Agents. Curr. Pharm. Des. 2013, 19, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anti Cancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Kang, J.S.; Kim, M.S.; Lee, K.P.; Lee, M.S. The study of doxorubicin and its complex with DNA by SERS and UV-resonance Raman spectroscopy. Bull Korean Chem. Soc. 2004, 25, 1211–1216. [Google Scholar]
- Bodley, A.; Liu, L.F.; Israel, M.; Seshadri, R.; Koseki, Y.; Giuliani, F.C.; Kirschenbaum, S.; Silber, R.; Potmesil, M. DNA topoisomerase II-mediated interaction of doxorubicin and daunorubicin congeners with DNA. Cancer Res. 1989, 49, 5969–5987. [Google Scholar]
- Elmore, R.H.; Wadkins, R.M.; Graves, D.E. Cooperative binding of m-AMSA to nucleic acids. Nucleic Acids Res. 1988, 16, 9707–9719. [Google Scholar] [CrossRef]
- Ketron, A.C.; Dennyl, W.A.; Graves, D.E.; Osheroff, N. Amsacrine as a Topo II Poison: Importance of Drug-DNA Interaction. Biochemistry 2012, 51, 1730–1739. [Google Scholar] [CrossRef]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The structure of DNA-bound human topoisomerase IIα: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef]
- Bollimpelli, V.S.; Dholaniya, P.S.; Kondapi, A.K. Topoisomerase IIβ and its role in different biological contexts. Arch. Biochem. Biophys. 2017, 633, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Ertan-Bolelli, T.; Bolelli, K. Discovery of New DNA Topoisomerase II Inhibitors using Structure Based Virtual Screening Method. J. Turk. Chem. Soc. 2019, 6, 71–78. [Google Scholar] [CrossRef]
- Adeniran, O.Y.; Metibemu, A.O.; Boboye, S.O. Virtual high-throughput screening (VHTS), three-dimensional quantitative structureactivity and relationship (3D-QSAR) and molecular docking studies of novel phytoinhibtors of topoisomerase II alpha. GSC Biol. Pharm. Sci. 2021, 15, 072–082. [Google Scholar] [CrossRef]
- Skok, Ž.; Durcik, M.; Skledar, D.G.; Barančoková, M.; Mašič, L.P.; Tomašič, T.; Zega, A.; Kikelj, D.; Zidar, N.; Ilaš, J. Discovery of new ATP-competitive inhibitors of human DNA topoisomerase IIα through screening of bacterial topoisomerase inhibitors. Bioorg. Chem. 2020, 102, 104049. [Google Scholar] [CrossRef] [PubMed]
- Knölker, H.-J.; Reddy, K.R. Isolation and Synthesis of Biologically Active Carbazole Alkaloids. Chem. Rev. 2002, 102, 4303–4428. [Google Scholar] [CrossRef]
- Kizek, R.; Adam, V.; Hrabeta, J.; Eckschlager, T.; Smutny, S.; Burda, J.V.; Frei, E.; Stiborova, M. Anthracyclines and ellipticines as DNA-damaging anticancer drugs: Recent advances. Pharmacol. Ther. 2012, 133, 26–39. [Google Scholar] [CrossRef]
- Stefania Sinicropi, M.; Lappano, R.; Caruso, A.; Francesca Santolla, M.; Pisano, A.; Rosano, C.; Capasso, A.; Panno, A.; Charles Lancelot, J.; Rault, S.; et al. (6-bromo-1, 4-dimethyl-9H-carbazol-3-yl-methylene)-hydrazine (carbhydraz) acts as a GPER agonist in breast cancer cells. Curr. Top. Med. Chem. 2015, 15, 1035–1042. [Google Scholar] [CrossRef]
- Saturnino, C.; Caruso, A.; Iacopetta, D.; Rosano, C.; Ceramella, J.; Muià, N.; Mariconda, A.; Bonomo, M.G.; Ponassi, M.; Rosace, G.; et al. Inhibition of Human Topoisomerase II by N,N,N-Trimethylethanammonium Iodide Alkylcarbazole Derivatives. Chemmedchem 2018, 13, 2635–2643. [Google Scholar] [CrossRef]
- Catanzaro, E.; Betari, N.; Arencibia, J.M.; Montanari, S.; Sissi, C.; De Simone, A.; Vassura, I.; Santini, A.; Andrisano, V.; Tumiatti, V.; et al. Targeting topoisomerase II with trypthantrin derivatives: Discovery of 7-((2-(dimethylamino)ethyl)amino)indolo [2,1-b]quinazoline-6,12-dione as an antiproliferative agent and to treat cancer. Eur. J. Med. Chem. 2020, 202, 112504. [Google Scholar] [CrossRef]
- Tosa, H.; Iinuma, M.; Tanaka, T.; Nozaki, H.; Ikeda, S.; Tsutsui, K.; Tsutsui, K.; Yamada, M.; Fujimori, S. Inhibitory activity of xanthone derivatives isolated from some guttiferaeous plants against DNA toposiomerases I and II. Chem. Pharm. Bull. 1997, 45, 418–420. [Google Scholar] [CrossRef]
- Mai, Y.-W.; Liang, C.-C.; Ou, J.-B.; Xie, H.-T.; Chen, S.-B.; Zhou, D.-C.; Yao, P.-F.; Huang, Z.-S.; Wang, H.; Huang, S.-L. 9-Bromo-2,3-diethylbenzo[de]chromene-7,8-dione (MSN54): A novel non-intercalative topoisomerase II catalytic inhibitor. Bioorg. Chem. 2021, 114, 105097. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.T.; Zhou, D.C.; Mai, Y.W.; Huo, L.; Yao, P.F.; Huang, S.L.; Wang, H.G.; Huang, Z.S.; Gu, L.Q. Construction of the oxaphenalene skeletons of mansonone F derivatives through C–H bond functionalization and their evaluation for anti-proliferative activities. RSC Adv. 2017, 7, 20919–20928. [Google Scholar] [CrossRef]
- Wu, W.-B.; Ou, J.-B.; Huang, Z.-H.; Chen, S.-B.; Ou, T.-M.; Tan, J.-H.; Li, D.; Shen, L.-L.; Huang, S.-L.; Gu, L.-Q.; et al. Synthesis and evaluation of mansonone F derivatives as topoisomerase inhibitors. Eur. J. Med. Chem. 2011, 46, 3339–3347. [Google Scholar] [CrossRef]
- Di Micco, S.; Masullo, M.; Bandak, A.F.; Berger, J.M.; Riccio, R.; Piacente, S.; Bifulco, G. Garcinol and Related Polyisoprenylated Benzophenones as Topoisomerase II Inhibitors: Biochemical and Molecular Modeling Studies. J. Nat. Prod. 2019, 82, 2768–2779. [Google Scholar] [CrossRef]
- Zidar, N.; Secci, D.; Tomašič, T.; Mašič, L.P.; Kikelj, D.; Passarella, D.; Argaez, A.N.G.; Hyeraci, M.; Via, L.D. Synthesis, Antiproliferative Effect, and Topoisomerase II Inhibitory Activity of 3-Methyl-2-phenyl-1H-indoles. ACS Med. Chem. Lett. 2020, 11, 691–697. [Google Scholar] [CrossRef]
- Zhou, D.-C.; Lu, Y.-T.; Mai, Y.-W.; Zhang, C.; Xia, J.; Yao, P.-F.; Wang, H.-G.; Huang, S.-L.; Huang, Z.-S. Design, synthesis and biological evaluation of novel perimidine o-quinone derivatives as non-intercalative topoisomerase II catalytic inhibitors. Bioorg. Chem. 2019, 91, 103131. [Google Scholar] [CrossRef]
- Sakr, H.; Ayyad, R.R.; El-Helby, A.A.; Khalifa, M.M.; Mahdy, H.A. Discovery of novel triazolophthalazine derivatives as DNA intercalators and topoisomerase II inhibitors. Arch. Pharm. 2021, 354, 2000456. [Google Scholar] [CrossRef]
- Pons, M.; Campayo, L.; Martinez-Balbás, M.; Azorín, F.; Navarro, P.; Giralt, E. A new ionizable chromophore of 1,4-bis (alkylamino) benzo [g] phthalazine which interacts with DNA by intercalation. J. Med. Chem. 1991, 34, 82–86. [Google Scholar] [CrossRef]
- Arencibia, J.M.; Brindani, N.; Franco-Ulloa, S.; Nigro, M.; Kuriappan, J.A.; Ottonello, G.; Bertozzi, S.M.; Summa, M.; Girotto, S.; Bertorelli, R.; et al. Design, Synthesis, Dynamic Docking, Biochemical Characterization, and In Vivo Pharmacokinetics Studies of Novel Topoisomerase II Poisons with Promising Antiproliferative Activity. J. Med. Chem. 2020, 63, 3508–3521. [Google Scholar] [CrossRef]
- Ibrahim, M.K.; Taghour, M.S.; Metwaly, A.M.; Belal, A.; Mehany, A.B.M.; Elhendawy, M.A.; Radwan, M.M.; Yassin, A.M.; El-Deeb, N.M.; Hafez, E.E.; et al. Design, synthesis, molecular modeling and anti-proliferative evaluation of novel quinoxaline derivatives as potential DNA intercalators and topoisomerase II inhibitors. Eur. J. Med. Chem. 2018, 155, 117–134. [Google Scholar] [CrossRef]
- El-Adl, K.; El-Helby, A.A.; Sakr, H.; Elwan, A. Design, synthesis, molecular docking and anti-proliferative evaluations of [1,2,4] triazolo [4,3-a] quinoline derivatives as DNA intercalators and Topoisomerase II inhibitors. Bioorg. Chem. 2020, 105, 104399. [Google Scholar] [CrossRef] [PubMed]
- Oyallon, B.; Brachet-Botineau, M.; Logé, C.; Bonnet, P.; Souab, M.; Robert, T.; Ruchaud, S.; Bach, S.; Berthelot, P.; Gouilleux, F.; et al. Structure-based design of novel quinoxaline-2-carboxylic acids and analogues as Pim-1 inhibitors. Eur. J. Med. Chem. 2018, 154, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-S.; Shin, W.-S.; Kim, C.-S.; Ahn, C.M.; Qi, X.-F.; Kim, S.-K. Molecular and cellular toxicological profiling of DNA bis-intercalator, quinoxaline compounds: Echinomycin as the versatile lead. Mol. Cell. Toxicol. 2018, 14, 9–18. [Google Scholar] [CrossRef]
- Eissa, I.H.; El-Naggar, A.M.; Abd El-Sattar, N.E.; Youssef, A.S. Design and Discovery of Novel Quinoxaline Derivatives as Dual DNA Intercalators and Topoisomerase II Inhibitors. Anti Cancer Agents Med. Chem. 2018, 18, 195–209. [Google Scholar] [CrossRef]
- Abbass, E.M.; Khalil, A.K.; Mohamed, M.M.; Eissa, I.H.; El-Naggar, A.M. Design, efficient synthesis, docking studies, and anticancer evaluation of new quinoxalines as potential intercalative Topo II inhibitors and apoptosis inducers. Bioorg. Chem. 2020, 104, 104255. [Google Scholar] [CrossRef] [PubMed]
- Bruno, P.M.; Lu, M.; Dennis, K.A.; Inam, H.; Moore, C.J.; Sheehe, J.; Elledge, S.J.; Hemann, M.T.; Pritchard, J.R. The primary mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is topoisomerase II poisoning. Proc. Natl. Acad. Sci. USA 2020, 117, 4053–4060. [Google Scholar] [CrossRef]
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11, eaaw8412. [Google Scholar] [CrossRef] [PubMed]
- Haddach, M.; Schwaebe, M.K.; Michaux, J.; Nagasawa, J.; O’Brien, S.E.; Whitten, J.P.; Pierre, F.; Kerdoncuff, P.; Darjania, L.; Stansfield, R.; et al. Discovery of CX-5461, the First Direct and Selective Inhibitor of RNA Polymerase I, for Cancer Therapeutics. ACS Med. Chem. Lett. 2012, 3, 602–606. [Google Scholar] [CrossRef]
- Ray, S.; Panova, T.; Miller, G.; Volkov, A.; Porter, A.C.; Russell, J.; Panov, K.I.; Zomerdijk, J.C. Topoisomerase IIα promotes activation of RNA polymerase I transcription by facilitating pre-initiation complex formation. Nat. Commun. 2013, 4, 1598. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Dos Santos, N.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Morotomi-Yano, K.; Yano, K.I. Nucleolar translocation of human DNA topoisomerase II by ATP depletion and its disruption by the RNA polymerase I inhibitor BMH-21. Sci. Rep. 2021, 11, 21533. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Ling, E.M.; Swan, R.L.; Brooks, M.L.; Austin, C.A. The Deubiquitinating Enzyme Inhibitor PR-619 is a Potent DNA Topoisomerase II Poison. Mol. Pharmacol. 2019, 96, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Altun, M.; Kramer, H.B.; Willems, L.I.; McDermott, J.L.; Leach, C.A.; Goldenberg, S.J.; Kumar, K.G.S.; Konietzny, R.; Fischer, R.; Kogan, E.; et al. Activity-Based Chemical Proteomics Accelerates Inhibitor Development for Deubiquitylating Enzymes. Chem. Biol. 2011, 18, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.-H.; Park, S.; Jang, H.J.; Hwang, S.-Y.; Shrestha, A.; Lee, E.-S.; Kwon, Y. AK-I-190, a New Catalytic Inhibitor of Topoisomerase II with Anti-Proliferative and Pro-Apoptotic Activity on Androgen-Negative Prostate Cancer Cells. Int. J. Mol. Sci. 2021, 22, 11246. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Matias-Barrios, V.M.; Radaeva, M.; Song, Y.; Alperstein, Z.; Lee, A.R.; Schmitt, V.; Lee, J.; Ban, F.; Xie, N.; Qi, J.; et al. Discovery of New Catalytic Topoisomerase II Inhibitors for Anticancer Therapeutics. Front. Oncol. 2021, 10, 633142. [Google Scholar] [CrossRef]
- Matias-Barrios, V.M.; Radaeva, M.; Ho, C.-H.; Lee, J.; Adomat, H.; Lallous, N.; Cherkasov, A.; Dong, X. Optimization of New Catalytic Topoisomerase II Inhibitors as an Anti-Cancer Therapy. Cancers 2021, 13, 3675. [Google Scholar] [CrossRef]
- Lee, M.-G.; Liu, Y.-C.; Lee, Y.-L.; El-Shazly, M.; Lai, K.-H.; Shih, S.-P.; Ke, S.-C.; Hong, M.-C.; Du, Y.-C.; Yang, J.-C.; et al. Heteronemin, a Marine Sesterterpenoid-Type Metabolite, Induces Apoptosis in Prostate LNcap Cells via Oxidative and ER Stress Combined with the Inhibition of Topoisomerase II and Hsp90. Mar. Drugs 2018, 16, 204. [Google Scholar] [CrossRef]
- Schumacher, M.; Cerella, C.; Eifes, S.; Chateauvieux, S.; Morceau, F.; Jaspars, M.; Dicato, M.; Diederich, M. Heteronemin, a spongean sesterterpene, inhibits TNF α-induced NF-κB activation through proteasome inhibition and induces apoptotic cell death. Biochem. Pharmacol. 2010, 79, 610–622. [Google Scholar] [CrossRef]
- Ortega, J.A.; Arencibia, J.M.; Minniti, E.; Byl, J.A.W.; Franco-Ulloa, S.; Borgogno, M.; Genna, V.; Summa, M.; Bertozzi, S.M.; Bertorelli, R.; et al. Novel, Potent, and Druglike Tetrahydroquinazoline Inhibitor That Is Highly Selective for Human Topoisomerase II α over β. J. Med. Chem. 2020, 63, 12873–12886. [Google Scholar] [CrossRef]
- Chen, J.; Li, D.; Li, W.; Yin, J.; Zhang, Y.; Yuan, Z.; Gao, C.; Liu, F.; Jiang, Y. Design, synthesis and anticancer evaluation of acridine hydroxamic acid derivatives as dual Topo and HDAC inhibitors. Bioorg. Med. Chem. 2018, 26, 3958–3966. [Google Scholar] [CrossRef] [PubMed]
- Sarcar, B.; Kahali, S.; Chinnaiyan, P. Vorinostat enhances the cytotoxic effects of the topoisomerase I inhibitor SN38 in glioblastoma cell lines. J. Neuro Oncol. 2010, 99, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.; Cubitt, C.L.; Zhang, S.; Chiappori, A. Combination of HDAC and topoisomerase inhibitors in small cell lung cancer. Cancer Biol. Ther. 2012, 13, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Oyedele, A.S.; Bogan, D.N.; Okoro, C.O. Synthesis, biological evaluation and virtual screening of some acridone derivatives as potential anticancer agents. Bioorg. Med. Chem. 2020, 28, 115426. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Y.; Xu, G.-S.; Li, X. A Unique Topoisomerase II Inhibitor with Dose-Affected Anticancer Mechanisms and Less Cardiotoxicity. Cells 2021, 10, 3138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, B.; Yang, T.; Wang, N.; Gao, C.; Tan, C.; Liu, H.; Jiang, Y. Synthesis and antiproliferative activity of 9-benzylamino-6-chloro-2-methoxy-acridine derivatives as potent DNA-binding ligands and topoisomerase II inhibitors. Eur. J. Med. Chem. 2016, 116, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Prasher, P.; Sharma, M. Medicinal chemistry of acridine and its analogues. MedChemComm 2018, 9, 1589–1618. [Google Scholar] [CrossRef]
- Nemr, M.T.M.; Sonousi, A.; Marzouk, A.A. Design, synthesis and antiproliferative evaluation of new tricyclic fused thiazolopyrimidines targeting topoisomerase II: Molecular docking and apoptosis inducing activity. Bioorg. Chem. 2020, 105, 104446. [Google Scholar] [CrossRef]
- Nemr, M.T.M.; AboulMagd, A.M. New fused pyrimidine derivatives with anticancer activity: Synthesis, topoisomerase II inhibition, apoptotic inducing activity and molecular modeling study. Bioorg. Chem. 2020, 103, 104134. [Google Scholar] [CrossRef]
- Khalila, O.M.; Gedawy, E.M.; El-Malaha, A.A.; Adly, M.E. Novel nalidixic acid derivatives targeting topoisomerase II enzyme; Design, synthesis, anticancer activity and effect on cell cycle profile. Bioorg. Chem. 2019, 83, 262–276. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, W.-J.; Li, P.-H.; Wang, J.; Dong, C.-Z.; Zhang, K.; Chen, H.-X.; Du, Z.-Y. Synthesis and biological evaluation of novel carbazole-rhodanine conjugates as topoisomerase II inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Jo, H.; Kwon, Y.; Lee, E.-S. Design, synthesis, and structure-activity relationships of new benzofuro [3,2-b] pyridine-7-ols as DNA topoisomerase II inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Oviatt, A.A.; Kuriappan, J.A.; Minniti, E.; Vann, K.R.; Onuorah, P.; Minarini, A.; De Vivo, M.; Osheroff, N. Polyamine-containing etoposide derivatives as poisons of human type II toposiomerases: Differential effects on topoisomerase IIα and IIβ. Bioorg. Med. Chem. Lett. 2018, 28, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Feng, S.; Feng, J.; Dong, J.; Yang, K.; Liu, Z.; Qiao, X. Synthesis and biological evaluation of novel pyrazoline derivatives containing indole skeleton as anti-cancer agents targeting topoisomerase II. Eur. J. Med. Chem. 2020, 200, 112459. [Google Scholar] [CrossRef] [PubMed]
- Bergant Loboda, K.; Janežič, M.; Štampar, M.; Žegura, B.; Filipic, M.; Perdih, A. Substituted 4,5′-Bithiazoles as Catalytic Inhibitors of Human DNA Topoisomerase IIα. J. Chem. Inf. Model. 2020, 60, 3662–3678. [Google Scholar] [CrossRef]
- Li, A.-L.; Hao, Y.; Wang, W.-Y.; Liu, Q.-S.; Sun, Y.; Gu, W. Design, Synthesis, and Anticancer Evaluation of Novel Indole Derivatives of Ursolic Acid as Potential Topoisomerase II Inhibitors. Int. J. Mol. Sci. 2020, 21, 2876. [Google Scholar] [CrossRef]
- Legina, M.S.; Nogueira, J.J.; Kandioller, W.; Jakupec, M.A.; González, L.; Keppler, B.K. Biological evaluation of novel thiomaltol-based organometallic complexes as topoisomerase IIα inhibitors. J. Biol. Inorg. Chem. 2020, 25, 451–465. [Google Scholar] [CrossRef]
- Hackl, C.M.; Legina, M.S.; Pichler, V.; Schmidlehner, M.; Roller, A.; Dömötör, O.; Enyedi, E.A.; Jakupec, M.A.; Kandioller, W.; Keppler, B.K. Thiomaltol-based organometallic complexes with 1 methylimidazole as leaving group: Synthesis, stability, and biological behavior. Chem. Eur. J. 2016, 22, 17269–17281. [Google Scholar] [CrossRef]
- Bau, J.T.; Kang, Z.; Austin, C.A.; Kurz, E.U. Salicylate, a catalytic Inhibitor of Topoisomerase II, Inhibits DNA Cleavage and Is Selective for the α-isoform. Mol. Pharmacol. 2014, 85, 198–207. [Google Scholar] [CrossRef]
- Kamila, B.; Anna, B.; Krzysztof, B.; Agnieszka, G. DNA topoisomerases as molecular targets for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2020, 35, 1781–1799. [Google Scholar]
- Sisodiya, S.; Paul, S.; Chaudhary, H.; Grewal, P.; Kumar, G.; Daniel, D.P.; Das, B.; Nayak, D.; Guchhait, S.K.; Kundu, C.N.; et al. Exploration of Benzo [b] carbazole-6,11-diones as anticancer agents: Synthesis and studies of hTopoIIα inhibition and apoptotic effects. Bioorg. Med. Chem. Lett. 2021, 49, 128274. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okoro, C.O.; Fatoki, T.H. A Mini Review of Novel Topoisomerase II Inhibitors as Future Anticancer Agents. Int. J. Mol. Sci. 2023, 24, 2532. https://doi.org/10.3390/ijms24032532
Okoro CO, Fatoki TH. A Mini Review of Novel Topoisomerase II Inhibitors as Future Anticancer Agents. International Journal of Molecular Sciences. 2023; 24(3):2532. https://doi.org/10.3390/ijms24032532
Chicago/Turabian StyleOkoro, Cosmas O., and Toluwase Hezekiah Fatoki. 2023. "A Mini Review of Novel Topoisomerase II Inhibitors as Future Anticancer Agents" International Journal of Molecular Sciences 24, no. 3: 2532. https://doi.org/10.3390/ijms24032532
APA StyleOkoro, C. O., & Fatoki, T. H. (2023). A Mini Review of Novel Topoisomerase II Inhibitors as Future Anticancer Agents. International Journal of Molecular Sciences, 24(3), 2532. https://doi.org/10.3390/ijms24032532