
Cytochrome P450 1B1 Expression Regulates Intracellular Iron Levels and Oxidative Stress in the Retinal Endothelium


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
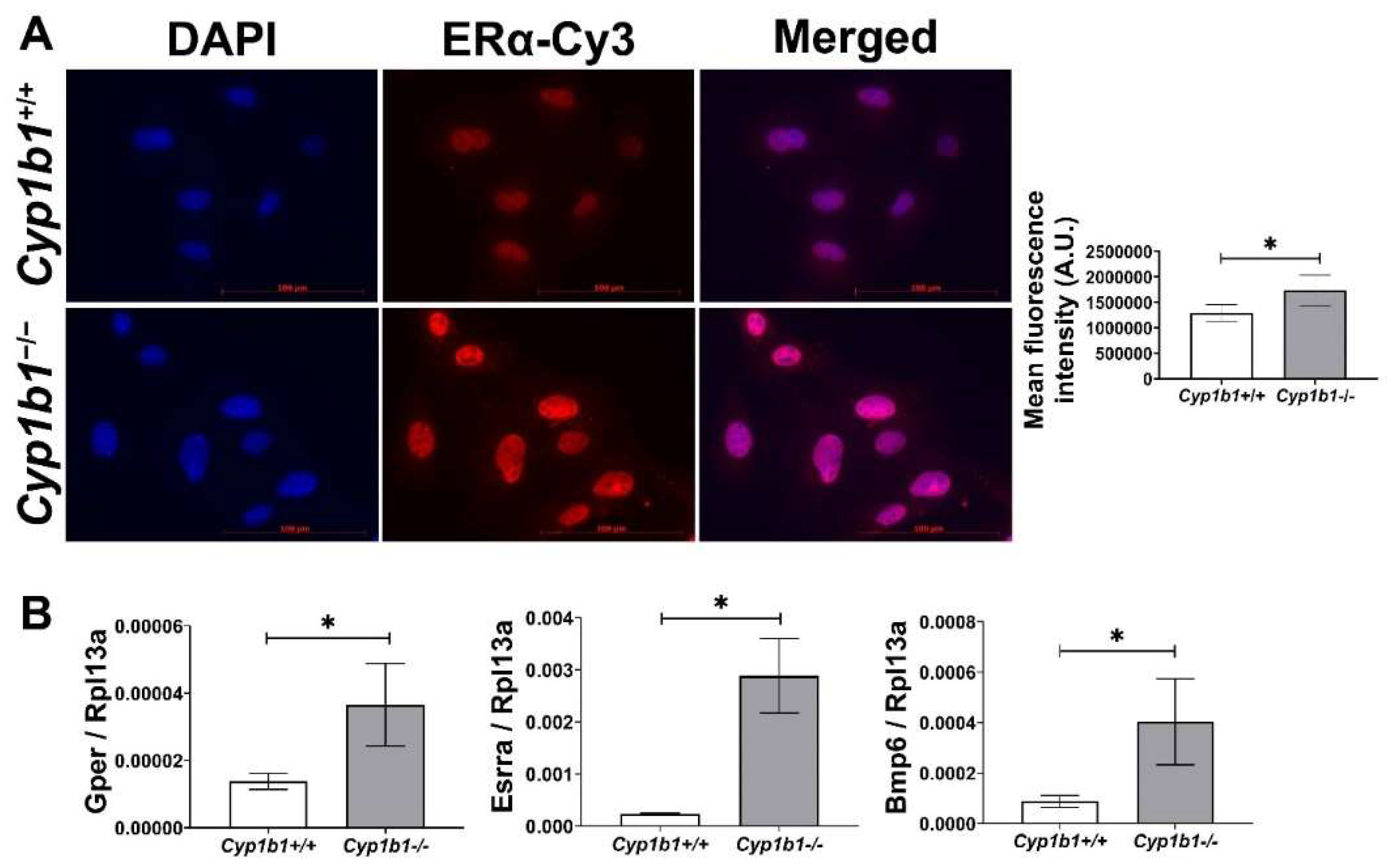
2.1. Increased ERα Nuclear Localization and Expression of ERα Target Genes in Cyp1b1−/− Retinal EC
2.2. Enhanced BMP6 Signaling in Cyp1b1−/− Retinal ECs
2.3. Increased Intracellular Fe2+ Levels in Cyp1b1−/− Retinal ECs
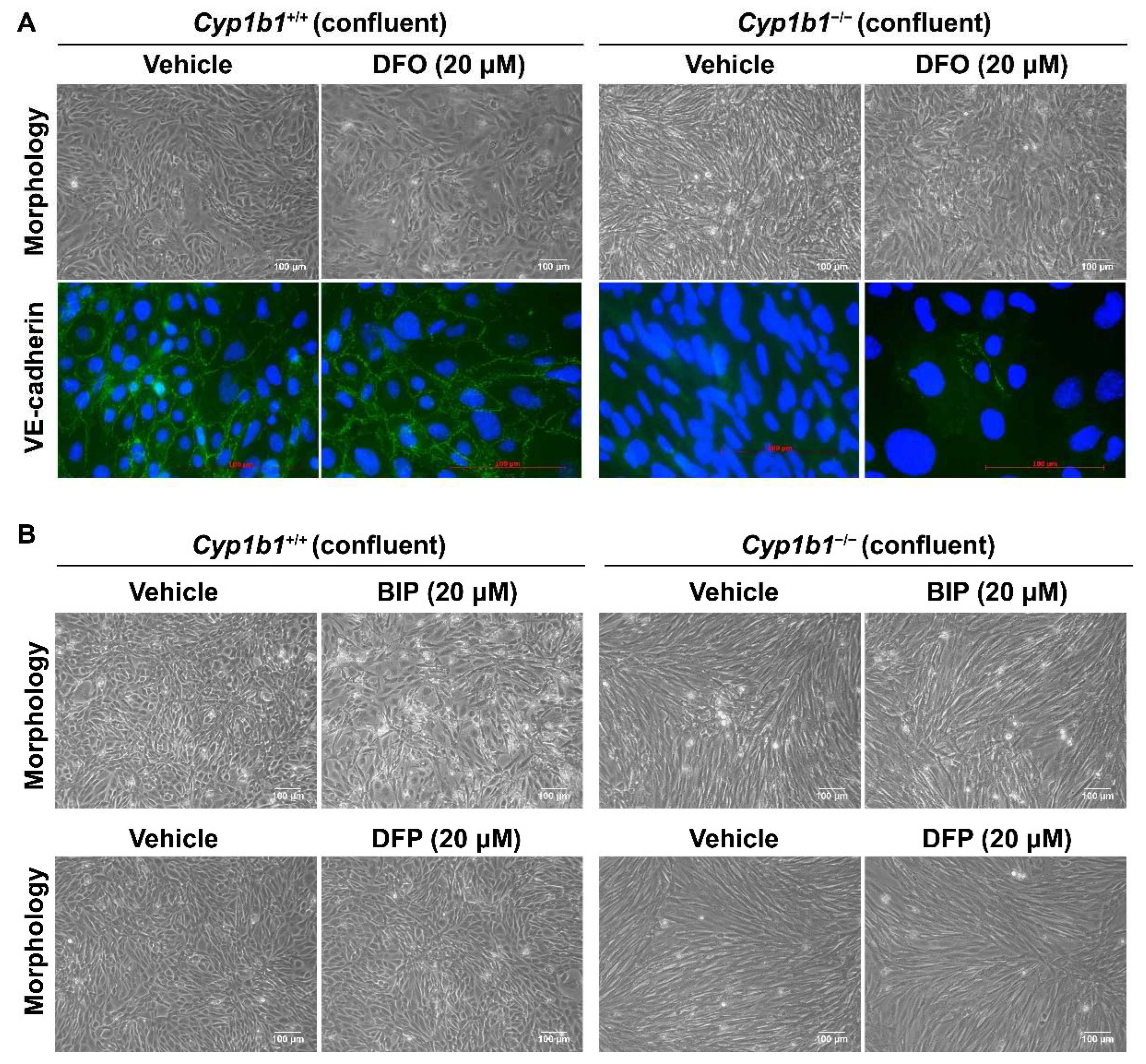
2.4. Removal of Excess Iron Reverses the Cellular Changes in Cyp1b1−/− Retinal ECs
2.5. Enhanced Lipid Peroxidation in Cyp1b1−/− Retinal ECs
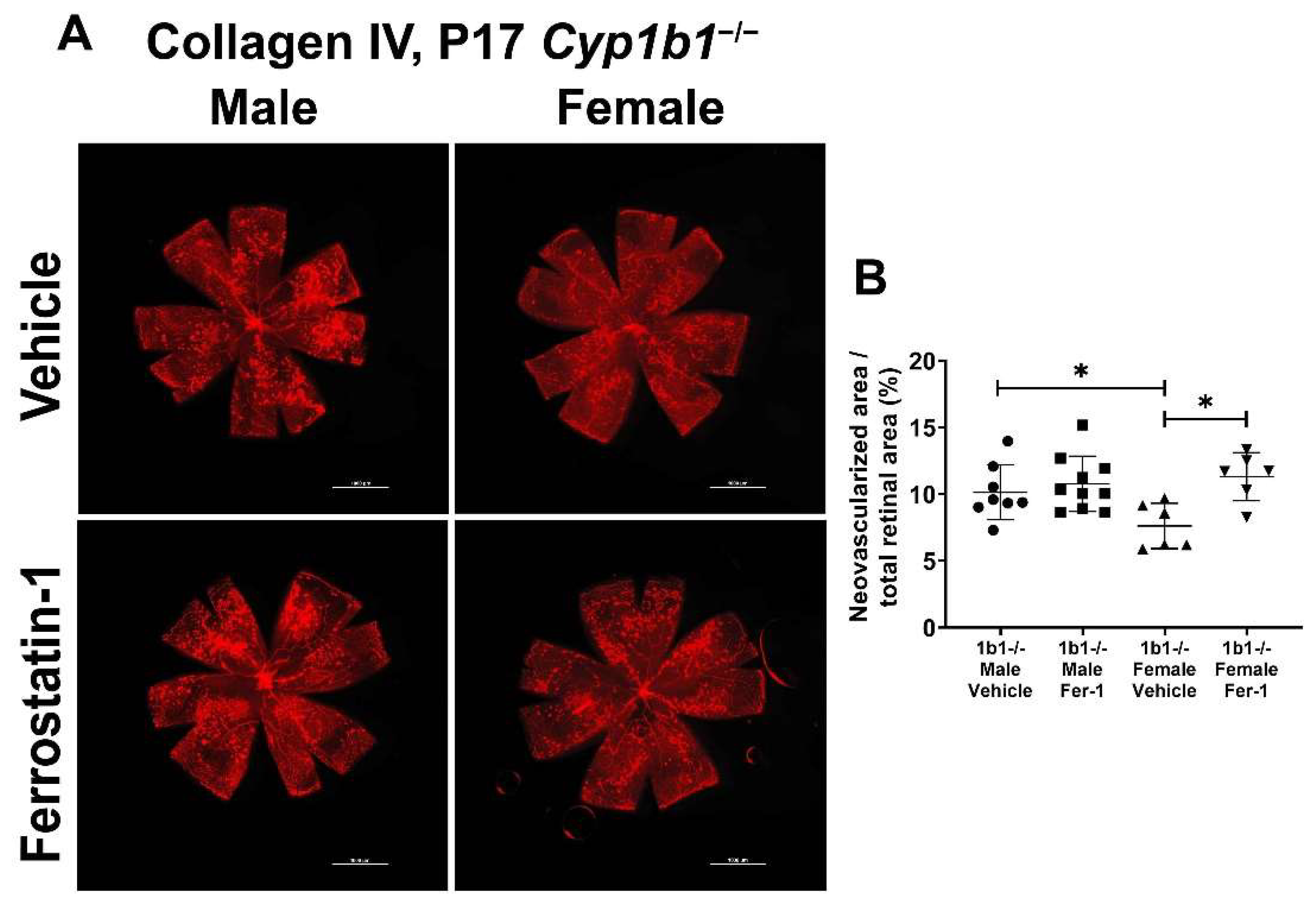
2.6. Increased Ferroptosis Sensitivity in Cyp1b1−/− Retinal ECs Is Associated with ERα Activity
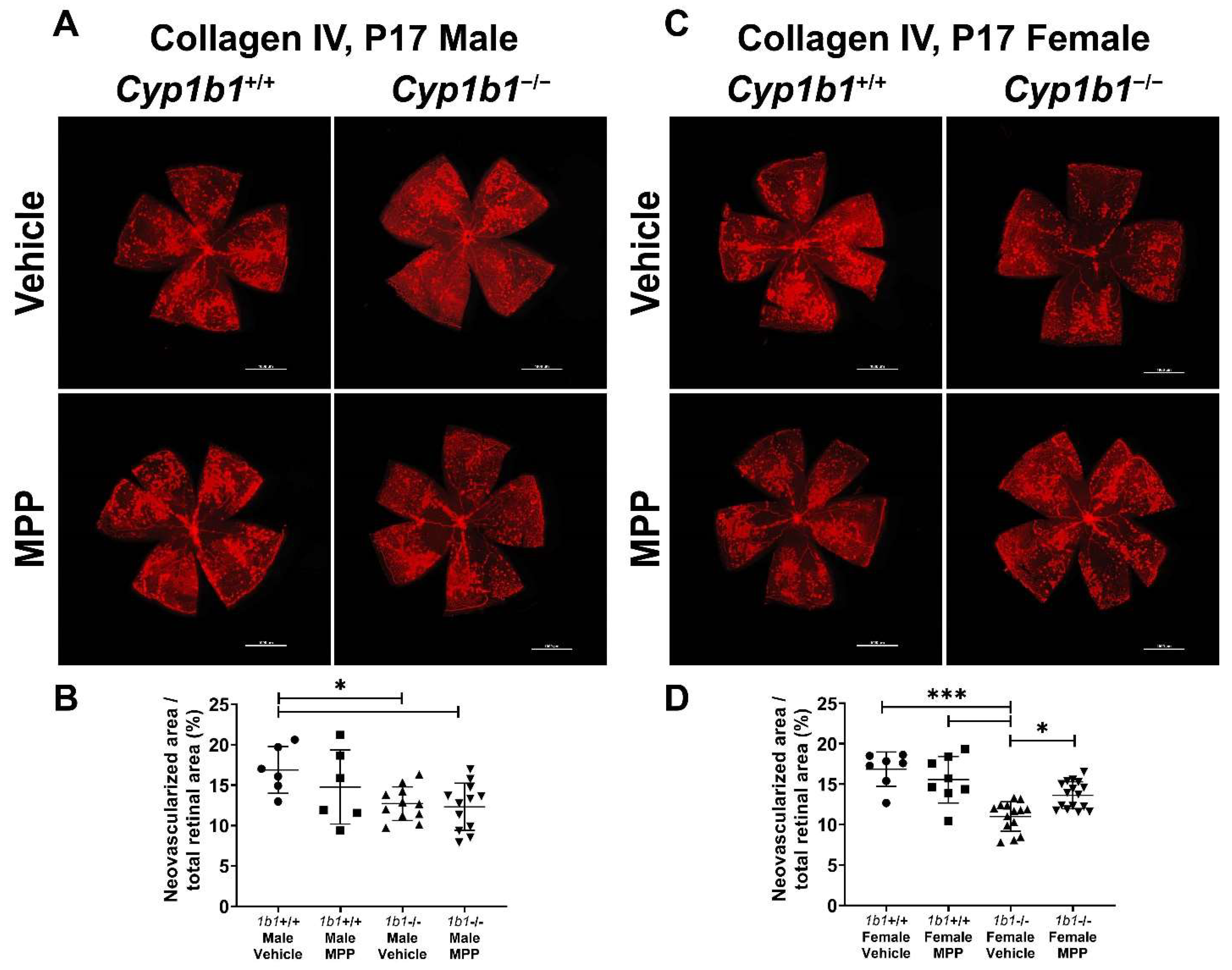
2.7. Restoration of Retinal Neovascularization in Cyp1b1−/− Mice by ERα Antagonist
2.8. Lipid Peroxide Chelation Restored Retinal Neovascularization in Cyp1b1−/− Mice
2.9. Effects of Iron Chelators on Retinal Neovascularization
2.10. Iron Supplementation Using Ferric Ammonium Citrate (FAC) Did Not Alter Retinal Neovascularization in Cyp1b1+/+ and Cyp1b1−/− Mice during OIR
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Cells
4.3. RNA Isolation, cDNA Preparation, and Real-Time Quantitative PCR (qPCR) Analysis
4.4. Western Blot Analysis
4.5. Indirect Immunofluorescence Staining
4.6. Intracellular Iron Level Analysis
4.7. Cell Viability Assay
4.8. Oxygen-Induced Ischemic Retinopathy
4.9. Retinal Wholemount Staining and Quantification of Neovascularization
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, S.; Sorenson, C.M.; Teixeira, L.; Dubielzig, R.R.; Peters, D.M.; Conway, S.J.; Jefcoate, C.R.; Sheibani, N. Cyp1b1 mediates periostin regulation of trabecular meshwork development by suppression of oxidative stress. Mol. Cell. Biol. 2013, 33, 4225–4240. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, D.; Jansson, I.; Rezaul, K.; Han, D.K.; Sarfarazi, M.; Schenkman, J.B. Cyp1b1 protein in the mouse eye during development: An immunohistochemical study. Drug Metab. Dispos. 2007, 35, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Hakkola, J.; Pasanen, M.; Pelkonen, O.; Hukkanen, J.; Evisalmi, S.; Anttila, S.; Rane, A.; Mantyla, M.; Purkunen, R.; Saarikoski, S.; et al. Expression of cyp1b1 in human adult and fetal tissues and differential inducibility of cyp1b1 and cyp1a1 by ah receptor ligands in human placenta and cultured cells. Carcinogenesis 1997, 18, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Bejjani, B.A.; Lewis, R.A.; Tomey, K.F.; Anderson, K.L.; Dueker, D.K.; Jabak, M.; Astle, W.F.; Otterud, B.; Leppert, M.; Lupski, J.R. Mutations in CYP1B1, the gene for cytochrome P4501B1, are the predominant cause of primary congenital glaucoma in Saudi Arabia. Am. J. Hum. Genet. 1998, 62, 325–333. [Google Scholar] [CrossRef]
- Stoilov, I.; Akarsu, A.N.; Sarfarazi, M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum. Mol. Genet. 1997, 6, 641–647. [Google Scholar] [CrossRef]
- Li, N.; Zhou, Y.; Du, L.; Wei, M.; Chen, X. Overview of Cytochrome P450 1B1 gene mutations in patients with primary congenital glaucoma. Exp. Eye Res. 2011, 93, 572–579. [Google Scholar] [CrossRef]
- Zhao, Y.; Sorenson, C.M.; Sheibani, N. Cytochrome P450 1B1 and Primary Congenital Glaucoma. J. Ophthalmic. Vis. Res. 2015, 10, 60–67. [Google Scholar] [CrossRef]
- Teixeira, L.; Zhao, Y.; Dubielzig, R.; Sorenson, C.; Sheibani, N. Ultrastructural abnormalities of the trabecular meshwork extracellular matrix in Cyp1b1-deficient mice. Vet. Pathol. 2015, 52, 397–403. [Google Scholar] [CrossRef]
- Alvarado, J.A.; Alvarado, R.G.; Yeh, R.F.; Franse-Carman, L.; Marcellino, G.R.; Brownstein, M.J. A new insight into the cellular regulation of aqueous outflow: How trabecular meshwork endothelial cells drive a mechanism that regulates the permeability of Schlemm’s canal endothelial cells. Br. J. Ophthalmol. 2005, 89, 1500–1505. [Google Scholar] [CrossRef]
- Kizhatil, K.; Ryan, M.; Marchant, J.K.; Henrich, S.; John, S.W. Schlemm’s canal is a unique vessel with a combination of blood vascular and lymphatic phenotypes that forms by a novel developmental process. PLoS Biol. 2014, 12, e1001912. [Google Scholar] [CrossRef] [PubMed]
- Eskin, S.G.; Turner, N.A.; McIntire, L.V. Endothelial cell cytochrome P450 1A1 and 1B1: Up-regulation by shear stress. Endothelium 2004, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chatzizisis, Y.S.; Coskun, A.U.; Jonas, M.; Edelman, E.R.; Feldman, C.L.; Stone, P.H. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: Molecular, cellular, and vascular behavior. J. Am. Coll. Cardiol. 2007, 49, 2379–2393. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Scheef, E.A.; Wang, S.; Sorenson, C.M.; Marcus, C.B.; Jefcoate, C.R.; Sheibani, N. CYP1B1 expression promotes the proangiogenic phenotype of endothelium through decreased intracellular oxidative stress and thrombospondin-2 expression. Blood 2009, 113, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Palenski, T.L.; Gurel, Z.; Sorenson, C.M.; Hankenson, K.D.; Sheibani, N. Cyp1B1 expression promotes angiogenesis by suppressing NF-kappaB activity. Am. J. Physiol. Cell Physiol. 2013, 305, C1170–C1184. [Google Scholar] [CrossRef]
- Tang, Y.; Scheef, E.A.; Gurel, Z.; Sorenson, C.M.; Jefcoate, C.R.; Sheibani, N. CYP1B1 and endothelial nitric oxide synthase combine to sustain proangiogenic functions of endothelial cells under hyperoxic stress. Am. J. Physiol. Cell Physiol. 2010, 298, C665–C678. [Google Scholar] [CrossRef]
- Bloodsworth, A.; O’Donnell, V.B.; Freeman, B.A. Nitric oxide regulation of free radical and enzyme-mediated lipid and lipoprotein oxidation. Arter. Thromb. Vasc. Biol. 2000, 20, 1707–1715. [Google Scholar] [CrossRef]
- Lepine, J.; Bernard, O.; Plante, M.; Tetu, B.; Pelletier, G.; Labrie, F.; Belanger, A.; Guillemette, C. Specificity and regioselectivity of the conjugation of estradiol, estrone, and their catecholestrogen and methoxyestrogen metabolites by human uridine diphospho-glucuronosyltransferases expressed in endometrium. J. Clin. Endocrinol. Metab. 2004, 89, 5222–5232. [Google Scholar] [CrossRef]
- Mookherjee, S.; Acharya, M.; Banerjee, D.; Bhattacharjee, A.; Ray, K. Molecular basis for involvement of CYP1B1 in MYOC upregulation and its potential implication in glaucoma pathogenesis. PLoS ONE 2012, 7, e45077. [Google Scholar] [CrossRef]
- Ong, D.B.; Colley, S.M.; Norman, M.R.; Kitazawa, S.; Tobias, J.H. Transcriptional regulation of a BMP-6 promoter by estrogen receptor alpha. J. Bone. Miner. Res. 2004, 19, 447–454. [Google Scholar] [CrossRef]
- Andriopoulos, B., Jr.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Annalora, A.J.; Marcus, C.B.; Jefcoate, C.R.; Sorenson, C.M.; Sheibani, N. Cytochrome P450 1B1: A Key Regulator of Ocular Iron Homeostasis and Oxidative Stress. Cells 2022, 11, 2930. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Cellular iron: Ferroportin is the only way out. Cell Metab. 2005, 1, 155–157. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Gruber, C.J.; Tschugguel, W.; Schneeberger, C.; Huber, J.C. Production and actions of estrogens. N. Engl. J. Med. 2002, 346, 340–352. [Google Scholar] [CrossRef]
- Gavin, K.M.; Seals, D.R.; Silver, A.E.; Moreau, K.L. Vascular endothelial estrogen receptor alpha is modulated by estrogen status and related to endothelial function and endothelial nitric oxide synthase in healthy women. J. Clin. Endocrinol. Metab. 2009, 94, 3513–3520. [Google Scholar] [CrossRef]
- Monje, P.; Zanello, S.; Holick, M.; Boland, R. Differential cellular localization of estrogen receptor α in uterine and mammary cells. Mol. Cell. Endocrinol. 2001, 181, 117–129. [Google Scholar] [CrossRef]
- Fan, D.X.; Yang, X.H.; Li, Y.N.; Guo, L. 17beta-Estradiol on the Expression of G-Protein Coupled Estrogen Receptor (GPER/GPR30) Mitophagy, and the PI3K/Akt Signaling Pathway in ATDC5 Chondrocytes In Vitro. Med. Sci. Monit. 2018, 24, 1936–1947. [Google Scholar] [CrossRef]
- Plante, B.J.; Lessey, B.A.; Taylor, R.N.; Wang, W.; Bagchi, M.K.; Yuan, L.; Scotchie, J.; Fritz, M.A.; Young, S.L. G protein-coupled estrogen receptor (GPER) expression in normal and abnormal endometrium. Reprod. Sci. 2012, 19, 684–693. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, Z.; Gladwell, W.; Teng, C.T. Estrogen Stimulates Estrogen-Related Receptor α Gene Expression through Conserved Hormone Response Elements. Endocrinology 2003, 144, 4894–4904. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Tajima, S.; Izawa-Ishizawa, Y.; Kihira, Y.; Ishizawa, K.; Tomita, S.; Tsuchiya, K.; Tamaki, T. Estrogen Regulates Hepcidin Expression via GPR30-BMP6-Dependent Signaling in Hepatocytes. PLoS ONE 2012, 7, e40465. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Dunmore, B.J.; Morrell, N.W. Bone morphogenetic protein type II receptor mutations causing protein misfolding in heritable pulmonary arterial hypertension. Proc. Am. Thorac. Soc. 2010, 7, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, V.M.; Salti, A.; Tilleman, H.; Zega, K.; Jukic, M.M.; Zou, H.; Friedel, R.H.; Prakash, N.; Blaess, S.; Edenhofer, F.; et al. BMP/SMAD Pathway Promotes Neurogenesis of Midbrain Dopaminergic Neurons In Vivo and in Human Induced Pluripotent and Neural Stem Cells. J. Neurosci. 2018, 38, 1662–1676. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, C.; Yoshikawa, H. Bone morphogenetic proteins. Nihon. Rinsho. 2004, 62 (Suppl. S2), 52–56. [Google Scholar] [PubMed]
- Ramey, G.; Deschemin, J.C.; Durel, B.; Canonne-Hergaux, F.; Nicolas, G.; Vaulont, S. Hepcidin targets ferroportin for degradation in hepatocytes. Haematologica 2010, 95, 501–504. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Knowles, H.J.; Raval, R.R.; Harris, A.L.; Ratcliffe, P.J. Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Cancer Res. 2003, 63, 1764–1768. [Google Scholar]
- Benn, A.; Bredow, C.; Casanova, I.; Vukičević, S.; Knaus, P. VE-cadherin facilitates BMP-induced endothelial cell permeability and signaling. J. Cell Sci. 2016, 129, 206–218. [Google Scholar] [CrossRef]
- Lin, Y.; Epstein, D.L.; Liton, P.B. Intralysosomal Iron Induces Lysosomal Membrane Permeabilization and Cathepsin D–Mediated Cell Death in Trabecular Meshwork Cells Exposed to Oxidative Stress. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6483–6495. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Biologically relevant metal ion-dependent hydroxyl radical generation. An update. FEBS Lett. 1992, 307, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Pu, F.; Chen, F.; Zhang, Z.; Shi, D.; Zhong, B.; Lv, X.; Tucker, A.B.; Fan, J.; Li, A.J.; Qin, K.; et al. Ferroptosis as a novel form of regulated cell death: Implications in the pathogenesis, oncometabolism and treatment of human cancer. Genes. Dis. 2022, 9, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Zhou, H.B.; Carlson, K.E.; Stossi, F.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Analogs of methyl-piperidinopyrazole (MPP): Antiestrogens with estrogen receptor alpha selective activity. Bioorganic. Med. Chem. Lett. 2009, 19, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Smith, L.E.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.; Sullivan, R.; D’Amore, P.A. Oxygen-induced retinopathy in the mouse. Investig. Ophthalmol. Vis. Sci. 1994, 35, 101–111. [Google Scholar]
- Falero-Perez, J.; Larsen, M.C.; Teixeira, L.B.C.; Zhang, H.F.; Lindner, V.; Sorenson, C.M.; Jefcoate, C.R.; Sheibani, N. Targeted deletion of Cyp1b1 in pericytes results in attenuation of retinal neovascularization and trabecular meshwork dysgenesis. Trends Dev. Biol. 2019, 12, 1–12. [Google Scholar]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.-M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox. Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Tajima, S.; Ikeda, Y.; Sawada, K.; Yamano, N.; Horinouchi, Y.; Kihira, Y.; Ishizawa, K.; Izawa-Ishizawa, Y.; Kawazoe, K.; Tomita, S.; et al. Iron reduction by deferoxamine leads to amelioration of adiposity via the regulation of oxidative stress and inflammation in obese and type 2 diabetes KKAy mice. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E77–E86. [Google Scholar] [CrossRef]
- Li, Y.; Pan, K.; Chen, L.; Ning, J.-l.; Li, X.; Yang, T.; Terrando, N.; Gu, J.; Tao, G. Deferoxamine regulates neuroinflammation and iron homeostasis in a mouse model of postoperative cognitive dysfunction. J. Neuroinflamm. 2016, 13, 268. [Google Scholar] [CrossRef]
- Li, Z.L.; Lam, S.; Tso, M.O. Desferrioxamine ameliorates retinal photic injury in albino rats. Curr. Eye Res. 1991, 10, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Suzuki, T.; Takahashi, K.; Koguchi, T.; Hirayama, T.; Mori, A.; Nakahara, T.; Nagasawa, H.; Ishii, K. Iron-chelating agents attenuate NMDA-Induced neuronal injury via reduction of oxidative stress in the rat retina. Exp. Eye Res. 2018, 171, 30–36. [Google Scholar] [CrossRef]
- Piga, A.; Gaglioti, C.; Fogliacco, E.; Tricta, F. Comparative effects of deferiprone and deferoxamine on survival and cardiac disease in patients with thalassemia major: A retrospective analysis. Haematologica 2003, 88, 489–496. [Google Scholar] [PubMed]
- Hadziahmetovic, M.; Pajic, M.; Grieco, S.; Song, Y.; Song, D.; Li, Y.; Cwanger, A.; Iacovelli, J.; Chu, S.; Ying, G.S.; et al. The Oral Iron Chelator Deferiprone Protects Against Retinal Degeneration Induced through Diverse Mechanisms. Transl. Vis. Sci. Amp. Technol. 2012, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Song, Y.; Hadziahmetovic, M.; Zhong, Y.; Dunaief, J.L. Systemic administration of the iron chelator deferiprone protects against light-induced photoreceptor degeneration in the mouse retina. Free Radic. Biol. Med. 2012, 53, 64–71. [Google Scholar] [CrossRef]
- Cui, Q.N.; Bargoud, A.R.; Ross, A.G.; Song, Y.; Dunaief, J.L. Oral administration of the iron chelator deferiprone protects against loss of retinal ganglion cells in a mouse model of glaucoma. Exp. Eye Res. 2020, 193, 107961. [Google Scholar] [CrossRef]
- Tenne, D.; Bogoslavsky, B.; Bino, A. Ferric Ammonium Citrate–What’s in It? Eur. J. Inorg. Chem. 2015, 2015, 4159–4161. [Google Scholar] [CrossRef]
- Messner, D.J.; Kowdley, K.V. Neoplastic transformation of rat liver epithelial cells is enhanced by non-transferrin-bound iron. BMC Gastroenterol. 2008, 8, 2. [Google Scholar] [CrossRef]
- Kir, D.; Saluja, M.; Modi, S.; Venkatachalam, A.; Schnettler, E.; Roy, S.; Ramakrishnan, S. Cell-permeable iron inhibits vascular endothelial growth factor receptor-2 signaling and tumor angiogenesis. Oncotarget 2016, 7, 65348–65363. [Google Scholar] [CrossRef]
- Hahn, P.; Qian, Y.; Dentchev, T.; Chen, L.; Beard, J.; Harris, Z.L.; Dunaief, J.L. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 13850–13855. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen–estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef]
- Yi, P.; Wang, Z.; Feng, Q.; Pintilie, G.D.; Foulds, C.E.; Lanz, R.B.; Ludtke, S.J.; Schmid, M.F.; Chiu, W.; O’Malley, B.W. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol. Cell 2015, 57, 1047–1058. [Google Scholar] [CrossRef]
- Ogueta, S.B.; Schwartz, S.D.; Yamashita, C.K.; Farber, D.B. Estrogen receptor in the human eye: Influence of gender and age on gene expression. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1906–1911. [Google Scholar]
- Munaut, C.; Lambert, V.; Noël, A.; Frankenne, F.; Deprez, M.; Foidart, J.-M.; Rakic, J.-M. Presence of oestrogen receptor type β in human retina. Br. J. Ophthalmol. 2001, 85, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Mangiamele, L.A.; Gomez, J.R.; Curtis, N.J.; Thompson, R.R. GPER/GPR30, a membrane estrogen receptor, is expressed in the brain and retina of a social fish (Carassius auratus) and colocalizes with isotocin. J. Comp. Neurol. 2017, 525, 252–270. [Google Scholar] [CrossRef]
- Parvathaneni, K.; Grigsby, J.G.; Betts, B.S.; Tsin, A.T. Estrogen-induced retinal endothelial cell proliferation: Possible involvement of pigment epithelium-derived factor and phosphoinositide 3-kinase/mitogen-activated protein kinase pathways. J. Ocul. Pharmacol. Ther. 2013, 29, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Sissung, T.M.; Price, D.K.; Sparreboom, A.; Figg, W.D. Pharmacogenetics and regulation of human cytochrome P450 1B1: Implications in hormone-mediated tumor metabolism and a novel target for therapeutic intervention. Mol. Cancer Res. 2006, 4, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Su, Y.-Q.; Eppig, J.J. Does bone morphogenetic protein 6 (BMP6) affect female fertility in the mouse? Biol. Reprod. 2010, 83, 997–1004. [Google Scholar] [CrossRef]
- Plant, A.; Tobias, J.H. Increased bone morphogenetic protein-6 expression in mouse long bones after estrogen administration. J. Bone Miner. Res. 2002, 17, 782–790. [Google Scholar] [CrossRef]
- Rickard, D.J.; Hofbauer, L.C.; Bonde, S.K.; Gori, F.; Spelsberg, T.C.; Riggs, B.L. Bone morphogenetic protein-6 production in human osteoblastic cell lines. Selective regulation by estrogen. J. Clin. Investig. 1998, 101, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yan, J.-D.; Zhang, L.; Wang, Q.; Lü, S.-J.; Zhang, J.; Zhu, T.-H. Activation of bone morphogenetic protein-6 gene transcription in MCF-7 cells by estrogen. Chin. Med. J. 2005, 118, 1629–1636. [Google Scholar] [PubMed]
- Gruber, R.; Graninger, W.; Bobacz, K.; Watzek, G.; Erlacher, L. BMP-6-induced osteogenic differentiation of mesenchymal cell lines is not modulated by sex steroids and resveratrol. Cytokine 2003, 23, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Jian, J.; Katz, S.; Abramson, S.B.; Huang, X. 17β-Estradiol inhibits iron hormone hepcidin through an estrogen responsive element half-site. Endocrinology 2012, 153, 3170–3178. [Google Scholar] [CrossRef] [PubMed]
- Gnana-Prakasam, J.P.; Martin, P.M.; Smith, S.B.; Ganapathy, V. Expression and function of iron-regulatory proteins in retina. IUBMB Life 2010, 62, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Chaudhary, S.; McDonald, D.; Kritikos, A.; Bhargava, D.; Singh, N. Local synthesis of hepcidin in the anterior segment of the eye: A novel observation with physiological and pathological implications. Exp. Eye Res. 2020, 190, 107890. [Google Scholar] [CrossRef]
- Gutteridge, J.; Richmond, R.; Halliwell, B. Inhibition of the iron-catalysed formation of hydroxyl radicals from superoxide and of lipid peroxidation by desferrioxamine. Biochem. J. 1979, 184, 469–472. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Non-Apoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Park, S.S. Cell Therapy Applications for Retinal Vascular Diseases: Diabetic Retinopathy and Retinal Vein Occlusion. Investig. Ophthalmol. Vis. Sci. 2016, 57. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kowluru, A.; Mishra, M.; Kumar, B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog. Retin. Eye Res. 2015, 48, 40–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, X.; Xu, K.; Wang, Y.; Wang, Y.; Liu, X.; Zhang, X.; Wang, L.; Li, X. 17β-estradiol ameliorates oxygen-induced retinopathy in the early hyperoxic phase. Biochem. Biophys. Res. Commun. 2015, 457, 700–705. [Google Scholar] [CrossRef]
- Miyamoto, N.; Mandai, M.; Takagi, H.; Suzuma, I.; Suzuma, K.; Koyama, S.; Otani, A.; Oh, H.; Honda, Y. Contrasting effect of estrogen on VEGF induction under different oxygen status and its role in murine ROP. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2007–2014. [Google Scholar]
- Taylor, N.L.; Day, D.A.; Millar, A.H. Targets of stress-induced oxidative damage in plant mitochondria and their impact on cell carbon/nitrogen metabolism. J. Exp. Bot. 2004, 55, 1–10. [Google Scholar] [CrossRef]
- Song, D.; Dunaief, J.L. Retinal iron homeostasis in health and disease. Front. Aging Neurosci. 2013, 5, 24. [Google Scholar] [CrossRef]
- Kwiatkowski, J.L.; Hamdy, M.; El-Beshlawy, A.; Ebeid, F.S.E.; Badr, M.; Alshehri, A.; Kanter, J.; Inusa, B.; Adly, A.A.M.; Williams, S.; et al. Deferiprone vs deferoxamine for transfusional iron overload in SCD and other anemias: A randomized, open-label noninferiority study. Blood Adv. 2022, 6, 1243–1254. [Google Scholar] [CrossRef]
- Hahn, P.; Ying, G.S.; Beard, J.; Dunaief, J.L. Iron levels in human retina: Sex difference and increase with age. Neuroreport 2006, 17, 1803–1806. [Google Scholar] [CrossRef]
- Hahn, P.; Song, Y.; Ying, G.S.; He, X.; Beard, J.; Dunaief, J.L. Age-dependent and gender-specific changes in mouse tissue iron by strain. Exp. Gerontol. 2009, 44, 594–600. [Google Scholar] [CrossRef]
- Baumann, B.H.; Shu, W.; Song, Y.; Sterling, J.; Kozmik, Z.; Lakhal-Littleton, S.; Dunaief, J.L. Liver-Specific, but Not Retina-Specific, Hepcidin Knockout Causes Retinal Iron Accumulation and Degeneration. Am. J. Pathol. 2019, 189, 1814–1830. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sorenson, C.M.; Sheibani, N. Attenuation of retinal vascular development and neovascularization during oxygen-induced ischemic retinopathy in Bcl-2-/- mice. Dev. Biol. 2005, 279, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.; Connor, K.M.; Sapieha, P.; Willett, K.L.; Krah, N.M.; Dennison, R.J.; Chen, J.; Guerin, K.I.; Smith, L.E.H. Computer-aided quantification of retinal neovascularization. Angiogenesis 2009, 12, 297–301. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.-S.; Zaitoun, I.S.; Wang, S.; Darjatmoko, S.R.; Sorenson, C.M.; Sheibani, N. Cytochrome P450 1B1 Expression Regulates Intracellular Iron Levels and Oxidative Stress in the Retinal Endothelium. Int. J. Mol. Sci. 2023, 24, 2420. https://doi.org/10.3390/ijms24032420
Song Y-S, Zaitoun IS, Wang S, Darjatmoko SR, Sorenson CM, Sheibani N. Cytochrome P450 1B1 Expression Regulates Intracellular Iron Levels and Oxidative Stress in the Retinal Endothelium. International Journal of Molecular Sciences. 2023; 24(3):2420. https://doi.org/10.3390/ijms24032420
Chicago/Turabian StyleSong, Yong-Seok, Ismail S. Zaitoun, Shoujian Wang, Soesiawati R. Darjatmoko, Christine M. Sorenson, and Nader Sheibani. 2023. "Cytochrome P450 1B1 Expression Regulates Intracellular Iron Levels and Oxidative Stress in the Retinal Endothelium" International Journal of Molecular Sciences 24, no. 3: 2420. https://doi.org/10.3390/ijms24032420
APA StyleSong, Y.-S., Zaitoun, I. S., Wang, S., Darjatmoko, S. R., Sorenson, C. M., & Sheibani, N. (2023). Cytochrome P450 1B1 Expression Regulates Intracellular Iron Levels and Oxidative Stress in the Retinal Endothelium. International Journal of Molecular Sciences, 24(3), 2420. https://doi.org/10.3390/ijms24032420