

Synthesis of Carvone Derivatives and In Silico and In Vitro Screening of Anti-Inflammatory Activity in Murine Macrophages

 ,
,  , and
, and 
Abstract
1. Introduction
2. Results
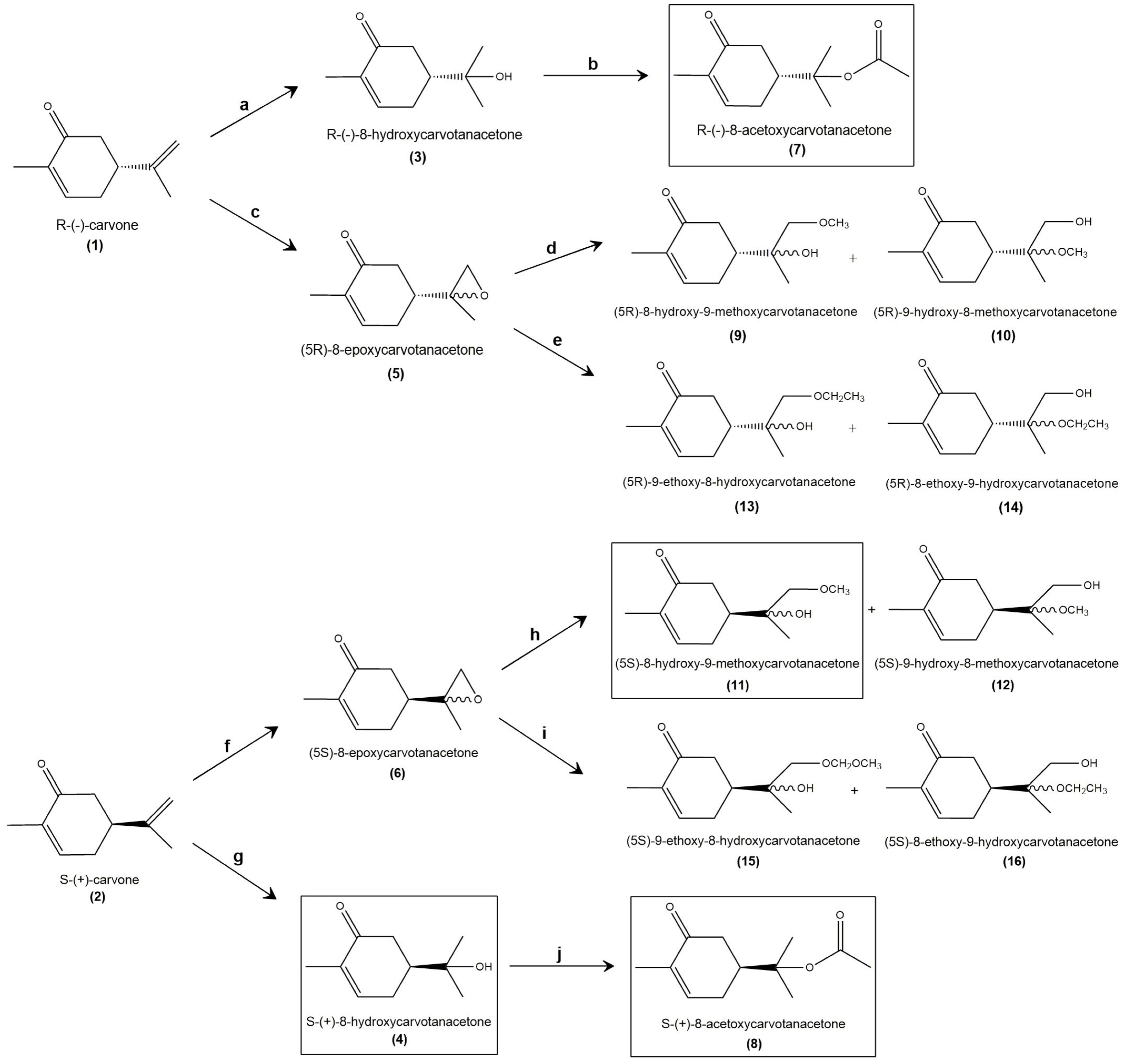
2.1. Synthesis and Structural Confirmation of the Carvone Derivatives
2.2. Cell Viability of Raw 264.7 Macrophages Treated with the Test Compounds
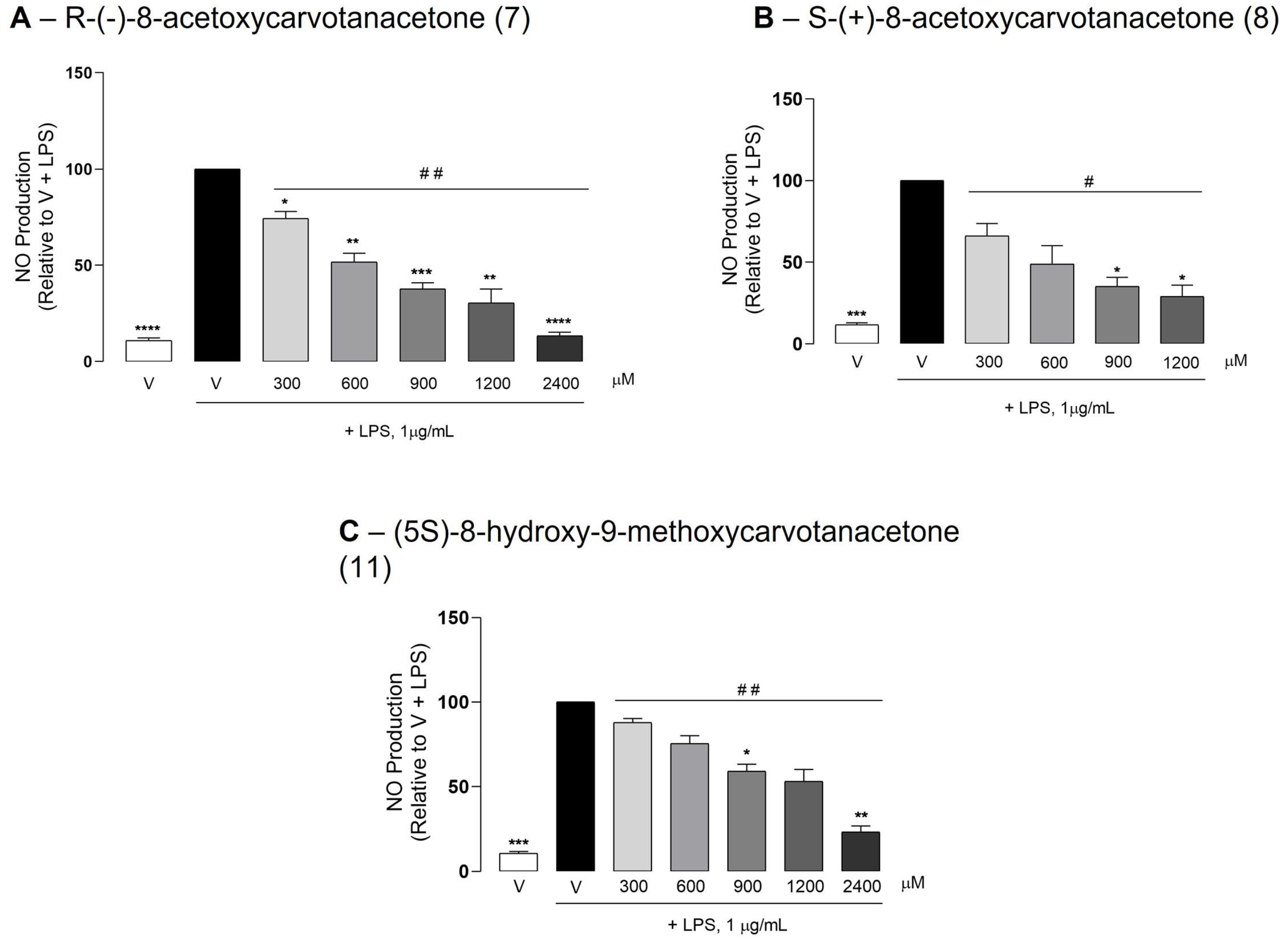
2.3. The Test Compounds Reduced LPS-Induced NO Production in Raw 264.7 Macrophages
2.4. Effect of the Test Compounds on LPS-Induced iNOS and IL-1β Expression in Raw 264.7 Macrophages
2.5. Predicted Properties of the Test Compounds
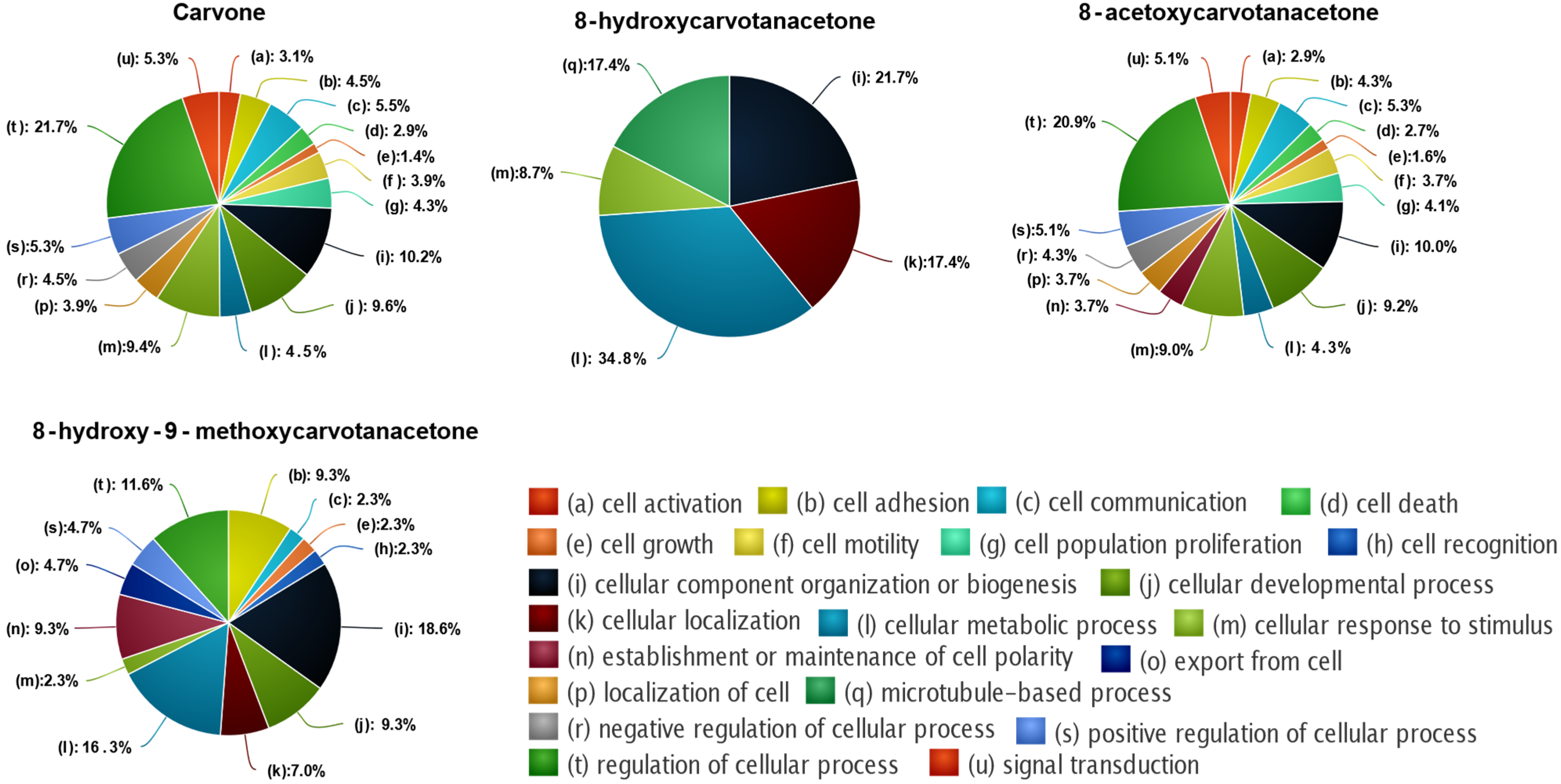
2.5.1. Enrichment Analysis and Target Prediction
2.5.2. ADME
3. Discussion
4. Materials and Methods
4.1. Semi-Synthesis of Carvone Derivatives
4.1.1. Chromatography
4.1.2. Nuclear Magnetic Resonance (NMR)
4.1.3. Infrared Spectroscopy (FT-IR)
4.1.4. Reagents and Solvents
4.1.5. Hydration Reaction
4.1.6. Esterification of Tertiary Alcohols: Synthesis of R-(−)-8-Acetoxycarvotanacetone (7) and S-(+)-8-Acetoxycarvotanacetone (8)
4.1.7. Regioselective Epoxidation of Carvone’s Exocyclic Double Bond: Synthesis of (5R)-8-Epoxycarvotanacetone (5) and (5S)-8-Epoxycarvotanacetone (6)
4.1.8. Catalytic Ring Opening of the Epoxide with Alcohols as Nucleophiles and Solvent
4.2. Cell Culture and Treatments
4.3. Selection of the Noncytotoxic Concentrations of the Test Compounds by the Resazurin Reduction Assay
4.4. Nitric Oxide Production
4.5. Western Blotting
4.6. ADME, Toxicity and Target Prediction Using Ligand Express
4.6.1. ADME Properties
4.6.2. Drug–Target Interaction (DTi) Prediction
4.6.3. Enrichment Analysis
4.7. Statistical Analysis of the In Vitro Results
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barbé-Tuana, F.; Funchal, G.; Schmitz, C.R.R.; Maurmann, R.M.; Bauer, M.E. The Interplay between Immunosenescence and Age-Related Diseases. Semin. Immunopathol. 2020, 42, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Candore, G.; Caruso, C.; Jirillo, E.; Magrone, T.; Vasto, S. Low Grade Inflammation as a Common Pathogenetic Denominator in Age-Related Diseases: Novel Drug Targets for Anti-Ageing Strategies and Successful Ageing Achievement. Curr. Pharm. Des. 2010, 16, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Mendes, A.F.; Cruz, M.T.; Gualillo, O. Editorial: The Physiology of Inflammation—The Final Common Pathway to Disease. Front. Physiol. 2018, 9, 1741. [Google Scholar] [CrossRef] [PubMed]
- Bektas, A.; Schurman, S.H.; Sen, R.; Ferrucci, L.; Branch, G.; Institutes, N. Aging, Inflammation and the Environment. Exp. Gerontol. 2019, 105, 10–18. [Google Scholar] [CrossRef]
- Calder, P.C.; Bosco, N.; Bourdet-Sicard, R.; Capuron, L.; Delzenne, N.; Doré, J.; Franceschi, C.; Lehtinen, M.J.; Recker, T.; Salvioli, S.; et al. Health Relevance of the Modification of Low Grade Inflammation in Ageing (Inflammageing) and the Role of Nutrition. Ageing Res. Rev. 2017, 40, 95–119. [Google Scholar] [CrossRef]
- Miguel, M.G. Antioxidant and Anti-Inflammatory Activities of Essential Oils: A Short Review. Molecules 2010, 15, 9252–9287. [Google Scholar] [CrossRef]
- Lagoumtzi, S.M.; Chondrogianni, N. Senolytics and Senomorphics: Natural and Synthetic Therapeutics in the Treatment of Aging and Chronic Diseases. Free Radic. Biol. Med. 2021, 171, 169–190. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural Products: A Continuing Source of Novel Drug Leads. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Kang, K.S. Phytochemical Constituents of Medicinal Plants for the Treatment of Chronic Inflammation. Biomolecules 2021, 11, 672. [Google Scholar] [CrossRef]
- Prasad, S.; Sung, B.; Aggarwal, B.B. Age-Associated Chronic Diseases Require Age-Old Medicine: Role of Chronic Inflammation. Prev. Med. 2012, 54, S29–S37. [Google Scholar] [CrossRef] [PubMed]
- Salmerón-Manzano, E.; Garrido-Cardenas, J.A.; Manzano-Agugliaro, F. Worldwide Research Trends on Medicinal Plants. Int. J. Environ. Res. Public Health 2020, 17, 3376. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Morris-Natschke, S.L.; Lee, K.-H. Strategies for the Optimization of Natural Leads to Anticancer Drugs or Drug Candidates. Med. Res. Rev. 2016, 36, 32–91. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.; Leitão, A.J.; Neves, B.M.; Judas, F.; Cavaleiro, C.; Mendes, A.F. Standardised Comparison of Limonene-Derived Monoterpenes Identifies Structural Determinants of Anti-Inflammatory Activity. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Agoni, C.; Olotu, F.A.; Ramharack, P.; Soliman, M.E. Druggability and Drug-Likeness Concepts in Drug Design: Are Biomodelling and Predictive Tools Having Their Say? J. Mol. Model. 2020, 26, 120. [Google Scholar] [CrossRef]
- Huo, M.; Cui, X.; Xue, J.; Chi, G.; Gao, R.; Deng, X.; Guan, S.; Wei, J.; Soromou, L.W.; Feng, H.; et al. Anti-Inflammatory Effects of Linalool in RAW 264.7 Macrophages and Lipopolysaccharide-Induced Lung Injury Model. J. Surg. Res. 2013, 180, e47–e54. [Google Scholar] [CrossRef]
- Ren, J.; Su, D.; Li, L.; Cai, H.; Zhang, M.; Zhai, J.; Li, M.; Wu, X.; Hu, K. Anti-Inflammatory Effects of Aureusidin in LPS-Stimulated RAW264.7 Macrophages via Suppressing NF-ΚB and Activating ROS- and MAPKs-Dependent Nrf2/HO-1 Signaling Pathways. Toxicol. Appl. Pharmacol. 2020, 387, 114846. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Sousa, C.; Neves, B.M.; Leitão, A.J.; Mendes, A.F. Elucidation of the Mechanism Underlying the Anti-Inflammatory Properties of (S)-(+)-Carvone Identifies a Novel Class of Sirtuin-1 Activators in a Murine Macrophage Cell Line. Biomedicines 2021, 9, 777. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as Regulators of Metabolism and Healthspan Europe PMC Funders Group. Nat. Rev. Mol. Cell Biol. 2016, 13, 225–238. [Google Scholar] [CrossRef]
- Sousa, C.; Mendes, A.F. Monoterpenes as Sirtuin-1 Activators: Therapeutic Potential in Aging and Related Diseases. Biomolecules 2022, 12, 921. [Google Scholar] [CrossRef] [PubMed]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry, 5th ed.; Springer International Publishing: Charlottesville, VA, USA, 2007; ISBN 978-0-387-44899-3. [Google Scholar]
- Büchi, G.; Wüest, H. New Synthesis of β-Agarofuran and of Dihydroagarofuran. J. Org. Chem. 1979, 44, 546–549. [Google Scholar] [CrossRef]
- Mandai, H.; Fujii, K.; Yasuhara, H.; Abe, K.; Mitsudo, K.; Korenaga, T.; Suga, S. Enantioselective Acyl Transfer Catalysis by a Combination of Common Catalytic Motifs and Electrostatic Interactions. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Mak, K.K.W.; Lai, Y.M.; Siu, Y.H. Regiospecific Epoxidation of Carvone: A Discovery-Oriented Experiment for Understanding the Selectivity and Mechanism of Epoxidation Reactions. J. Chem. Educ. 2006, 83, 1058–1061. [Google Scholar] [CrossRef]
- Sawano, T.; Yamamoto, H. Regio-and Enantioselective Substrate-Directed Epoxidation. Eur. J. Org. Chem. 2020, 2020, 2369–2378. [Google Scholar] [CrossRef]
- Smith, J.G. Synthetically Useful Reactions of Epoxides. Synthesis 1984, 1984, 629–656. [Google Scholar] [CrossRef]
- Stead, P.; Marley, H.; Mahmoudian, M.; Webb, G.; Noble, D.; Ip, Y.T.; Piga, E.; Rossi, T.; Roberts, S.; Dawson, M.J. Efficient Procedures for the Large-Scale Preparation of (1S,2S)-Trans-2-Methoxycyclohexanol, a Key Chiral Intermediate in the Synthesis of Tricyclic β-Lactam Antibiotics. Tetrahedron Asymmetry 1996, 7, 2247–2250. [Google Scholar] [CrossRef]
- Leitão, A.J.L.; Salvador, J.A.R.; Pinto, R.M.A.; Sá e Melo, M.L. Hydrazine Sulphate: A Cheap and Efficient Catalyst for the Regioselective Ring-Opening of Epoxides. A Metal-Free Procedure for the Preparation of β-Alkoxy Alcohols. Tetrahedron Lett. 2008, 49, 1694–1697. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2014, 39, 1003–1018. [Google Scholar] [CrossRef]
- Fromm, M.F. Importance of P-Glycoprotein at Blood–Tissue Barriers. Trends Pharmacol. Sci. 2004, 25, 423–429. [Google Scholar] [CrossRef]
- Molecular Modeling Group of the SIB. Swiss Institute of Bioinformatics SwissADME. Available online: http://www.swissadme.ch/ (accessed on 13 January 2023).
- Pires, D.E.V.; Ascher, D.B. PkCSM—Pharmacokinetics. Available online: https://biosig.lab.uq.edu.au/pkcsm/prediction (accessed on 13 January 2023).
- Mendes, A.F.; Caramona, M.M.; Carvalho, A.P.; Lopes, M.C. Differential Roles of Hydrogen Peroxide and Superoxide in Mediating IL-1-Induced NF-κB Activation and INOS Expression in Bovine Articular Chondrocytes. J. Cell. Biochem. 2003, 88, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic Inflammation in the Etiology of Disease across the Life Span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, S.A.C.; Salvador, J.A.R.; Cortés, R.; Cascante, M. Novel Celastrol Derivatives with Improved Selectivity and Enhanced Antitumour Activity: Design, Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2017, 138, 422–437. [Google Scholar] [CrossRef]
- Eckrich, R.; Neumann, B.; Stammler, H.G.; Kuck, D. Stereoselective Epoxidation of Cyclohexa-Anellated Triquinacenes with Iodine/Silver(I) Oxide as Compared to m-Chloroperbenzoic Acid. J. Org. Chem. 1996, 61, 3839–3843. [Google Scholar] [CrossRef] [PubMed]
- Präbst, K.; Engelhardt, H.; Ringgeler, S.; Hübner, H. Basic Colorimetric Proliferation Assays: MTT, WST, and Resazurin. Method. Mol. Biol. 2017, 1601, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.S.; Grisham, M.B. Methods to Detect Nitric Oxide and Its Metabolites in Biological Samples. Free Radic. Biol. Med. 2007, 43, 645–657. [Google Scholar] [CrossRef]
- Brereton, A.E.; MacKinnon, S.; Safikhani, Z.; Reeves, S.; Alwash, S.; Shahani, V.; Windemuth, A. Predicting Drug Properties with Parameter-Free Machine Learning: Pareto-Optimal Embedded Modeling (POEM). Mach. Learn. Sci. Technol. 2020, 1, 025008. [Google Scholar] [CrossRef]
- Sugiyama, M.G.; Cui, H.; Redka, D.S.; Karimzadeh, M.; Rujas, E.; Maan, H.; Hayat, S.; Cheung, K.; Misra, R.; McPhee, J.B.; et al. Multiscale Interactome Analysis Coupled with Off-Target Drug Predictions Reveals Drug Repurposing Candidates for Human Coronavirus Disease. Sci. Rep. 2021, 11, 23315. [Google Scholar] [CrossRef]
- Yan, L. Ggvenn: Draw Venn Diagram by “Ggplot2”; CRAN: Vienna, Austria, 2022. [Google Scholar]
- R Core Team R. A Language and Environment for Statistical Computing; CRAN: Vienna, Austria, 2018. [Google Scholar]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omi. J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Grote, S. GOfuncR: Gene Ontology Enrichment Using FUNC; CRAN: Vienna, Austria, 2021. [Google Scholar]
- Available online: https://www.Meta-Chart.Com/ (accessed on 30 December 2022).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carvone | 8-Hydroxy-Carvotanacetone | 8-Acetoxy-Carvotanacetone | 8-Hydroxy-9-Methoxy-Carvotanacetone | |
|---|---|---|---|---|
| Name | Prediction | Prediction | Prediction | Prediction |
| BBB | yes | yes | yes | yes |
| Caco2 | yes | yes | yes | yes |
| HIA | yes | yes | yes | yes |
| PgpI | no | no | no | no |
| PgpR | no | no | no | no |
| 5HT1B | inactive | inactive | inactive | inactive |
| 5HT7R | active | active | active | active |
| AA1R | inactive | inactive | active | inactive |
| AA2AR | active | active | inactive | inactive |
| ACE | active | active | active | active |
| ACES | inactive | inactive | inactive | inactive |
| ACM1 | inactive | active | active | active |
| ACM3 | active | active | active | active |
| ACM4 | inactive | inactive | inactive | inactive |
| ACM5 | inactive | inactive | inactive | inactive |
| ADA1A | active | active | active | active |
| ADA1B | inactive | inactive | active | active |
| ADA2A | inactive | inactive | active | active |
| ADRB1 | active | active | active | active |
| ADRB2 | active | active | active | active |
| AGTR1 | inactive | active | inactive | inactive |
| AMES | No | No | No | No |
| ANDR | active | active | inactive | active |
| AOFA | active | active | inactive | inactive |
| BKRB2 | inactive | inactive | inactive | inactive |
| CALRL | active | active | active | active |
| CNR2 | active | active | active | active |
| DRD1 | inactive | inactive | inactive | inactive |
| DRD2 | active | inactive | inactive | inactive |
| EDNRA | active | active | active | active |
| EDNRB | inactive | inactive | inactive | inactive |
| ERa | yes | yes | yes | yes |
| GCR | active | active | active | active |
| hERG | known inactive | known inactive | known inactive | known inactive |
| HRH1 | active | active | inactive | inactive |
| HRH2 | inactive | inactive | inactive | inactive |
| NK1R | active | inactive | inactive | inactive |
| NK2R | inactive | inactive | inactive | inactive |
| NPY1R | active | active | active | active |
| PE2R2 | active | inactive | active | inactive |
| PGH2 | active | active | active | active |
| PPARG | active | active | inactive | inactive |
| SC6A2 | inactive | inactive | inactive | inactive |
| SC6A3 | inactive | inactive | inactive | inactive |
| UR2R | active | active | inactive | active |
| V1AR | active | active | inactive | inactive |
| Property | 2 | 4 | 8 | 11 |
|---|---|---|---|---|
| H-bond donors (<5) | 0 | 1 | 0 | 1 |
| H-bond acceptors (<10) | 1 | 2 | 3 | 3 |
| Mol. weight (160 to 480) | 150 | 168 | 210 | 198 |
| Log P (−0.4 to +5.6) 1 | 2.49 | 1.68 | 2.25 | 1.31 |
| Log P (−0.4 to +5.6) 2 | 2.43 | 1.57 | 2.08 | 1.35 |
| Heavy atoms (20 to 70) | 11 | 12 | 15 | 14 |
| Molar refractivity (40 to 130) 2 | 47 | 49 | 59 | 55 |
| Surface area 1 | 68 | 73 | 90 | 85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moço, G.; Sousa, C.; Capitão, A.; MacKinnon, S.S.; Leitão, A.J.; Mendes, A.F. Synthesis of Carvone Derivatives and In Silico and In Vitro Screening of Anti-Inflammatory Activity in Murine Macrophages. Int. J. Mol. Sci. 2023, 24, 2263. https://doi.org/10.3390/ijms24032263
Moço G, Sousa C, Capitão A, MacKinnon SS, Leitão AJ, Mendes AF. Synthesis of Carvone Derivatives and In Silico and In Vitro Screening of Anti-Inflammatory Activity in Murine Macrophages. International Journal of Molecular Sciences. 2023; 24(3):2263. https://doi.org/10.3390/ijms24032263
Chicago/Turabian StyleMoço, Gabriela, Cátia Sousa, Ana Capitão, Stephen Scott MacKinnon, Alcino Jorge Leitão, and Alexandrina Ferreira Mendes. 2023. "Synthesis of Carvone Derivatives and In Silico and In Vitro Screening of Anti-Inflammatory Activity in Murine Macrophages" International Journal of Molecular Sciences 24, no. 3: 2263. https://doi.org/10.3390/ijms24032263
APA StyleMoço, G., Sousa, C., Capitão, A., MacKinnon, S. S., Leitão, A. J., & Mendes, A. F. (2023). Synthesis of Carvone Derivatives and In Silico and In Vitro Screening of Anti-Inflammatory Activity in Murine Macrophages. International Journal of Molecular Sciences, 24(3), 2263. https://doi.org/10.3390/ijms24032263