
Alterations in Lymphocytic Metabolism—An Emerging Hallmark of MS Pathophysiology?


Abstract
1. Introduction
1.1. Production of Cellular Energy
1.2. Role of Mitochondria in Energy Production
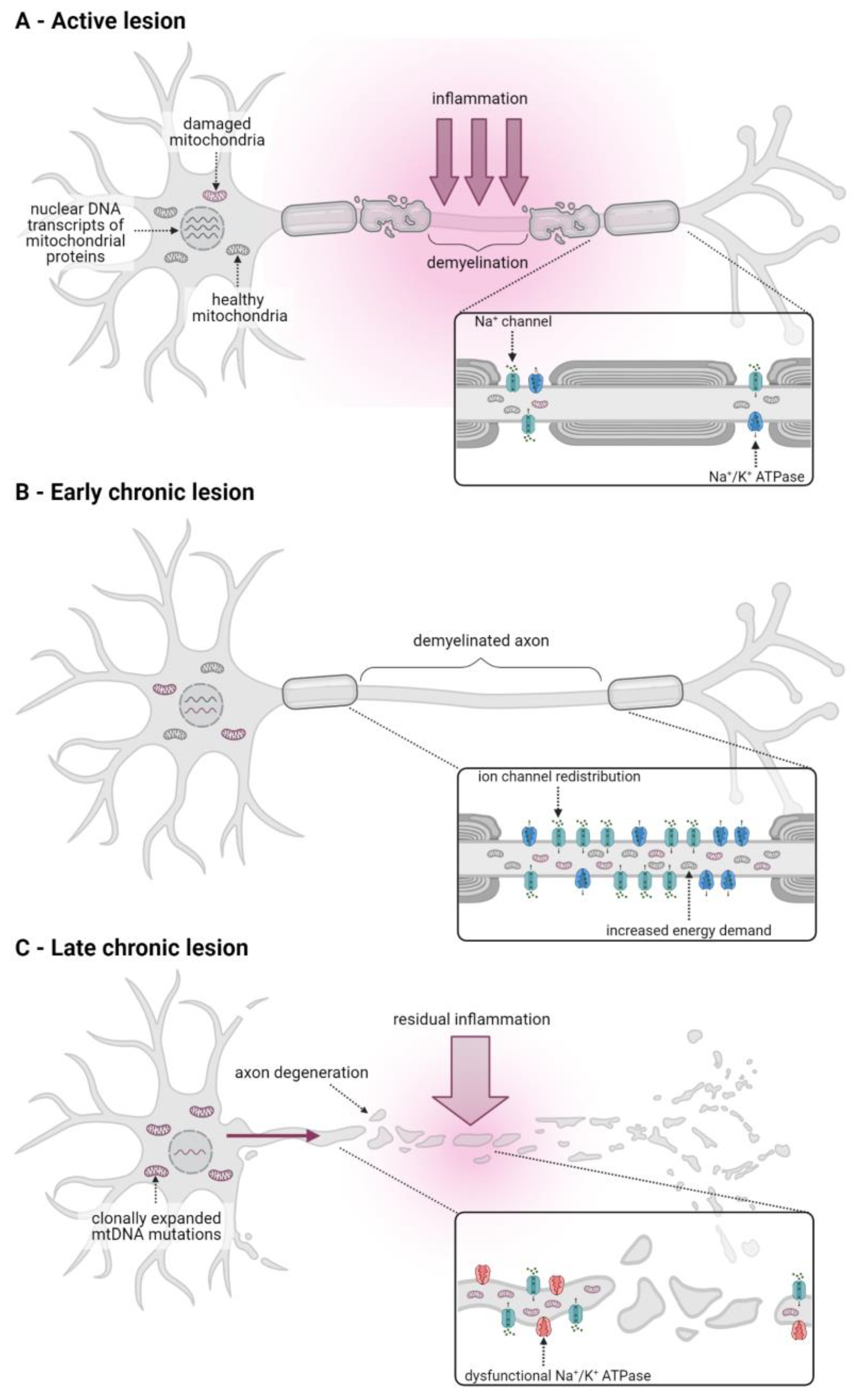
2. Mitochondrial Dysfunction Is Involved in CNS Degenerative Processes in Multiple Sclerosis
3. Lymphocyte Activation and Their Role in MS and EAE
3.1. Lymphocytic Metabolism under Non-Autoimmune Conditions
3.2. The Role of Lymphocytes in MS and EAE
4. Metabolic Alterations in Lymphocytes of MS Patients and in EAE Mice
4.1. Mitochondrial OxPhos Activity Is Decreased in PBMCs of MS Patients and in EAE
4.2. Oxidative Stress Is Increased in PBMCs of MS Patients and in EAE
4.3. Glycolytic Activity Is Altered in Lymphocytes of MS Patients and in EAE
4.4. Modulation of Glycolytic Activity Ameliorates EAE
4.5. Common RRMS Treatments Can Restore Metabolic Alterations in PBMCs of MS Patients
5. Conclusions
- Do immunometabolic alterations in CD4+ T-cells contribute to disease progression or are they secondary to other MS pathophysiological processes?
- Are immunometabolic alterations present in all CD4+ T cells or are there differences in Teffs and Tregs as suggested by EAE studies? Can similar changes be observed in other immune cells implicated in MS pathophysiology, such as B-cells?
- What is the effect of altered metabolism on lymphocytic activation state and function, specifically with regard to Treg suppressive capacity?
- Is there a correlation between immunometabolic alterations in PBMCs and RRMS disease activity and does it differ between RRMS and progressive forms of the disease?
- What are the underlying molecular mechanisms responsible for altered immunometabolism in PBMCs? What is the role of ROS?
- What is the mechanism by which common pharmacological treatments restore altered immunometabolism in peripheral lymphocytes?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lassmann, H. Pathogenic Mechanisms Associated with Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2018, 9, 3116. [Google Scholar] [CrossRef] [PubMed]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Wallin, M.T.; Culpepper, W.J.; Nichols, E.; Bhutta, Z.A.; Gebrehiwot, T.T.; Hay, S.I.; Khalil, I.A.; Krohn, K.J.; Liang, X.; Naghavi, M.; et al. Global, regional, and national burden of multiple sclerosis 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Disanto, G.; Berlanga, A.J.; Handel, A.E.; Para, A.E.; Burrell, A.M.; Fries, A.; Handunnetthi, L.; De Luca, G.C.; Morahan, J.M. Heterogeneity in multiple sclerosis: Scratching the surface of a complex disease. Autoimmune Dis. 2010, 2011, 932351. [Google Scholar] [CrossRef]
- Kister, I.; Bacon, T.E.; Chamot, E.; Salter, A.R.; Cutter, G.R.; Kalina, J.T.; Herbert, J. Natural history of multiple sclerosis symptoms. Int. J. MS Care 2013, 15, 146–158. [Google Scholar] [CrossRef]
- Belbasis, L.; Bellou, V.; Evangelou, E.; Ioannidis, J.P.; Tzoulaki, I. Environmental risk factors and multiple sclerosis: An umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 2015, 14, 263–273. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics, C.; Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Electronic address, c. c. y. e.; International Multiple Sclerosis Genetics, C. Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk. Cell 2018, 175, 1679–1687.e7. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Frisullo, G.; Plantone, D.; Marti, A.; Iorio, R.; Damato, V.; Nociti, V.; Patanella, A.K.; Bianco, A.; Batocchi, A.P. Type 1 immune response in progressive multiple sclerosis. J. Neuroimmunol. 2012, 249, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Balashov, K.E.; Smith, D.R.; Khoury, S.J.; Hafler, D.A.; Weiner, H.L. Increased interleukin 12 production in progressive multiple sclerosis: Induction by activated CD4+ T cells via CD40 ligand. Proc. Natl. Acad. Sci. USA 1997, 94, 599–603. [Google Scholar] [CrossRef]
- Karni, A.; Abraham, M.; Monsonego, A.; Cai, G.; Freeman, G.J.; Hafler, D.; Khoury, S.J.; Weiner, H.L. Innate immunity in multiple sclerosis: Myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J. Immunol. 2006, 177, 4196–4202. [Google Scholar] [CrossRef] [PubMed]
- Dargahi, N.; Katsara, M.; Tselios, T.; Androutsou, M.E.; de Courten, M.; Matsoukas, J.; Apostolopoulos, V. Multiple Sclerosis: Immunopathology and Treatment Update. Brain Sci. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Derfuss, T.; Mehling, M.; Papadopoulou, A.; Bar-Or, A.; Cohen, J.A.; Kappos, L. Advances in oral immunomodulating therapies in relapsing multiple sclerosis. Lancet Neurol. 2020, 19, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Mahad, D.J.; Lassmann, H.; van Horssen, J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med. 2014, 20, 179–187. [Google Scholar] [CrossRef]
- Shulman, R.G.; Rothman, D.L.; Behar, K.L.; Hyder, F. Energetic basis of brain activity: Implications for neuroimaging. Trends Neurosci. 2004, 27, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell. Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Brooks, G.A. Lactate as a fulcrum of metabolism. Redox. Biol. 2020, 35, 101454. [Google Scholar] [CrossRef]
- Yin, M.; O’Neill, L.A.J. The role of the electron transport chain in immunity. FASEB J. 2021, 35, e21974. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolanos, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 2018, 25, 542–572. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- TeSlaa, T.; Teitell, M.A. Techniques to monitor glycolysis. Methods Enzymol. 2014, 542, 91–114. [Google Scholar] [PubMed]
- van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef]
- Campbell, G.; Mahad, D.J. Mitochondrial dysfunction and axon degeneration in progressive multiple sclerosis. FEBS Lett. 2018, 592, 1113–1121. [Google Scholar] [CrossRef]
- Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef]
- Blagov, A.V.; Sukhorukov, V.N.; Orekhov, A.N.; Sazonova, M.A.; Melnichenko, A.A. Significance of Mitochondrial Dysfunction in the Progression of Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 12725. [Google Scholar] [CrossRef]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012, 135, 886–899. [Google Scholar] [CrossRef]
- Lu, F.; Selak, M.; O’Connor, J.; Croul, S.; Lorenzana, C.; Butunoi, C.; Kalman, B. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J. Neurol. Sci. 2000, 177, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Bo, L.; Rodenburg, R.J.; Belien, J.A.; Musters, R.; Hazes, T.; Wintjes, L.T.; Smeitink, J.A.; Geurts, J.J.; De Vries, H.E.; et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J. Pathol. 2009, 219, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Zambonin, J.L.; Zhao, C.; Ohno, N.; Campbell, G.R.; Engeham, S.; Ziabreva, I.; Schwarz, N.; Lee, S.E.; Frischer, J.M.; Turnbull, D.M.; et al. Increased mitochondrial content in remyelinated axons: Implications for multiple sclerosis. Brain 2011, 134, 1901–1913. [Google Scholar] [CrossRef] [PubMed]
- Craner, M.J.; Newcombe, J.; Black, J.A.; Hartle, C.; Cuzner, M.L.; Waxman, S.G. Molecular changes in neurons in multiple sclerosis: Altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc. Natl. Acad. Sci. USA 2004, 101, 8168–8173. [Google Scholar] [CrossRef]
- Campbell, G.; Licht-Mayer, S.; Mahad, D. Targeting mitochondria to protect axons in progressive MS. Neurosci. Lett. 2019, 710, 134258. [Google Scholar] [CrossRef]
- Broadwater, L.; Pandit, A.; Clements, R.; Azzam, S.; Vadnal, J.; Sulak, M.; Yong, V.W.; Freeman, E.J.; Gregory, R.B.; McDonough, J. Analysis of the mitochondrial proteome in multiple sclerosis cortex. Biochim. Biophys. Acta 2011, 1812, 630–641. [Google Scholar] [CrossRef]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef]
- Witte, M.E.; Nijland, P.G.; Drexhage, J.A.; Gerritsen, W.; Geerts, D.; van Het Hof, B.; Reijerkerk, A.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Reduced expression of PGC-1alpha partly underlies mitochondrial changes and correlates with neuronal loss in multiple sclerosis cortex. Acta Neuropathol. 2013, 125, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Pandit, A.; Vadnal, J.; Houston, S.; Freeman, E.; McDonough, J. Impaired regulation of electron transport chain subunit genes by nuclear respiratory factor 2 in multiple sclerosis. J. Neurol. Sci. 2009, 279, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Boothby, M.R.; Chi, H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 2020, 20, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef]
- Benova, K.; Hanckova, M.; Koci, K.; Kudelova, M.; Betakova, T. T cells and their function in the immune response to viruses. Acta Virol. 2020, 64, 131–143. [Google Scholar]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef]
- Kinnunen, T.; Chamberlain, N.; Morbach, H.; Cantaert, T.; Lynch, M.; Preston-Hurlburt, P.; Herold, K.C.; Hafler, D.A.; O’Connor, K.C.; Meffre, E. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J. Clin. Investig. 2013, 123, 2737–2741. [Google Scholar] [CrossRef]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders—Advances and challenges. Hum. Vaccin. Immunother. 2022, 18, 2035117. [Google Scholar] [CrossRef]
- Chapman, N.M.; Chi, H. Metabolic adaptation of lymphocytes in immunity and disease. Immunity 2022, 55, 14–30. [Google Scholar] [CrossRef]
- Menk, A.V.; Scharping, N.E.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018, 22, 1509–1521. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Bantug, G.R.; Galluzzi, L.; Kroemer, G.; Hess, C. The spectrum of T cell metabolism in health and disease. Nat. Rev. Immunol. 2018, 18, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; Acin-Perez, R.; Villarroya-Beltri, C.; Mazzeo, C.; Nunez-Andrade, N.; Gabande-Rodriguez, E.; Ledesma, M.D.; Blazquez, A.; Martin, M.A.; Falcon-Perez, J.M.; et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef]
- Manosalva, C.; Quiroga, J.; Hidalgo, A.I.; Alarcon, P.; Anseoleaga, N.; Hidalgo, M.A.; Burgos, R.A. Role of Lactate in Inflammatory Processes: Friend or Foe. Front. Immunol. 2021, 12, 808799. [Google Scholar] [CrossRef] [PubMed]
- Pucino, V.; Certo, M.; Bulusu, V.; Cucchi, D.; Goldmann, K.; Pontarini, E.; Haas, R.; Smith, J.; Headland, S.E.; Blighe, K.; et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4(+) T Cell Metabolic Rewiring. Cell Metab. 2019, 30, 1055–1074.e8. [Google Scholar] [CrossRef]
- Yan, Y.; Huang, L.; Liu, Y.; Yi, M.; Chu, Q.; Jiao, D.; Wu, K. Metabolic profiles of regulatory T cells and their adaptations to the tumor microenvironment: Implications for antitumor immunity. J. Hematol. Oncol. 2022, 15, 104. [Google Scholar] [CrossRef]
- Kishore, M.; Cheung, K.C.P.; Fu, H.; Bonacina, F.; Wang, G.; Coe, D.; Ward, E.J.; Colamatteo, A.; Jangani, M.; Baragetti, A.; et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity 2017, 47, 875–889.e10. [Google Scholar]
- Cluxton, D.; Petrasca, A.; Moran, B.; Fletcher, J.M. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front. Immunol. 2019, 10, 115. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef]
- Fu, Z.; Ye, J.; Dean, J.W.; Bostick, J.W.; Weinberg, S.E.; Xiong, L.; Oliff, K.N.; Chen, Z.E.; Avram, D.; Chandel, N.S.; et al. Requirement of Mitochondrial Transcription Factor A in Tissue-Resident Regulatory T Cell Maintenance and Function. Cell Rep. 2019, 28, 159–171.e4. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martinez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 2019, 565, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.R.; Ahsan, F.M.; Wolf, D.M.; Shirihai, O.; Teitell, M.A. Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience 2018, 5, 99–109. [Google Scholar] [CrossRef]
- Meiser, J.; Kramer, L.; Sapcariu, S.C.; Battello, N.; Ghelfi, J.; D’Herouel, A.F.; Skupin, A.; Hiller, K. Pro-inflammatory Macrophages Sustain Pyruvate Oxidation through Pyruvate Dehydrogenase for the Synthesis of Itaconate and to Enable Cytokine Expression. J. Biol. Chem. 2016, 291, 3932–3946. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Boitard, C.; Yasunami, R.; Dardenne, M.; Bach, J.F. T cell-mediated inhibition of the transfer of autoimmune diabetes in NOD mice. J. Exp. Med. 1989, 169, 1669–1680. [Google Scholar] [CrossRef]
- Morgan, M.E.; Flierman, R.; van Duivenvoorde, L.M.; Witteveen, H.J.; van Ewijk, W.; van Laar, J.M.; de Vries, R.R.; Toes, R.E. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum. 2005, 52, 2212–2221. [Google Scholar] [CrossRef]
- Kohm, A.P.; Carpentier, P.A.; Anger, H.A.; Miller, S.D. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol. 2002, 169, 4712–4716. [Google Scholar] [CrossRef]
- Zhang, X.; Koldzic, D.N.; Izikson, L.; Reddy, J.; Nazareno, R.F.; Sakaguchi, S.; Kuchroo, V.K.; Weiner, H.L. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int. Immunol. 2004, 16, 249–256. [Google Scholar] [PubMed]
- Verma, N.D.; Lam, A.D.; Chiu, C.; Tran, G.T.; Hall, B.M.; Hodgkinson, S.J. Multiple sclerosis patients have reduced resting and increased activated CD4(+)CD25(+)FOXP3(+)T regulatory cells. Sci. Rep. 2021, 11, 10476. [Google Scholar] [CrossRef] [PubMed]
- Alissafi, T.; Kalafati, L.; Lazari, M.; Filia, A.; Kloukina, I.; Manifava, M.; Lim, J.H.; Alexaki, V.I.; Ktistakis, N.T.; Doskas, T.; et al. Mitochondrial Oxidative Damage Underlies Regulatory T Cell Defects in Autoimmunity. Cell Metab. 2020, 32, 591–604.e7. [Google Scholar] [CrossRef] [PubMed]
- Putheti, P.; Pettersson, A.; Soderstrom, M.; Link, H.; Huang, Y.M. Circulating CD4+CD25+ T regulatory cells are not altered in multiple sclerosis and unaffected by disease-modulating drugs. J. Clin. Immunol. 2004, 24, 155–161. [Google Scholar] [CrossRef]
- Haas, J.; Hug, A.; Viehover, A.; Fritzsching, B.; Falk, C.S.; Filser, A.; Vetter, T.; Milkova, L.; Korporal, M.; Fritz, B.; et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur. J. Immunol. 2005, 35, 3343–3352. [Google Scholar] [CrossRef]
- Delbarba, A.; Abate, G.; Prandelli, C.; Marziano, M.; Buizza, L.; Arce Varas, N.; Novelli, A.; Cuetos, F.; Martinez, C.; Lanni, C.; et al. Mitochondrial Alterations in Peripheral Mononuclear Blood Cells from Alzheimer’s Disease and Mild Cognitive Impairment Patients. Oxid Med. Cell Longev. 2016, 2016, 5923938. [Google Scholar] [CrossRef]
- Fearon, U.; Canavan, M.; Biniecka, M.; Veale, D.J. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 385–397. [Google Scholar] [CrossRef]
- Morel, L. Immunometabolism in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2017, 13, 280–290. [Google Scholar] [CrossRef]
- Smith, A.M.; Depp, C.; Ryan, B.J.; Johnston, G.I.; Alegre-Abarrategui, J.; Evetts, S.; Rolinski, M.; Baig, F.; Ruffmann, C.; Simon, A.K.; et al. Mitochondrial dysfunction and increased glycolysis in prodromal and early Parkinson’s blood cells. Mov. Disord. 2018, 33, 1580–1590. [Google Scholar] [CrossRef]
- De Riccardis, L.; Rizzello, A.; Ferramosca, A.; Urso, E.; De Robertis, F.; Danieli, A.; Giudetti, A.M.; Trianni, G.; Zara, V.; Maffia, M. Bioenergetics profile of CD4(+) T cells in relapsing remitting multiple sclerosis subjects. J. Biotechnol. 2015, 202, 31–39. [Google Scholar] [CrossRef]
- De Riccardis, L.; Ferramosca, A.; Danieli, A.; Trianni, G.; Zara, V.; De Robertis, F.; Maffia, M. Metabolic response to glatiramer acetate therapy in multiple sclerosis patients. BBA Clin. 2016, 6, 131–137. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, V.; Galgani, M.; Porcellini, A.; Colamatteo, A.; Santopaolo, M.; Zuchegna, C.; Romano, A.; De Simone, S.; Procaccini, C.; La Rocca, C.; et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 2015, 16, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, H.; Nogueras, L.; Gil-Sanchez, A.; Hervas, J.V.; Valcheva, P.; Gonzalez-Mingot, C.; Martin-Gari, M.; Canudes, M.; Peralta, S.; Solana, M.J.; et al. Impairment of Mitochondrial Redox Status in Peripheral Lymphocytes of Multiple Sclerosis Patients. Front. Neurosci. 2019, 13, 938. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Eschborn, M.; Lindner, M.; Liebmann, M.; Herold, M.; Janoschka, C.; Torres Garrido, B.; Schulte-Mecklenbeck, A.; Gross, C.C.; Breuer, J.; et al. Teriflunomide treatment for multiple sclerosis modulates T cell mitochondrial respiration with affinity-dependent effects. Sci. Transl. Med. 2019, 11, eaao5563. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, C.; Carbone, F.; De Rosa, V.; Colamatteo, A.; Galgani, M.; Perna, F.; Lanzillo, R.; Brescia Morra, V.; Orefice, G.; Cerillo, I.; et al. Immunometabolic profiling of T cells from patients with relapsing-remitting multiple sclerosis reveals an impairment in glycolysis and mitochondrial respiration. Metabolism 2017, 77, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, M.; Korn, L.; Janoschka, C.; Albrecht, S.; Lauks, S.; Herrmann, A.M.; Schulte-Mecklenbeck, A.; Schwab, N.; Schneider-Hohendorf, T.; Eveslage, M.; et al. Dimethyl fumarate treatment restrains the antioxidative capacity of T cells to control autoimmunity. Brain 2021, 144, 3126–3141. [Google Scholar] [CrossRef]
- Tänzer, A. Molecular Mechanisms of Immunometabolic Dysfunction in Multiple Sclerosis; Humboldt-Universität zu Berlin: Berlin, Germany, 2019. [Google Scholar]
- Zahoor, I.; Suhail, H.; Datta, I.; Ahmed, M.E.; Poisson, L.M.; Waters, J.; Rashid, F.; Bin, R.; Singh, J.; Cerghet, M.; et al. Blood-based untargeted metabolomics in relapsing-remitting multiple sclerosis revealed the testable therapeutic target. Proc. Natl. Acad. Sci. USA 2022, 119, e2123265119. [Google Scholar] [CrossRef]
- De Biasi, S.; Simone, A.M.; Bianchini, E.; Lo Tartaro, D.; Pecorini, S.; Nasi, M.; Patergnani, S.; Carnevale, G.; Gibellini, L.; Ferraro, D.; et al. Mitochondrial functionality and metabolism in T cells from progressive multiple sclerosis patients. Eur. J. Immunol. 2019, 49, 2204–2221. [Google Scholar]
- Armon-Omer, A.; Neuman, H.; Sharabi-Nov, A.; Shahien, R. Mitochondrial activity is impaired in lymphocytes of MS patients in correlation with disease severity. Mult. Scler. Relat. Disord. 2020, 41, 102025. [Google Scholar] [CrossRef]
- Hargreaves, I.; Mody, N.; Land, J.; Heales, S. Blood Mononuclear Cell Mitochondrial Respiratory Chain Complex IV Activity Is Decreased in Multiple Sclerosis Patients: Effects of beta-Interferon Treatment. J. Clin. Med. 2018, 7, 36. [Google Scholar] [CrossRef]
- Sun, Y.; Tian, T.; Gao, J.; Liu, X.; Hou, H.; Cao, R.; Li, B.; Quan, M.; Guo, L. Metformin ameliorates the development of experimental autoimmune encephalomyelitis by regulating T helper 17 and regulatory T cells in mice. J. Neuroimmunol. 2016, 292, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Wang, C.; Fessler, J.; DeTomaso, D.; Avila-Pacheco, J.; Kaminski, J.; Zaghouani, S.; Christian, E.; Thakore, P.; Schellhaass, B.; et al. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell 2021, 184, 4168–4185.e21. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Hollinshead, K.E.R.; Hao, Y.; Au, C.; Kroehling, L.; Ng, C.; Lin, W.Y.; Li, D.; Silva, H.M.; Shin, J.; et al. Niche-Selective Inhibition of Pathogenic Th17 Cells by Targeting Metabolic Redundancy. Cell 2020, 182, 641–654.e20. [Google Scholar] [CrossRef]
- Seki, S.M.; Stevenson, M.; Rosen, A.M.; Arandjelovic, S.; Gemta, L.; Bullock, T.N.J.; Gaultier, A. Lineage-Specific Metabolic Properties and Vulnerabilities of T Cells in the Demyelinating Central Nervous System. J. Immunol. 2017, 198, 4607–4617. [Google Scholar] [CrossRef]
- Karmaus, P.W.F.; Chen, X.; Lim, S.A.; Herrada, A.A.; Nguyen, T.M.; Xu, B.; Dhungana, Y.; Rankin, S.; Chen, W.; Rosencrance, C.; et al. Metabolic heterogeneity underlies reciprocal fates of T(H)17 cell stemness and plasticity. Nature 2019, 565, 101–105. [Google Scholar] [CrossRef]
- Payne, K.J.; Crooks, G.M. Immune-cell lineage commitment: Translation from mice to humans. Immunity 2007, 26, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef]
- Schwarz, A.; Schumacher, M.; Pfaff, D.; Schumacher, K.; Jarius, S.; Balint, B.; Wiendl, H.; Haas, J.; Wildemann, B. Fine-tuning of regulatory T cell function: The role of calcium signals and naive regulatory T cells for regulatory T cell deficiency in multiple sclerosis. J. Immunol. 2013, 190, 4965–4970. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Reference | Sample Size (MS Phenotype Where Known) | Medication | Cell Type | Main Findings (MS Patients Compared to Controls) |
|---|---|---|---|---|
| Immunometabolism in RRMS | ||||
| Alissafi et al., 2020 [73] | 11 MS (RRMS, all active) | N/A | Tregs |
|
| De Riccardis et al., 2015 [80] | 25 MS (RRMS) 8 HC | 25 without treatment | CD4+ T cells |
|
| De Riccardis et al., 2016 [81] | 20 MS (RRMS) 20 HC | 20 without treatment, 20 GA * | CD4+ T cells |
|
| De Rosa et al., 2015 [82] | MS (RRMS) HC (unclear participant numbers) | Without treatment | CD4+ conventional T cells |
|
| Gonzalo et al., 2019 [83] | 34 MS (RRMS) 24 HC | 26 various IM, 8 without treatment | PBMCs |
|
| Klotz et al., 2019 [84] | 49 MS (RRMS; 25 in remission, 24 with relapse) 24 HC | 49 without treatment, 49 TF ** | CD4+ T cells |
|
| La Rocca et al., 2017 [85] | 71 MS (RRMS) 57 HC | 32 without treatment, 39 IFN-β | PBMCs CD4+ T cells |
|
| Liebmann et al., 2021 [86] | 42 MS (RRMS) 15 HC | DMF | CD4+ T cells CD8+ T cells |
|
| Tänzer, 2019 [87] | 31 MS (RRMS, in remission) 31 HC | 27 various IM, 4 without treatment | CD4+ T cells CD8+ T cells |
|
| Zahoor et al., 2022 [88] | 35 MS (RRMS) 14 HC | N/A | PBMCs |
|
| Immunometabolism in Progressive MS | ||||
| De Biasi et al., 2019 [89] | 53 MS (22 PPMS, 31 SPMS) 20 HC | 53 without treatment | CD4+ T cells (naïve and memory cells) |
|
| Immunometabolism in unspecified MS subtypes | ||||
| Armon-Omer et al., 2020 [90] | 62 MS 83 HC | 49 various IM, 13 without treatment | PBMCs |
|
| Hargreaves et al., 2018 [91] | 11 MS 24 HC | 4 IFN-β, 7 without treatment | PBMCs |
|
| Reference | Modulation | Effects |
|---|---|---|
| Alissafi et al., 2020 [73] | Genetic overexpression of mitochondria-specific superoxide scavenger specifically in Tregs. | Protected Treg mitochondria from oxidative stress and reduced Th1 and Th17 activity. |
| Gerriets et al., 2015 [63] | DCA inhibition of PDHK1. | Decreased Th17 and increased Tregs; protected against EAE. |
| Karmaus et al., 2019 [97] | 2-DG to inhibit hexokinase; Raptor knockout. | Reduced CNS inflammation. |
| Seki et al., 2017 [96] | 3-BrPa inhibition of hexokinase and GAPDH. | Blocked Th1 release of IFNγ and Th17 release of IL-17; reduced EAE severity. |
| Shi et al., 2011 [94] | 2-DG to inhibit hexokinase. | Increased Treg differentiation, and reduced EAE severity. |
| Sun et al., 2016 [92] | Metformin to inhibit AMPK. | Attenuation of EAE, Th17 proliferation, CNS immune cells infiltration and the release of proinflammatory cytokines; increased Treg proliferation and the release of anti-inflammatory cytokines. |
| Wagner et al., 2021 [97] | Disruption of polyamine pathway. | Increased frequency of Tregs and alleviated EAE symptoms. |
| Wu et al., 2020 [95] | Gpi1 knockout to suppress glycolysis. | Demonstrated requirement of proinflammatory Th17 cells for EAE induction. |
| Zahoor et al., 2022 [88] | 2-DG to inhibit hexokinase. | Decreased disease severity, immune cell infiltration into the CNS, demyelination and the release of proinflammatory cytokines. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greeck, V.B.; Williams, S.K.; Haas, J.; Wildemann, B.; Fairless, R. Alterations in Lymphocytic Metabolism—An Emerging Hallmark of MS Pathophysiology? Int. J. Mol. Sci. 2023, 24, 2094. https://doi.org/10.3390/ijms24032094
Greeck VB, Williams SK, Haas J, Wildemann B, Fairless R. Alterations in Lymphocytic Metabolism—An Emerging Hallmark of MS Pathophysiology? International Journal of Molecular Sciences. 2023; 24(3):2094. https://doi.org/10.3390/ijms24032094
Chicago/Turabian StyleGreeck, Viktoria B., Sarah K. Williams, Jürgen Haas, Brigitte Wildemann, and Richard Fairless. 2023. "Alterations in Lymphocytic Metabolism—An Emerging Hallmark of MS Pathophysiology?" International Journal of Molecular Sciences 24, no. 3: 2094. https://doi.org/10.3390/ijms24032094
APA StyleGreeck, V. B., Williams, S. K., Haas, J., Wildemann, B., & Fairless, R. (2023). Alterations in Lymphocytic Metabolism—An Emerging Hallmark of MS Pathophysiology? International Journal of Molecular Sciences, 24(3), 2094. https://doi.org/10.3390/ijms24032094