Abstract
Primary CNS neoplasms are responsible for considerable mortality and morbidity, and many therapies directed at primary brain tumors have proven unsuccessful despite their success in preclinical studies. Recently, the tumor immune microenvironment has emerged as a critical aspect of primary CNS neoplasms that may affect their malignancy, prognosis, and response to therapy across patients and tumor grades. This review covers the tumor microenvironment of various primary CNS neoplasms, with a focus on glioblastoma and meningioma. Additionally, current therapeutic strategies based on elements of the tumor microenvironment, including checkpoint inhibitor therapy and immunotherapeutic vaccines, are discussed.
1. Introduction
Primary central nervous system (CNS) neoplasms represent the 13th most common type of cancer worldwide and are responsible for over 250,000 deaths annually [1]. CNS neoplasms arise from a number of progenitor cell types, with the most common malignancies being meningiomas and gliomas. While CNS malignancies are responsible for considerable morbidity and mortality, the development of novel therapies for these tumors has proven to be difficult due to challenges such as the blood–brain barrier, and promising pre-clinical treatments have often failed to produce meaningful differences in clinical trials.
In recent years, as our understanding of the diversity and complexity of tumors has evolved, there has been an increasing focus on the sub-characterization of tumors to optimize therapy selection, as well as the identification of novel therapeutic targets to improve outcomes for otherwise treatment-resistant cancers. One particular tumor feature that has emerged at the forefront of these efforts is the immune microenvironment, defined by the specific mix of immune cells associated with a tumor and the extensive crosstalk that occurs between tumor cells and these immune cells.
While the tumor microenvironment (TME) is known to be an important factor in the pathogenesis of systemic malignancies, the CNS was historically considered to have a paucity of immune activity [2]. However, recent developments have shown that the CNS contains a distinctive immune environment that exerts both immunosuppressive and anti-tumor effects on CNS neoplasms throughout the course of their pathogenesis. This review summarizes our current understanding of the TMEs of various primary brain tumors, focusing primarily on high and low-grade gliomas and meningiomas. Additionally, we discuss various therapeutic strategies that harness or target the unique TME elements present in CNS malignancies.
2. Tumor Microenvironment of Various Intracranial Primary Brain Tumors
2.1. Glioblastoma
Glioblastoma (GBM) is the most malignant form of glioma and carries a very poor prognosis, with a median survival of 15 months [3]. GBM is notable for its particularly immunosuppressive microenvironment, leading to tumor evasion of immune responses and the failure of a number of attempted therapies. This immunosuppressive environment is complex and multifactorial, resulting from the recruitment of immunosuppressive cell populations, high levels of lymphocyte exhaustion, immunometabolism alterations in the local environment, and structural changes in the local tumor environment. Key cell types within the immune microenvironment of GBM include tumor infiltrating lymphocytes (TILs), natural killer (NK) cells, tumor associated macrophages (TAMs), microglia, myeloid-derived suppressor cells (MDSCs), neutrophils, dendritic cells, and fibroblasts [4] (Table 1, Figure 1).

Table 1.
Summary of immune cell infiltration of GBM.
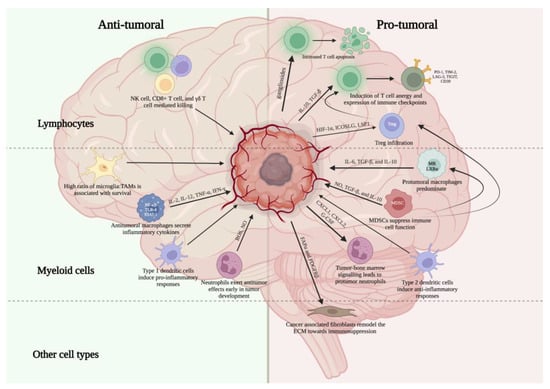
Figure 1.
Overview of key pro- and anti-tumor immune cell populations within the glioblastoma microenvironment. Created with Biorender.com.
2.1.1. Key Immune Cell Populations
Lymphoid Cells
Cytotoxic T cell dysfunction contributes to high-grade glioma pathogenesis. Compared to healthy controls, a higher proportion of CD4+ and CD8+ T cells infiltrating GBM tumors are apoptotic, particularly activated T cells expressing the Fas ligand [5]. Numerous mechanisms, including the induction of T cell apoptosis via the secretion of gangliosides by GBM tumor cells and the activation of TAMs that disrupt T cell function, may underlie GBM-driven T cell dysfunction and apoptosis [70,71]. Additionally, patients with GBM have been shown to have limited T-cell clone diversity compared to healthy patients [6]. The TME contains a number of immunosuppressive cytokines that may be responsible for T cell dysfunction. For example, glioma cells secrete or induce secretion of the immunosuppressive cytokines IL-10 and TGF-β, which can increase the threshold for T cell activation, and directly suppress their anti-tumor activity, allowing for tumor proliferation [7]. In addition to tumor cells, Ravi et al. demonstrated that HMOX1+ myeloid cells can also secrete IL-10, causing T cell dysfunction and immune evasion [48]. Paradoxically, IL-10 has also been demonstrated to have anti-glioma effects in a mouse model when expressed in conjunction with other cytokines, such as IL-2 [7,72]. The contradictory roles of IL-10 are likely contingent upon environmental cues and individual tumor characteristics, further complicating potential therapeutic interventions aimed at altering IL-10 pathways. Notably, the cytokine TGF-β also has a significant immunosuppressive role and has been shown to inhibit the cytotoxic abilities of various immune cell types, including T cells [7,73]. In preclinical models of glioma, siRNA silencing of TGF-β led to increased immune cell lysis of glioma. Furthermore, the inhibition of the TGF-β receptor in a murine model of glioma led to higher levels of CD8+ T cells and increased survival [74,75].
In addition to an increased barrier for initial activation, T cell overstimulation and exhaustion is also a defining feature of the GBM immune microenvironment. Tumor infiltrating lymphocytes (TILs) in GBM have been shown to express PD-1, a marker of exhaustion, at higher rates than peripheral T cells in the bloodstream [8]. Glioma-associated IL-10 upregulates the expression of PD-L1 on TAMs and peripheral monocytes, which can then bind to and stimulate PD-1 on TILs, leading to T cell anergy [9,10]. Notably, Davidson et al. found that PD-1+ T cells in patients with malignant glioma have decreased diversity compared to PD-1- T cells [8]. Woroniecka et al. further characterized T cell exhaustion in glioblastoma, reporting that multiple immune checkpoints, in addition to PD-1, such as TIM-2, LAG-3, TIGIT, and CD39, were expressed by TILs [76]. These TILs secreted lower levels of immune-stimulating factors, including IFN-γ, IL-2, and TNF-α, than TILs from control blood or patient blood [76]. Notably, TILs that were positive for PD-1, TIM-3, and LAG-3 were less functional than PD-1 single positive cells, demonstrating a joint role for these immune checkpoint molecules in inducing T cell exhaustion in GBM [76]. Overall, cytotoxic T cell dysfunction in the GBM TME contributes significantly to tumor immune escape and results from increased barriers to activation, decreased anti-tumor function, and high levels of exhaustion.
FOXP3+ T regulatory cell (Treg) enrichment has also been observed in glioblastoma and other high-grade tumor types [11,12]. Treg infiltration of the GBM TME may be mediated by numerous pathways, including hypoxia inducible factor 1α (HIF-1α), inducible T cell co-stimulator ligand (ICOSLG), lymphocyte-specific protein 1 (LSP1), and tolerogenic IDO-1+ dendritic cells [77,78,79,80]. While these cells likely contribute to immunosuppressive tumor environments, studies have failed to show a significant correlation between Treg presence and GBM patient survival [13,14,15]. Antunes et al. suggest that this may be due to the presence of distinct Treg subtypes, such as tumor-specific subtypes that are not present in the periphery, which differentially contribute to the immunosuppressive environment in GBM [4,81]. For example, CCR8 is a marker of multiclonal Tregs in tumors, which may play an immunosuppressive role, and the targeted depletion of CCR8-expressing cells may have potent anti-tumor effects [82]. Furthermore, Treg accumulation in GBM may be due to the interaction of CCR4, which is highly expressed on Tregs, and CCL22, which is a CCR4 ligand secreted by GBM tumor cells [13]. Further studies are needed to characterize the immunosuppressive contributions of different Treg subsets to the TME in GBM and identify potential therapeutic targets.
Recent studies have also examined the role of γδ T cells in GBM pathogenesis and prognosis [77]. γδ T cells are unique in that they can recognize and bind to antigens in an MHC-independent fashion. Lee et al. identified γδ T cells located near TAMs in GBM tumor tissue from four patients and reported that γδ T cell immune-related gene expression was similar to that of cytotoxic T cells and anti-tumor macrophages, suggesting a potential anti-tumor role [16]. Additionally, γδ T cells can eliminate GBM cells in a murine model via a granzyme-mediated mechanism when sensitized by the cytokine IL-21 [17]. Notably, Chauvin et al. reported that γδ T cells eliminate mesenchymal GBM cells via the NKG2D pathway, and the bisphosphonates zoledronate and minodronate have been shown to exert anti-tumor effects in vitro via the stimulation of γδ T cells [18,19,20]. Given that γδ T cells may be depleted in GBM patients compared to healthy patients, increasing γδ T cell infiltration and activation may be a promising immunotherapeutic strategy [21].
Natural killer (NK) cells have also been characterized as a component of the GBM TME. A number of studies investigating the immune gene expression profile in gliomas have reported that activated NK cells are associated with a better prognosis for glioma patients [22,23,24,25]. Interestingly, however, one study also reported that activated NK cells were predictive of malignant transformation of low-grade glioma [25,83]. Studies in mice support the finding that NK cells have cytotoxic effects against GBM tumor cells, with one study reporting that the inactivation of NK cells promoted lung metastases in a murine GBM model [84,85]. Furthermore, a study investigating the relationship between immune phenotypes and GBM patient prognosis found that activated NK cells were associated with improved patient survival [86]. However, the anti-tumor effect of NK cells may differ according to the level of tumor differentiation. Kozlawska et al. demonstrated that while activated NK cells have a demonstrated cytotoxic effect against GBM stem cells, NK cells may also contribute to GBM differentiation via interferon gamma (IFN-γ) signaling [26]. While differentiated GBM tumor cells are less susceptible to NK-cell mediated killing, they also have limited growth potential and may be more susceptible to chemotherapy, suggesting a role for a multimodal therapeutic approach [26,27]. A potential therapeutic role for NK cells is supported by reports that both intracranially and systemically delivered NK cells can home to GBM tumor and exert a cytotoxic effect in a murine model [87].
Myeloid Cells
Myeloid cells are the largest immune component of the GBM TME and play a significant role in both the overall immune response and efficacy of therapies for GBM patients [88]. Overall, the myeloid compartment of the TME is viewed as immunosuppressive, and a number of emerging therapeutic pathways are aimed at either reducing this immunosuppression or re-programming myeloid cells to have anti-tumor responses [89]. However, the myeloid compartment of the GBM TME is actually made up of a number of different cell types with both pro- and anti-tumor functions, including macrophages (TAMs), microglia, myeloid-derived suppressor cells (MDSCs), neutrophils, and dendritic cells.
TAMs are a major type of immune cell within the GBM TME and exert both pro-tumor and anti-tumor effects [28]. Historically, TAMs have been categorized as either M1 (pro-inflammatory) or M2 (anti-inflammatory), with the former suppressing tumor progression and the latter contributing to immunosuppressive environments that promote tumor growth. For example, M1 macrophages were thought to release inflammatory cytokines (such as IL-2, IL-12, IFN-γ and TNF-α), express a distinct set of metalloproteinases, and suppress angiogenesis, while M2 macrophages were through to release immunosuppressive cytokines (such as IL-6, IL-10 and TGF-β) and promote angiogenesis [7,90,91,92,93,94].
However, macrophage polarization in CNS tumors is likely more complex than previously thought, with recent studies suggesting that the M1/M2 stratification may be oversimplified [28,29,30,31]. In fact, recent single-cell profiling studies in gliomas have uncovered a diversity of myeloid cell populations, none of which approximate the classic M1 or M2 phenotypes [32,33,34]. Instead, TAMs develop disease-specific alterations and are capable of adapting to various immunogenic cues in the local microenvironment (such as chemokines, cytokines, metabolic changes, microRNA, oxygen availability, and pH) [35,36,37]. For example, Friebel et al. performed single-cell mapping of immune cells in gliomas and reported that the signature trajectory of TAMs was driven by the pathogenic insult, i.e., tumor type, rather than by CNS tissue [32]. Lin et al. also reported the presence of monocytes with stem cell-like properties in tumor tissues that may exist and proliferate in a self-renewing state without differentiating, and numerous studies have identified heterogeneous populations of macrophages expressing markers that were classically associated with both the M1 and M2 phenotype [38,39,40,41,42]. Furthermore, Zilionis et al. utilized single cell transcriptomics to analyze tumor-invading myeloid cells and defined 14 different monocyte/macrophage transcriptional states [95]. Thus, while past studies have utilized the M1/M2 distinction, future studies should focus on analyzing macrophages by genotype, the produced cytokine, or functionality.
Given the evolving views regarding the validity of M1 vs. M2 polarization, researchers have also begun exploring other ways to define sub-populations of TAMs within the GBM TME. For example, a number of studies have characterized the developmental origins of TAMs in GBM. Chen et al. reported that 85% of TAMs were monocyte-derived cells from the bone marrow, whereas 15% were resident microglia [42]. Furthermore, they found that monocyte-derived macrophages and resident microglia had differing localization and gene-expression patterns, suggesting different contributory roles of these TAM subtypes to GBM pathogenesis and progression [42,96]. Woolf et al. investigated the role of microglia in GBM via single-cell image analysis of resected tissues and found that a greater ratio of microglia to TAMs was correlated with an increase in patient survival [43]. Overall, TAMs derived from bone marrow seem to express immunosuppressive proteins at higher levels than microglia when stimulated in vitro, but in vivo studies remain inconclusive regarding differences in function due to the difficulty in reliably characterizing and observing these cell populations [97]. Thus, modulating the ability of monocyte-derived macrophages to migrate to the CNS may present a potential therapeutic avenue. For example, monocyte migration is driven by the monocyte chemoattractant protein CCL2 via the CCR2 receptor, and CCL2 downregulation resulted in reduced monocyte migration and prolonged survival in a murine model of glioblastoma [98].
Studies specifically investigating resident microglia in glioblastoma suggest that these “professional phagocytes” of the central nervous system do not successfully phagocytose invading glioma cells. This may be due to increased expression of CD47, an anti-phagocytotic surface protein, on tumor cells [44]. A high level of leucine rich repeats and immunoglobulin like domains 2 (LRIG2) expression, which is linked with GBM progression and poor prognosis, has been shown to increase CD47 expression, and CD47 expression has been shown to be associated with a higher tumor grade and worse clinical outcomes [99,100]. Furthermore, Hutter et al. and Gholamin et al. reported that anti-CD47 reduced tumor growth in orthotopic xenograft models of glioblastoma and various pediatric brain tumor types, respectively [45,46].
Researchers have also begun investigating the signaling pathways altered in pro-tumor microglia. For example, a tumorigenic microglial phenotype may also be induced by the upregulation of the mammalian target of rapamycin (mTOR) pathway in GBM and by the downregulation of microglial sensome genes, such as sialic acid-binding immunoglobulin-like lectin-H (Siglech); these are critical for sensing tumor cells and their byproducts [101,102]. Along with phagocytic dysfunction, tumor-induced microglial changes may also inhibit the recruitment of effector immune cells, thus promoting immune evasion [101].
Myeloid-derived suppressor cells (MDSCs) also contribute significantly to the overall immunosuppressive environment of glioblastoma [47,103]. MDSCs mediate immunosuppression by suppressing the function of cytotoxic T cells, NK cells, anti-inflammatory macrophages, and dendritic cells through the production of nitric oxide and various immunosuppressive cytokines, such as IL-10 and TGF-β [48,49,50] Further, they promote the recruitment of Tregs, B cells and M2 macrophages, which contribute to high-grade glioma progression [49,51]. Lee-Chang et al. also investigated the role of MDSCs in promoting B-cell mediated immunosuppression in GBM and reported that MDSCs promote B cell regulatory function via the transfer of membrane-bound PD-L1, which gives regulatory B cells the ability to mediate immunosuppression via the inhibition of CD8+ T cell activation [51]. Studies have also reported both increased numbers of monocytic-MDSCs in the peripheral blood of patients with high-grade gliomas and higher levels of activation in these MDSCs compared to healthy controls [104,105]. Furthermore, tumor infiltration and immunosuppression by MDSCs, which are dependent on tumor cell expression of macrophage migration inhibitory factor (MIF), have been directly associated with poor prognosis [104]. Given these findings, MDSCs may have a potential future role as biomarkers for GBM detection and prognosis [104,106].
Neutrophils exhibit both anti-tumor and pro-tumor phenotypes within the TME [52,53]. Cues secreted by the TME may influence neutrophil polarization; for example, IFN-β induces anti-tumor activity, whereas TGF-β induces pro-tumor activity [52,53,55]. Notably, like TAMS, neutrophils were previously thought to have a binary anti-tumor (N1) or pro-tumor (N2) phenotype, but recent work has demonstrated that they actually have a wider spectrum of activation states [53]. Specifically, at least three subsets of neutrophils have been identified: mature high-density neutrophils (HDNs), immature low-density neutrophils (LDNs), and mature low-density neutrophils [53,107,108,109,110]. HDNs appear to have anti-tumor functions, whereas LDNs may have immunosuppressive functions [53,107,108,109,110]. To better understand the influence of neutrophils on GBM progression, Magod et al. aimed to characterize neutrophil infiltration into the glioma microenvironment temporally and reported that the ability of neutrophils to inhibit tumor growth was lost during GBM tumor progression in a murine model [54]. Furthermore, the authors described a tumor–bone-marrow signaling pathway that resulted in the development of neutrophils with pro-tumor activity from the bone marrow as tumors progressed [54]. Specifically, pro-tumor neutrophils in this study were shown to suppress cytotoxic T cell function and support tumor angiogenesis [54]. By contrast, proinflammatory neutrophils can attract CD8+ T cells via the production of proinflammatory cytokines and chemokines (IL-12, VEGF, TNF-α, GM-CSF, CCL3, CXCL9, and CXCL10) [55,56,57]. Various neutrophil-mediated tumor cell-killing mechanisms have been described, including direct contact and the generation of ROS, NO production, and the induction of calcium influx into tumor cells via the TRPM2 channel [53,111,112,113]. However, some neutrophil-mediated tumor deaths may actually be detrimental to overall patient outcomes. For example, tumor cell ferroptosis, induced by neutrophils via the transfer of granules containing myeloperoxidase, is associated with greater tumor necrosis and poor patient outcomes [58]. Additionally, neutrophils have been implicated in the mechanism of biopsy-associated GBM tumor spreading indirectly via macrophage recruitment and directly via the secretion of soluble factors to induce tumor cell migration [114]. Notably, neutrophil to lymphocyte (NLR) in the periphery may also a prognostic marker for patient outcome. One study demonstrated that a high NLR is associated with tumor progression, decreased survival, and poor prognosis in patients with GBM [115].
Dendritic cells represent another important component of the immune microenvironment in GBM. While not present in normal brain parenchyma, dendritic cells may migrate via afferent lymphatics to the brain in pathological states and present tumor antigens in order to coordinate anti-tumor T cell responses [59,116]. Uncommitted dendritic cells may mature into type-1 or type-2 polarized effector cells, the former of which induce proinflammatory TH1 responses and play a role in anti-tumor immunity [59]. For example, these dendritic cells can present tumor antigens to naïve CD8+ T lymphocytes at tumor-draining lymph nodes and produce cytokines, such as CCL9, CCL10, and IL-12, that enhance the anti-tumor activity of other immune cells, including T cells and NK cells [59,60,61,62,63,64]. The anti-tumor properties of dendritic cells have led to studies investigating their therapeutic applications. For example, Flores et al. reported that hematopoietic stem cell-derived dendritic cells, isolated from GBM, induced anti-tumor immunity in mice when delivered via vaccine. In conjunction with other studies, they also found that dendritic cell presence, induction, and/or activation can enhance the response to immune checkpoint blockade therapy in rodent glioblastoma models [117,118,119]. However, the immunosuppressive environment of high-grade gliomas, including vascular endothelial growth factor (VEGF), prostaglandin E2 (PGE2), and IL-10 expression, may induce a regulatory or tolerant dendritic cell phenotype [65]. Studies suggest that these immunosuppressive factors may suppress dendritic cell maturation via over-expression of the transcription factor Nrf, which regulates the cellular response to oxidative stress [59,120]. Indeed, inhibiting the Nrf pathway has been shown to enhance dendritic cell maturation and promote the cytotoxic T cell response in an in vitro glioma cell preparation [59]. Regulatory dendritic cells, in turn, secrete the immunosuppressive cytokines IL-10 and TGF-β, which promote the activation of Tregs and decrease the recruitment of cytotoxic T cells [65,121,122,123,124].
Other Key Cell Populations
Cancer-associated fibroblasts (CAFs) have recently been shown to modify both metabolic and immunologic aspects of the TME in GBM. Through remodeling of the extracellular matrix, CAFs may contribute to the development of an immunosuppressive TME. The expression of proteins involved in the activation of fibroblasts, including fibroblast activation protein α (FAPα) and PDGFRβ, has been shown in both murine models and human GBM [66]. Additionally, Chen et al. established an association between the expression of six CAFs-related genes (ABCC3, CTHRC1, MSR1, PDLIM1, TNFRSF12A, and CHI3L2) and higher risk in gliomas by analyzing the cancer genome atlas and the Chinese glioma genome atlas. The high-risk group included higher tumor grade and poor prognosis [68]. Furthermore, Kim et al. consistently identified small quantities (<1%) of fibroblasts in resected human GBM tumor samples. They also demonstrated that the intracranial implantation of NIH3T3 embryonic fibroblasts led to the development of larger GL261 glioma tumors in a murine model [67]. Additionally, Trylcova et al. demonstrated that CAF-conditioned media increased the chemotactic migration of glioma cells in vitro [69]. These findings support a pro-tumor role for CAFs and identify them as a potential target for future therapies.
2.1.2. Metabolic and Structural Changes within the TME
There are also numerous metabolic alterations within the GBM TME that significantly alter local immune cell functions. Hypoxia is a key feature of the GBM TME, caused primarily by increased demand from tumor cells and errant or insufficient neovascularization. Among these changes is the upregulation of hypoxia inducible factor (HIF) expression, which promotes the differentiation of immunosuppressive cell populations, such as Tregs, and the up-regulation of immune checkpoint ligands, such as PD-L1 [125,126,127]. Hypoxic conditions also alter the cytokine milieu within the TME, increasing the secretion of immunosuppressive cytokines, such as IL-6 and IL-8, by GBM cells [128,129].
In addition to low oxygen levels, reduced glucose levels also promote immunosuppression within the GBM TME. Glycolytic pathways are upregulated by a number of cells within the GBM TME, including tumor cells and metabolically active immunosuppressive cell populations [126,130]. This greatly decreases the available glucose for anti-tumor immune cell populations, which may inhibit a number of key immune functions. For example, studies in other tumor types have demonstrated that decreased glucose availability hinders glycolysis-dependent TCR signaling and may promote exhaustion in T cell populations [131,132]. In addition to the lack of available glucose, the byproducts of glycolysis may also directly exert immunosuppressive effects. Specifically, lactate can cause the apoptosis of CD8+ lymphocytes and NK cells, and local acidification has been shown to promote M2 polarization within macrophage populations [133].
The catabolism of amino acids has also emerged as a key metabolic immunomodulatory factor within the GBM TME. Tryptophan catabolism, through IDO1, IDO2, and TDO2, has been well characterized in the context of GBM and seems to contribute to both immunosuppression and tumor growth [126]. Specifically, the downstream metabolites of tryptophan, such as kynurenine, can promote tumor cell survival and suppress the proliferation of both lymphoid and myeloid cell populations [134]. Interestingly, IDO inhibitor monotherapy did not significantly improve outcomes in pre-clinical studies, but IDO treatment may potentiate radiotherapy [135]. Arginine metabolism is also altered in the context of GBM. GBM cells are dependent on extracellular arginine availability for proliferation, and thus upregulate amino acid transporters and deplete the local environment [126,136]. This arginine depletion directly hinders the function of TILs, which rely on arginine for proliferation and cytotoxic functions [136]. Furthermore, arginine is differentially metabolized by pro-tumor and anti-tumor macrophages. Specifically, M1 polarized TAMs metabolize arginine primarily via nitric oxide synthase and M2 polarized TAMs primarily utilize arginase [137]. Thus, the alteration of arginine availability and metabolism has been proposed as a therapeutic strategy, and arginine deprivation has been noted to potentiate a response to radiotherapy in pre-clinical models [138].
In addition to metabolic alterations, the GBM environment is also characterized by structural changes, such as deficiencies in waste clearance and fluid balance via the glymphatic system. In preclinical studies, impaired lymphatic function has been shown to reduce CSF outflow in glioma-bearing mice [139]. Toh et al. investigated glymphatic function, measured by the index for diffusivity along the perivascular space (ALPS index), in 201 glioma patients and found that IDH-wild type gliomas had a lower ALPS index than IDH-mutant gliomas [140]. They also found that greater volumes of peritumoral edema were associated with a lower ALPS index, suggesting that glymphatic dysfunction may contribute to malignancy [140].
2.1.3. Systemic Immunosuppression
In addition to the profound local immunosuppression within the GBM TME described above, patients with GBM also have significant systemic immunosuppression [141]. Studies have noted that treatment-naïve patients with GBM have decreased systemic lymphocyte levels, seen both as profound CD4+ count depression, as well as atrophic, T cell-depleted lymphoid organs [142,143]. Interestingly, naïve T cells were instead sequestered within the bone marrow, seemingly due to internalization of the key lymphocyte trafficking receptor, S1P1 [143]. Additionally, many widely-used therapies for GBM can worsen this immunosuppression. For example, treatment with dexamethasone further decreases CD4+ counts in GBM patients [144]. Dexamethasone treatment also induces the development of HLA-DR low/- monocytes, which are unable to develop into mature dendritic cells and inhibit T cell proliferation [144]. While the exact mechanisms for the systemic immunosuppression seen in GBM are poorly understood, Ayasoufi et al. demonstrated that serum from mice implanted with GL261 cells hindered T cell proliferation in vitro [142]. Furthermore, they demonstrated that the serum components driving this immunosuppression have a molecular weight of >100 kDa, suggesting that the immunosuppression is not due to circulating steroids, but rather due to larger molecules [142].
2.2. Other Gliomas
Key differences have been noted between the TME and immune cell infiltrates of high- and low-grade (IDH-mutant) gliomas, with low-grade gliomas generally having less immune cell infiltration than high-grade gliomas [145] (Table 2). Within low-grade gliomas, the degree of infiltration by specific immune cell types may be of prognostic value. Yang et al. quantitatively characterized the degree of immune cell infiltration (ICI) in low-grade gliomas and found that low ICI scores, associated with higher levels of Tregs, resting NK cells, and M2 macrophages and lower levels of CD8+ T cells, were predictive of poorer survival outcomes [146].
Table 2.
Key differences in the immune cell infiltration of high- and low-grade gliomas.
Lymphocyte infiltration has been shown to differ between high- and low-grade gliomas, with studies demonstrating decreased numbers of TILs in low-grade compared to high-grade gliomas [35,145]. Studies investigating the mechanism of lower TIL recruitment have noted that the chemokines CXCL9 and CXCL10, which are implicated in T cell trafficking, are expressed at lower levels in low-grade compared to high-grade gliomas [147,148]. Furthermore, IDH mutations directly increase the production of R-2-hydroxyglutarate (2HG), which may be responsible for the observed decreased CXCL10 levels in low-grade tumors [148].
Immunosuppressive T cells seem to be less prominent in the low-grade glioma TME when compared to high-grade tumors. For example, Garber et al. reported lower levels of PD-1-expressing TILs in low-grade gliomas, suggesting a less inflammatory and subsequently exhausted phenotype in these tumors [149,150,151]. Lower levels of Tregs have also been reported in low-grade gliomas [14,32]. Overall, the differences in T cell characteristics between low- and high-grade gliomas likely contribute significantly to the differential immunosuppression seen between the two tumor types.
IDH mutations may also influence NK cell recruitment in low-grade glioma [152]. Ren et al. reported higher levels of NK cell infiltration in IDH mutant vs. IDH wildtype tumors, which was also associated with improved prognosis in glioma patients [152]. This increase in NK cell infiltration is likely mediated via an IDH-mutation-induced increase in the expression of the chemokine CX3CL1, which is involved in NK cell recruitment [152,153].
While TAMs are the dominant immune cell type in both IDH-mutant and IDH-wild type gliomas, there are important differences in TAM phenotypes among glioma grades. Vidyarthi et al. investigated macrophage polarization in gliomas and reported fewer M2 and more M1 macrophages in low- vs. high-grade gliomas [154]. Furthermore, the dominant developmental pathway for TAMs also differs, with monocyte-derived macrophages dominating over microglia in IDH-wild type gliomas [40,155]. Additionally, tumor progression is associated with a greater macrophage to microglia ratio in the TME [155].
Myeloid cell infiltrate also differs significantly among various low-grade gliomas. For example, astrocytomas are associated with greater numbers of macrophages and microglia than oligodendrogliomas [155]. Differences in immune cell gene expression of different low-grade tumors were noted to be comparable to or even exceed differences between the gene expression programs of oligodendroglioma and astrocytoma tumor cells themselves, suggesting a critical role for the immune microenvironment in determining the unique pathology and prognosis of these tumors despite their shared glial cell origin [155].
2.3. Meningioma
Meningiomas are neoplasms that arise from brain or spinal cord meninges [156]. While meningiomas are typically considered benign, these tumors also carry a significant rate of recurrence [157,158]. Unlike tumors arising from brain parenchyma, meningiomas are not protected by the blood–brain barrier or specialized CNS immunoregulatory mechanisms, and are thus more accessible to immune cells in the periphery [159,160]. The immune microenvironment of meningiomas is diverse, featuring lymphocytes (TILs), macrophages (TAMs), microglia, myeloid-derived suppressor cells (MDSCs), dendritic cells, and mast cells [161] (Table 3).
Table 3.
Summary of immune cell infiltration of meningiomas.
2.3.1. Key Immune Cell Populations
Lymphoid Cells
Lymphocytes, predominantly T cells, significantly contribute to the immune cell composition of meningiomas and typically cluster around perivascular spaces [164]. Fang et al. characterized the lymphocytic infiltrate of 28 meningiomas, of which 61% harbored B cells, 28.5% harbored antibody-producing plasma cells, and 100% harbored T cells [164]. High levels of tumor infiltrating CD3+CD8+FOXP3− lymphocytes were associated with improved progression free survival in high-grade meningiomas [162]. Additionally, Tregs were enriched in high-grade compared to low-grade meningiomas [163], whereas the expression of CXCL16, a T cell and monocyte chemoattractant, was increased in low-grade meningiomas [178]. T cells expressing PD-1, indicative of exhaustion, were shown to be enriched in tumors compared to periphery, suggesting that these cells along with Tregs may contribute to an immunosuppressive microenvironment in meningiomas [164]. PD-L1 and TILs were also specifically noted in neurofibromatosis type 2 meningiomas [179]. Furthermore, Li et al. found that PD-L1-expressing tumor cells were increased in proportion to the WHO tumor grade, suggesting a role for exhaustion in tumor progression [172]. The TME may also influence the likelihood of recurrence, as recurrent meningiomas are characterized by lower numbers of TILs [162].
Notably, meningiomas arising from different locations may exhibit different lymphocytic infiltration profiles. Cavernous sinus meningiomas were found to have decreased overall immune infiltration compared to convexity meningiomas, which may be a result of reduced VEGF signaling in these tumors [180]. Furthermore, Zador et al. reported that skull base meningiomas, which tend to display more benign characteristics, exhibit high levels of γδ T cell infiltration. In contrast, convexity tumors are dominated by mast cells and neutrophils, which may contribute to the more aggressive nature of these tumors [165].
Myeloid Cells
TAMs have been reported as the most prevalent immune cell type in the meningioma TME, constituting 18–44% of all cells in meningioma samples [157,166,167,168,169]. A correlation between macrophage infiltration and the expression of the monocyte chemoattractant protein-1 (MCP-1/CCL2) by meningiomas has been demonstrated, suggesting a role for this chemokine in the mechanism of meningioma infiltration by TAMs [181]. Notably, Proctor et al. reported that over 80% of macrophages in meningiomas are of the immunosuppressive M2 phenotype, evidenced by increased levels of CD163 and CD206 expression and low levels of CD80 and CD86 expression [166]. Furthermore, M2 macrophage infiltration has been shown to correlate with tumor size, grade, and recurrence across meningioma subtypes [166,170]. Tumor-associated factors, such as hypoxia, which is characteristic of tumor necrosis, have been noted to promote M2 polarization. In contrast, a higher ratio of M1 to M2 macrophage has been associated with increased progression-free survival [166]. Notably, one study demonstrated that TAMs of meningiomas with a monosomic chromosome 22 deletion had an M1-dominant phenotype [167]. This suggests that the deletion of genes located on this chromosome (such as the MIF gene) may have anti-tumoral effects [167]. To better characterize meningioma-associated macrophages and microglia, Grund et al. specifically investigated the infiltration of microglia and macrophages at the border between meningioma tumors and normal brain [171]. Overall, 25% of meningiomas had microglia or macrophagic cells at the tumor–brain border [171]. Furthermore, the presence of these cells was associated with malignant tumor grade and the loss of the pial-glial basement membrane, suggesting that these immune cells may be causally linked to more invasive tumor behavior [171]. Additionally, Woolf et al. investigated myeloid cell populations in resected meningioma tissue and found that microglia were scarce, whereas TAMs were abundant, likely reflecting the non-brain origin of these tumors [43].
MDSCs have also been identified in meningiomas, with increased numbers of MDSCs reported in high-grade, compared to low-grade, meningiomas [172]. Additionally, Pinton et al. observed that while peripheral MDSCs in patients with meningiomas caused minimal immunosuppression, infiltrating MDSCs reduced activated T cell activity [173]. The authors suggest that this may indicate a role for the tumor-induced differentiation of MDSCs into macrophages with immunosuppressive activity [173].
Dendritic cell infiltrate into meningiomas may also offer prognostic insight, though studies are limited. Chen et al. investigated the association between immune cell infiltration profiles and survival in meningioma patients and found that lower levels of dendritic cell infiltration were associated with better survival outcomes [174]. The authors suggest that this survival advantage may be due to a reduction in B-cell receptor signaling in tumors with greater dendritic cell infiltration [174,182].
Other Key Immune Cell Populations
Mast cells are also a prominent cell type in the TME of meningiomas. Immunohistochemical staining for tryptase, a mast cell marker, has demonstrated that these cells are present in 32–40% of low-grade (grade 1) and 86–90% of high-grade (grade 2 and 3) meningiomas [175,176]. However, the increased presence of mast cells in high-grade tumors has not been shown consistently in the literature; for example, Jabini et al. did not find an association between mast cells and the meningioma tumor grade [183]. Mast cells are typically located adjacent to blood vessels but may also be located more centrally and diffusely within higher grade tumors [161,175,176,184]. Notably, mast cells have been found to be associated with peritumoral edema, which can significantly contribute to the morbidity of these tumors by complicating surgery and increasing hospitalization times. High numbers of mast cells are also associated with cystic changes in meningiomas [177], and increased numbers of mast cells have been found in secretory compared to non-secretory meningiomas [185]. Furthermore, Tirokatai et al. found a potential relationship between VEGF-positive mast cells and edema in secretory meningiomas, suggesting a potential contributory role for mast cells in the development of edema [186]. Peritumoral brain edema in meningiomas has also been associated with IL-6 expression, suggesting a relationship between the immune characteristics and pathogenesis of these tumors [187].
2.3.2. Structural Changes within the TME
The role of glymphatic dysfunction in meningiomas has also been investigated. The volume of peritumoral edema in meningiomas has been found to be inversely associated with the ALPS index, which serves as a measure of glymphatic function [188]. These findings correspond to findings in gliomas as well, suggesting that glymphatics play a similar role in brain tumor immunity across tumor types.
2.4. Other
2.4.1. Chordoma
Chordomas are malignant sarcomas that occur in the skull base, vertebral bodies, or sacrum [189]. Dridi et al. conducted an immunohistochemical analysis of 81 chordomas and found that macrophages and CD4+ T cells predominated, with CD8+ T cells, CD20+ B cells, and high endothelial venules present in lesser numbers [190]. These immune infiltrates likely create an anti-inflammatory microenvironment, which is in line with the fact that M2 macrophages outnumber M1 macrophages in nearly all types of sarcomas and that these tumor cells express CD47, which downregulates proinflammatory macrophage activity [191]. Furthermore, both Mathios et al. and Dridi et al. reported that, while chordoma tumor cells do not themselves express PD-L1, this marker is expressed by infiltrating immune cells in a subset of chordomas. In these studies, PD-LI+ immune cells were associated with larger tumor size and may be negative prognostic indicators [190,192]. Yet, studies on PD-L1 expression on chordoma tumor cells have not been consistent, with Zou et al. and Feng et al. previously reporting PD-L1 expression in 66.7% and 94.9% of cases, respectively [193,194]. Further investigation is necessary to characterize the TME of these indolent tumors.
2.4.2. Sellar Tumors
Lu et al. conducted a histological analysis of 35 pituitary adenomas and found that the quantity of CD68+ macrophages was positively correlated with the size and invasiveness of adenomas [195]. Further, they reported that while overall T lymphocyte infiltration in pituitary adenomas was sparse, growth-hormone-secreting adenomas had higher levels of CD4+ and CD8+ T cell infiltration than non-growth-hormone-secreting adenomas. Yagnik et al. also investigated macrophage infiltration in non-functioning pituitary tumors and found that tumors that invaded the cavernous sinus had M2/M1 macrophage ratios greater than one, whereas tumors that did not invade had M2/M1 macrophage ratios less than one [196]. This finding persisted when the authors cultured M1 and M2 macrophages with non-functioning pituitary adenoma cells and found that M2 macrophages promoted greater invasion and proliferation of adenoma cells than M1 macrophages [196].
3. Therapeutic Targets within the TME
The TME plays a key role in determining the malignancy and progression of CNS tumors and is relatively genetically stable compared to tumor cells [28,197]. Thus, various immunotherapeutic strategies are being considered to target the TME of CNS tumors.
3.1. Checkpoint Inhibitor Therapy
Given their efficacy in other cancer types, including breast, bladder, and lung cancers, immune checkpoint inhibitors have been extensively studied for use in CNS neoplasms [198]. In a murine orthotopic glioma model, combined radiation and anti-PD-1 therapy was shown to increase survival, and the survival advantage was shown to be dependent on the presence of functional CD8+ T cells [199]. Additionally, intravenously injected NK cells treated with PD-1 antibodies increased survival in a glioma mouse model, compared to treatment with NK cells alone [200]. Furthermore, Mathios et al. demonstrated that combined local chemotherapy and anti-PD-1 blockade improved survival in glioblastoma-bearing mice, compared to systemic chemotherapy or local chemotherapy alone [201].
In a small phase 1 clinical trial, Cloughesy et al. demonstrated in 35 patients that neoadjuvant therapy with PD-1 blockade for recurrent GBM was correlated with an increased progression-free and overall survival [202]. In a subsequent study using proteomics and transcriptomics to evaluate immune cell infiltration before and after neoadjuvant PD-1 blockade, it was found that the neoadjuvant PD-1 blockade increased the infiltration of CD8+ T cells but did not modify immunosuppressive macrophages [203]. In the checkmate-143 phase 3 clinical trial, using the monoclonal PD-1 antibody nivolumab, no survival advantage was demonstrated over the VEGF inhibitor bevacizumab [204]. However, post hoc subgroup analyses for this trial suggested that nivolumab may be beneficial for patients without baseline corticosteroid use and with MGMT promoter methylation [204]. Subsequent trials have separately evaluated nivolumab in patients with and without MGMT promoter methylation [205]. In the checkmate-498 phase 3 trial, nivolumab was found to have no survival advantage over temozolomide in patients without MGMT promoter methylation [205]. In the checkmate-548 trial investigating standard-of-care in combination with nivolumab or placebo in patients with newly diagnosed GBM and methylated or indeterminate MGMT promoters, no improvement in survival was demonstrated [206]. Notably, however, patients with PD-L1 expression levels greater than 5% in this study displayed a trend toward improved progression-free survival [206]. Further clinical trials investigating PD-1 blockade in specific patient populations with GBM are underway and may offer further insight into the subtypes that may benefit from this therapy (Supplementary Table S1).
One potential marker for response is a high mutational burden, which has generally been correlated with a better response to checkpoint blockade therapy in other cancers. [207]. However, understanding the cause of an increased mutational burden for each patient is critical, as different underlying mutational processes result in differing neoantigen qualities and treatment efficacies [208,209]. For example, Tuoat et al. reported no difference in survival between GBM patients with and without temozolomide-induced hypermutation who were treated with PD-1 blockade [210]. However, patients with other underlying causes of high mutational burden, such as biallelic mismatch repair deficiency and germline POLE deficiency, have demonstrated favorable responses to PD-1 blockade therapy [211,212]. Thus, the identification of the specific drivers of hypermutation in brain cancer patients may be necessary to elucidate the relationship between mutational burden and responsiveness to checkpoint inhibitor therapy.
CTLA-4, a glycoprotein on T cells, which blocks activation and is constitutively expressed on Tregs, also offers a potential target for immune checkpoint therapy that has been validated in other cancer types [213]. In a murine glioma model, CTLA-4 blockade was shown to enhance anti-tumor immunity via improved CD4+ T cell function and resistance to Foxp3+ Tregs [214]. However, in exploratory phase 1 cohorts of the checkmate 143 trial (investigating nivolumab with or without the CTLA-4 inhibitor ipilimumab), the combination therapy was not well-tolerated by patients [215].
Lymphocyte activation gene 3 (LAG-3) is another target for checkpoint inhibitor therapy. Opduoalag, which combines nivolumab (anti-PD-1) with the anti-LAG-3 drug Relatlimab, was recently approved by the FDA for use in advanced melanoma. LAG-3 expression has been demonstrated in human glioblastoma, and LAG-3 inhibition, or knockout, has been shown to have anti-GBM effects in a murine model [216]. LAG-3+ TILs are sparse in GBM and have not been shown to correlate with survival, but preclinical studies suggest that targeting LAG-3 may be a promising therapeutic strategy [216,217]. Other possible targets for checkpoint inhibitor therapy in glioblastoma include Indoleamine-pyrrole 2,3-dioxygenase (IDO), T-cell immunoglobulin mucin-3 (TIM-3), Killer-cell immunoglobulin-like receptors (KIRs), and 4-1BB (for activation rather than blockade), all of which have been investigated to varying degrees [216,218,219,220,221,222].
Notably, while in-human trials evaluating singular checkpoint-inhibitors have been thus far unable to demonstrate improvements in survival in patients with GBM, targeting more than one checkpoint and combination with other treatments may be a promising therapeutic strategy. Monotherapies with immune checkpoint inhibitors may result in the upregulation of other checkpoints, thus limiting their efficacy [204]. For example, in a murine model of lung adenocarcinoma, Koyoma et al. demonstrated that TIM-3 was upregulated after PD-1 blockade therapy, and that targeting TIM-3, following a PD-1 blockade failure, offered a survival advantage [223]. Preclinical studies in glioma models, using combination therapies, have also produced promising results [220,224]. Kim et al. evaluated a combination PD-1 and TIM-3 blockade in a murine glioma model and found that combination therapy, along with focal radiation, significantly improved long-term survival [220]. Furthermore, Belcaid et al. reported that activation of the costimulatory signal 4-1BB, in combination with CTLA-4 blockade and focal radiotherapy, improved survival in a glioma mouse model in a CD4+ T cell-dependent manner [224]. However, as in the case of combination nivolumab and ipilimumab in the checkmate-143 phase 1 cohort, combination therapies may be associated with significant toxicities [204]. Nevertheless, combination therapies may be critical in overcoming resistance, recurrence, and progression of CNS malignancies. All current clinical trials investigating checkpoint inhibitor therapy in GBM are listed in Supplementary Table S1.
While the majority of work has focused on checkpoint inhibition in gliomas, checkpoint inhibitor therapy has also been studied for potential use in other CNS neoplasms. In a meningioma mouse model, administration of the PD-1 checkpoint inhibitor avelumab along with infusion of highly active NK cells resulted in improved survival [225]. In 2017, Gelerstein et al. noted a significant reduction in the size of an intracranial meningioma in a patient being treated with the PD-1 blockade therapy nivolumab for advanced lung cancer [226]. Nidamanuri et al. performed a retrospective chart review of meningiomas in patients treated with PD-1 inhibitors at their institution and concluded that there may be a role for immune checkpoint therapy in patients with recurrent, high-grade meningiomas [227]. Currently, seven clinical trials are investigating a potential role for PD-1 or combined PD-1 and CTLA-4 blockade in recurrent or refractory meningiomas [159,227,228] (Supplementary Table S2). Phase 2 trial results from the clinical trial NCT02648997 demonstrated that, while nivolumab was well-tolerated, it did not improve six-month progression free survival [228]. Notably, however, two patients with high tumor mutational burdens were found to significantly benefit from nivolumab therapy and had substantial macrophage and lymphocytic infiltration, demonstrating a potential role for checkpoint inhibition in this patient subset [211,228].
3.2. Anti-Tumor Vaccines
Vaccines against brain tumors are also a promising immunotherapeutic strategy that allow for adaptive immune surveillance against cancer cells. Vaccine development for CNS neoplasms, to date, has focused on gliomas [229,230]. Peptide-based brain tumor vaccines are designed to deliver tumor specific antigens in order to activate an immune response against cancer cells [229]. One vaccine that has been extensively investigated in preclinical and clinical models is rindopepimut, which targets epidermal growth factor receptor variant III (EGFRvIII) [231]. EGFRvIII is exclusively but heterogeneously found on glioblastoma cells [229,231]. Based on success in preclinical models and improved progression free survival in three phase II clinical trials, an international phase III trial of 745 primary GBM patients was initiated to investigate combination rindopepimut and temozolomide [231]. While patients in the experimental group had a strong humoral response, no improvement in survival was noted [231]. Rindopepimut has also been investigated for recurrent GBM in combination with bevacizumab, an angiogenesis inhibitor [232]. In a randomized phase II study, combination rindopepimut and bevacizumab was found to induce regression and improve the 24-month survival rate in recurrent GBM, from 3% in controls to 20% in the treatment group, suggesting that the combination of immunotherapeutic vaccines with VEGF inhibitors may be promising [232]. Brain tumor vaccines targeting mutant IDH, which is characteristic of low-grade gliomas and has high levels of penetrance in tumor cells, have also been investigated. Schumacher et al. demonstrated the potential utility of IDH1-R132H-mutated peptide in a mouse model, in which vaccine administration induced a CD4+ T cell immune response [233]. Subsequently, the phase 1 NOA-16 and RESIST clinical trials were initiated to evaluate IDH-1R132H peptide vaccines in patients with newly diagnosed and recurrent glioma, respectively [234] (Supplementary Table S3). The majority of patients in both trials demonstrated anti-IDH1 R132H immune responses. Furthermore, in the NOA-16 trial, those who had vaccine-induced T-cell and/or B cell immune responses had a two-year progression-free survival rate of 0.82, whereas those without an immune response showed progression within two years [234]. While further results are needed to evaluate the efficacy of the vaccine in these trials, both suggest a potential therapeutic role for the IDH1-R132H vaccine in IDH-mutant gliomas.
Other peptide antigens are also being investigated for utilization in anti-glioma vaccines, including Wilms tumor 1, cytomegalovirus peptide pp6547, survivin, and telomerase reverse transcriptase [235]. Notably, given the high probability of immune escape from a single antigen, anti-tumor immunotherapeutic vaccines have also been engineered to include more than one antigen. One such vaccine is the ICT-107 vaccine, created by incubation of an autologous dendritic cell with six peptides (AIM-2, gp100, HER2, IL13Rα2, MAGE-1, and TRP-2) that are over-expressed in GBM tumor cells [236]. In a phase II study, the ICT-107 vaccine was shown to increase progression free survival by 2.2 months when compared to unpulsed dendritic cells in newly diagnosed GBM patients. Interestingly, the subset of patients with HLA-A2 antigen expression had a higher immune response via Elispot, an assay that detects functional TILs [236]. Another multipeptide vaccine, TAS0313 (including the 8 antigens EGFR, KUA, LCK, MRP3, PTHRP, SART2, SART3, and WHSC2), has also demonstrated both tolerability and immunogenicity, indicated by increased levels of IgG and cytotoxic T cells, in phase I/II studies [237]. Effector T cell responses and tolerability were also confirmed for the IMA950 multipeptide vaccine (including the 11 antigens derived from the BCAN, CSPG4, FABP7, IGF2BP3, NRCAM, NLGN4X, PTPRZ1, TNC, c-met, survivin, and HBV core antigen proteins in phase I and phase I/II trials); however, it was not shown to be effective in combination with the VEGF-inhibitor bevacizumab in a study performed by Boydell et al. [238,239,240]. Finally, the multipeptide vaccine EO2401, which features peptides derived from the microbiome that mimics tumor antigens, is currently being investigated in phase I/II trials (Supplementary Table S3).
Gliomas have also been targeted via tumor cell lysate-based vaccines, which involve the delivery of multiple immunogenic antigens from an allogeneic tumor cell lysate [241]. Ogino et al. investigated the therapeutic potential of injecting low-grade glioma patients with glioma-associated antigens in the GBM6-AD stem cell line, derived from a patient diagnosed with GBM, in order to induce an immune response and prevent the malignant transformation of low-grade gliomas [241]. They found that vaccine administration both initiated a peripheral CD8+ immune response and induced CD8+ T cell migration into the glioma microenvironment [241]. Another tumor cell-lysate based vaccine, DCVax-L, took a different approach, using autologous tumor lysate from patients’ resected glioblastoma tissue [242]. A strong T cell response was elicited by the DCVax-L in preclinical studies, and phase I trials demonstrated that patients with low TGF-β expression may be more likely to benefit from the vaccine [242]. In phase 3 trials, interim results suggested that patients were living longer, as evidenced by long tails on Kaplan–Meier survival curves (estimated median survival of 88.2 months) [243]. However, further investigation is needed to validate these findings [243]. All current clinical trials investigating immunotherapeutic vaccines in GBM are listed in Supplementary Table S3.
3.3. Other Therapeutic Approaches
Numerous other approaches have also been investigated as potential brain cancer therapies. Chimeric antigen receptor (CAR) T cell therapy, which involves engineering T cells to express tumor-specific chimeric antigen receptors and was approved by the Food and Drug Administration in 2017, represents a potential avenue for brain tumor therapy [244]. Notably, T cells can be designed via the introduction of costimulatory signals to have an activated phenotype, allowing them to potentially overcome the immunosuppressive brain tumor microenvironment that is characteristic of many brain tumor neoplasms [244]. As with vaccines, CAR-T cell therapy in brain cancer has largely focused on glioma [245].
To date, the targets of CAR-T cell therapy for glioblastoma include EGFRvIII, HER2, and IL13Rα2 [245]. EGFRvIII-targeting CAR-T cells have had limited success in clinical trials. In a phase I trial, the infusion of EGFRvIII-targeting CAR-T cells led to an adaptive immunosuppressive tumor response characterized by FOX3P+ Treg infiltration and the upregulation of immunosuppressive markers, including IDO-1, IL-10, PD-L1, and TGF-β [246]. In a subsequent phase I pilot trial, the administration of EGFRvIII-targeting CAR-T cells demonstrated dose-dependent adverse events, but no clinical benefit [247]. A phase I trial has also been conducted for HER-2 specific CAR-T cells. In this study, Ahmed et al. administered HER-2-specific CAR-T cells, which were constructed from virus-specific T cells in order to potentially elicit a costimulatory effect via latent virus antigens [248]. In total, eight out of seventeen patients in this trial benefitted from the treatment, achieving either a partial response or stable disease, and three patients demonstrated no progression at a 29-month follow-up [248]. CAR-T cell therapy with IL13Rα2 may also warrant further investigation, with two out of three patients in a pilot study demonstrating transient anti-glioma T-cell responses [245,249]. Other potential targets that are being investigated for use in CAR-T cell therapy in clinical studies include B7-H3, CD147, GD2, MMP2, and NKG2D, with additional targets including CAIX, CD70, CSPG4, EPhA2, and TROP2 being investigated in preclinical studies [245]. All current clinical trials investigating CAR-T cell therapy in GBM are listed in Supplementary Table S4.
CAR-T cell therapy has also been investigated in preclinical studies in other primary brain tumor types, including anaplastic meningiomas and chordomas [250,251]. Tang et al. investigated the local delivery of B7-H3-targeted CAR-T cells in a patient with recurrent anaplastic meningioma and reported that the treatment demonstrated both tolerability and local bioactivity, with decreased B7-H3 expression detected in the cavity after treatment [250]. Tumor bulk was also noted to be stable near the site of delivery for the duration of treatment [250]. B7-H3 has also been identified as a potential target for CAR-T therapy in chordoma [251]. Long et al. investigated the expression of several CAR-T targets in skull-based chordomas and found that B7-H3 was expressed in the greatest quantities. Furthermore, B7-H3-targeting CAR-T cells were demonstrated to have anti-tumor effects against in vitro tumor spheres derived from surgically resected chordoma tissue, demonstrating a potential role for CAR-T cell therapy in patients expressing this antigen [251]. Further studies are required to validate targets and evaluate the applicability of CAR-T cell therapy in brain tumor patients.
As numerous T-cell-based therapies continue to be investigated for GBM and other primary brain cancers, novel assays are needed to identify targets for T-cell anti-tumor immune responses. One such assay is the mutation-associated neoantigen functional expansion of specific T cells (MANAFEST) assay, which represents a novel strategy for identifying tumor biomarkers and predicting patients’ response to immunotherapies. Notably, this assay uses TCR sequencing and bioinformatics to identify tumor-specific TCR Vβ clonotypes and provides critical information about the anti-tumor T cell response over time, with high sensitivity and specificity [252]. This data can then be used to determine particular genomic features that are related to tumor immunogenicity and identify molecular motifs that predict patients’ innate immune responses, as well as responses to immunotherapies [252]. Other strategies to define therapeutic targets include the characterization of the tumor cell surface proteins via spatial proteomics, which can identify distinct topographic histologic patterns that are of particular interest in heterogenous tumors, such as GBM [253,254]. For example, Rose et al. utilized spatial proteomics and MALDI-mass spectrometry to identify 11 proteins that may be potential targets for the future development of cancer vaccines or CAR-T immunotherapy [253]. Advancements in antigen discovery platforms will enable the appropriate targeting of novel therapeutic modalities and further personalized brain tumor care.
As discussed, immunotherapeutic approaches against brain tumors have largely focused on restoring or engineering anti-tumor T cell responses. However, numerous other immune cell types contribute to the immunosuppressive microenvironment in brain tumors, and thus may also be viable targets. One approach being investigated is to alter the differentiation of tumor-associated macrophages (TAMs) from a pro-tumor to an anti-tumor phenotype. Specifically, preclinical studies have demonstrated that the inhibition of the colony stimulating factor 1 receptor (CSF-1), which is involved in inducing macrophage differentiation and proliferation, may be promising [255,256]. Pyonteck et al. evaluated the efficacy of CSF-1 blockade in a murine glioma model and found that CSF-1 inhibition improved survival. Specifically, the vehicle-treated cohort had a median survival of 5.7 weeks, whereas 64.3% of CSF-1-blockade-treated mice were alive at the 26-week endpoint. The CSF-1 blockade also led to tumor regression and slowed the growth of patient-derived xenografts [255]. Additionally, Stafford et al. investigated the role of CSF-1 inhibition after treatment with ionizing radiation (IR), which was shown to upregulate CSF-IR expression in GBM xenografts [256]. They found that the CSF-1R blockade prevented the differentiation of monocytes into immunosuppressive M2 polarized macrophages [256]. However, in phase II studies of the small molecule CSF-1 and KIT ligand inhibitor PLX3397, there was no improvement in the primary endpoint of 6-month progression free survival [257]. The lack of success of anti-CSF-1 monotherapy beyond preclinical models may be due to the induction of acquired resistance mechanisms, such as FOXP3+ Treg infiltration and MDSC recruitment [258,259,260]. Combining multiple therapeutic strategies may help overcome the shortcomings of monotherapy, and studies have demonstrated that the CSF-1R blockade may enhance the anti-tumoral T cell responses induced by checkpoint inhibitor therapy [259,261,262]. For example, Przystal et al. demonstrated that the CSF-1R blockade may improve the effect of anti-PD1 therapy in glioblastoma [261]. These findings support the further investigation of combinatory therapy for brain cancer.
Additionally, novel nanomedicine technologies have emerged as a potential way to modulate the immune microenvironment to slow tumor growth. For example, M1 macrophage-derived extracellular vesicles (M1EVs) delivery is another TAM-focused strategy that has been investigated recently in GBM. Wang et al. demonstrated that functionalized M1EVs were capable of penetrating the blood–brain barrier, inducing M2 to M1 polarization, and delivering and activating the chemotherapeutic agent banoxantrone [263]. Furthermore, they demonstrated that M1EVs had therapeutic effects in cell-derived and patient-derived xenografts [263]. These findings demonstrate a complementary role for nanomedicine and immunotherapeutics in the experimental treatment of glioblastoma.
Lastly, CSF-1/CSF-1R signaling has also been targeted as a therapy in high-grade meningiomas to reduce the activity of M2 phenotype immunosuppressive macrophages [264]. In a murine meningioma model, Yueng et al. demonstrated that treatment with anti-CSF-1R monoclonal antibodies, and not anti-PD-1 therapy, led to a reduction in tumor growth [264]. Thus, macrophage remodeling strategies to overcome the immunosuppressive TME may be relevant across brain tumor types.
4. Conclusions
The TME of CNS tumors significantly impacts the overall biology of the tumor, with important implications for patient prognosis and treatment. Research to date has allowed the sub-characterization of tumors based on TME features, as well as the development of novel therapeutic modalities targeted towards specific components of the TME. Yet, while novel therapies have produced notable results in pre-clinical studies, a majority of clinical trials to date have failed to demonstrate a significant impact on patient outcomes. However, ongoing work to advance our understanding of the complexities of the CNS TME will allow for optimized and individualized therapy selection, as well as the development of new therapeutic strategies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24032020/s1.
Author Contributions
Conceptualization, A.L.K., P.P.S. and M.L.; methodology, A.L.K., P.P.S. and M.L.; validation, A.L.K., P.P.S. and M.L.; writing—original draft preparation, A.L.K., P.P.S. and M.L.; writing—review and editing, A.L.K., P.P.S. and M.L.; visualization, A.L.K., P.P.S. and M.L.; supervision, M.L.; project administration, A.L.K., P.P.S. and M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors thank Dimitrios Mathios for his contributions to this manuscript.
Conflicts of Interest
A.L.K. and P.P.S. report no conflicts. M.L. reports research support from Arbor, BMS, Accuray, Biohaven, and Urogen. He is a consultant for VBI, InCephalo Therapeutics, Merck, Pyramid Bio, Insightec, Biohaven, Sanianoia, Hemispherian, Novocure, Noxxon, InCando, Century Therapeutics, CraniUs, MediFlix, and Stryker. He is a shareholder in Egret Therapeutics, and holds patents for focused radiation and checkpoint inhibitors, local chemotherapy and checkpoint inhibitors, and checkpoints for neuro-Inflammation. He is on the data safety and monitoring board for Cellularity.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS immune privilege: Hiding in plain sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef]
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neurooncol. 2012, 107, 207–212. [Google Scholar] [CrossRef]
- Pombo Antunes, A.R.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife 2020, 9, e52176. [Google Scholar] [CrossRef]
- Walker, D.G.; Chuah, T.; Rist, M.J.; Pender, M.P. T-cell apoptosis in human glioblastoma multiforme: Implications for immunotherapy. J. Neuroimmunol. 2006, 175, 59–68. [Google Scholar] [CrossRef]
- Zhang, J.L.; Zhong, X.S.; Yang, S.B.; Kang, X.; Li, Y.; Chen, J.X.; Li, W.B. Features and therapeutic potential of T-cell receptors in high-grade glioma. Chin. Med. J. 2019, 132, 1435–1440. [Google Scholar] [CrossRef]
- Perng, P.; Lim, M. Immunosuppressive Mechanisms of Malignant Gliomas: Parallels at Non-CNS Sites. Front. Oncol. 2015, 5, 153. [Google Scholar] [CrossRef]
- Davidson, T.B.; Lee, A.; Hsu, M.; Sedighim, S.; Orpilla, J.; Treger, J.; Mastall, M.; Roesch, S.; Rapp, C.; Galvez, M.; et al. Expression of PD-1 by T Cells in Malignant Glioma Patients Reflects Exhaustion and Activation. Clin. Cancer Res. 2019, 25, 1913–1922. [Google Scholar] [CrossRef]
- Wilmotte, R.; Burkhardt, K.; Kindler, V.; Belkouch, M.C.; Dussex, G.; Tribolet, N.; Walker, P.R.; Dietrich, P.Y. B7-homolog 1 expression by human glioma: A new mechanism of immune evasion. Neuroreport 2005, 16, 1081–1085. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef]
- Tumangelova-Yuzeir, K.; Naydenov, E.; Ivanova-Todorova, E.; Krasimirova, E.; Vasilev, G.; Nachev, S.; Kyurkchiev, D. Mesenchymal Stem Cells Derived and Cultured from Glioblastoma Multiforme Increase Tregs, Downregulate Th17, and Induce the Tolerogenic Phenotype of Monocyte-Derived Cells. Stem Cells Int. 2019, 2019, 6904638. [Google Scholar] [CrossRef]
- DiDomenico, J.; Lamano, J.B.; Oyon, D.; Li, Y.; Veliceasa, D.; Kaur, G.; Ampie, L.; Choy, W.; Lamano, J.B.; Bloch, O. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology 2018, 7, e1448329. [Google Scholar] [CrossRef]
- Jacobs, J.F.; Idema, A.J.; Bol, K.F.; Grotenhuis, J.A.; de Vries, I.J.; Wesseling, P.; Adema, G.J. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J. Neuroimmunol. 2010, 225, 195–199. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Abou-Ghazal, M.; Reina-Ortiz, C.; Yang, D.S.; Sun, W.; Qiao, W.; Hiraoka, N.; Fuller, G.N. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin. Cancer Res. 2008, 14, 5166–5172. [Google Scholar] [CrossRef]
- El Andaloussi, A.; Lesniak, M.S. CD4+ CD25+ FoxP3+ T-cell infiltration and heme oxygenase-1 expression correlate with tumor grade in human gliomas. J. Neurooncol. 2007, 83, 145–152. [Google Scholar] [CrossRef]
- Lee, M.; Park, C.; Woo, J.; Kim, J.; Kho, I.; Nam, D.H.; Park, W.Y.; Kim, Y.S.; Kong, D.S.; Lee, H.W.; et al. Preferential Infiltration of Unique Vgamma9Jgamma2-Vdelta2 T Cells Into Glioblastoma Multiforme. Front. Immunol. 2019, 10, 555. [Google Scholar] [CrossRef]
- Joalland, N.; Chauvin, C.; Oliver, L.; Vallette, F.M.; Pecqueur, C.; Jarry, U.; Scotet, E. IL-21 Increases the Reactivity of Allogeneic Human Vgamma9Vdelta2 T Cells Against Primary Glioblastoma Tumors. J. Immunother. 2018, 41, 224–231. [Google Scholar] [CrossRef]
- Chauvin, C.; Joalland, N.; Perroteau, J.; Jarry, U.; Lafrance, L.; Willem, C.; Retiere, C.; Oliver, L.; Gratas, C.; Gautreau-Rolland, L.; et al. NKG2D Controls Natural Reactivity of Vgamma9Vdelta2 T Lymphocytes against Mesenchymal Glioblastoma Cells. Clin. Cancer Res. 2019, 25, 7218–7228. [Google Scholar] [CrossRef]
- Nakazawa, T.; Nakamura, M.; Park, Y.S.; Motoyama, Y.; Hironaka, Y.; Nishimura, F.; Nakagawa, I.; Yamada, S.; Matsuda, R.; Tamura, K.; et al. Cytotoxic human peripheral blood-derived gammadeltaT cells kill glioblastoma cell lines: Implications for cell-based immunotherapy for patients with glioblastoma. J. Neurooncol. 2014, 116, 31–39. [Google Scholar] [CrossRef]
- Nakazawa, T.; Nakamura, M.; Matsuda, R.; Nishimura, F.; Park, Y.S.; Motoyama, Y.; Hironaka, Y.; Nakagawa, I.; Yokota, H.; Yamada, S.; et al. Antitumor effects of minodronate, a third-generation nitrogen-containing bisphosphonate, in synergy with gammadeltaT cells in human glioblastoma in vitro and in vivo. J. Neurooncol. 2016, 129, 231–241. [Google Scholar] [CrossRef]
- Bryant, N.L.; Suarez-Cuervo, C.; Gillespie, G.Y.; Markert, J.M.; Nabors, L.B.; Meleth, S.; Lopez, R.D.; Lamb, L.S., Jr. Characterization and immunotherapeutic potential of gammadelta T-cells in patients with glioblastoma. Neuro Oncol. 2009, 11, 357–367. [Google Scholar] [CrossRef]
- Vauleon, E.; Tony, A.; Hamlat, A.; Etcheverry, A.; Chiforeanu, D.C.; Menei, P.; Mosser, J.; Quillien, V.; Aubry, M. Immune genes are associated with human glioblastoma pathology and patient survival. BMC Med. Genom. 2012, 5, 41. [Google Scholar] [CrossRef]
- Zhong, Q.Y.; Fan, E.X.; Feng, G.Y.; Chen, Q.Y.; Gou, X.X.; Yue, G.J.; Zhang, G.H. A gene expression-based study on immune cell subtypes and glioma prognosis. BMC Cancer 2019, 19, 1116. [Google Scholar] [CrossRef]
- Bockmayr, M.; Klauschen, F.; Maire, C.L.; Rutkowski, S.; Westphal, M.; Lamszus, K.; Schuller, U.; Mohme, M. Immunologic Profiling of Mutational and Transcriptional Subgroups in Pediatric and Adult High-Grade Gliomas. Cancer Immunol. Res. 2019, 7, 1401–1411. [Google Scholar] [CrossRef]
- Zhu, C.; Zou, C.; Guan, G.; Guo, Q.; Yan, Z.; Liu, T.; Shen, S.; Xu, X.; Chen, C.; Lin, Z.; et al. Development and validation of an interferon signature predicting prognosis and treatment response for glioblastoma. Oncoimmunology 2019, 8, e1621677. [Google Scholar] [CrossRef]
- Kozlowska, A.K.; Tseng, H.C.; Kaur, K.; Topchyan, P.; Inagaki, A.; Bui, V.T.; Kasahara, N.; Cacalano, N.; Jewett, A. Resistance to cytotoxicity and sustained release of interleukin-6 and interleukin-8 in the presence of decreased interferon-gamma after differentiation of glioblastoma by human natural killer cells. Cancer Immunol. Immunother. 2016, 65, 1085–1097. [Google Scholar] [CrossRef]
- Tseng, H.C.; Bui, V.; Man, Y.G.; Cacalano, N.; Jewett, A. Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells through Cell-Cell Contact and Secreted Factors. Front. Immunol. 2014, 5, 269. [Google Scholar] [CrossRef]
- Andersen, R.S.; Anand, A.; Harwood, D.S.L.; Kristensen, B.W. Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers 2021, 13, 4255. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef]
- Gregoire, H.; Roncali, L.; Rousseau, A.; Cherel, M.; Delneste, Y.; Jeannin, P.; Hindre, F.; Garcion, E. Targeting Tumor Associated Macrophages to Overcome Conventional Treatment Resistance in Glioblastoma. Front. Pharmacol. 2020, 11, 368. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Friebel, E.; Kapolou, K.; Unger, S.; Nunez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Sankowski, R.; Bottcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Arrieta, V.A.; Najem, H.; Petrosyan, E.; Lee-Chang, C.; Chen, P.; Sonabend, A.M.; Heimberger, A.B. The Eclectic Nature of Glioma-Infiltrating Macrophages and Microglia. Int. J. Mol. Sci. 2021, 22, 13382. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef]
- Lin, J.D.; Nishi, H.; Poles, J.; Niu, X.; McCauley, C.; Rahman, K.; Brown, E.J.; Yeung, S.T.; Vozhilla, N.; Weinstock, A.; et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight 2019, 4, e124574. [Google Scholar] [CrossRef]
- Sorensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef]
- Muller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Woolf, Z.; Swanson, M.E.V.; Smyth, L.C.; Mee, E.W.; Schweder, P.; Heppner, P.; Kim, B.J.H.; Turner, C.; Oldfield, R.L.; Curtis, M.A.; et al. Single-cell image analysis reveals a protective role for microglia in glioblastoma. Neurooncol. Adv. 2021, 3, vdab031. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPalpha antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef]
- Alban, T.J.; Bayik, D.; Otvos, B.; Rabljenovic, A.; Leng, L.; Jia-Shiun, L.; Roversi, G.; Lauko, A.; Momin, A.A.; Mohammadi, A.M.; et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front. Immunol. 2020, 11, 1191. [Google Scholar] [CrossRef]
- Ravi, V.M.; Neidert, N.; Will, P.; Joseph, K.; Maier, J.P.; Kuckelhaus, J.; Vollmer, L.; Goeldner, J.M.; Behringer, S.P.; Scherer, F.; et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat. Commun. 2022, 13, 925. [Google Scholar] [CrossRef]
- Lakshmanachetty, S.; Cruz-Cruz, J.; Hoffmeyer, E.; Cole, A.P.; Mitra, S.S. New Insights into the Multifaceted Role of Myeloid-Derived Suppressor Cells (MDSCs) in High-Grade Gliomas: From Metabolic Reprograming, Immunosuppression, and Therapeutic Resistance to Current Strategies for Targeting MDSCs. Cells 2021, 10, 893. [Google Scholar] [CrossRef]
- Jia, W.; Jackson-Cook, C.; Graf, M.R. Tumor-infiltrating, myeloid-derived suppressor cells inhibit T cell activity by nitric oxide production in an intracranial rat glioma + vaccination model. J. Neuroimmunol. 2010, 223, 20–30. [Google Scholar] [CrossRef]
- Lee-Chang, C.; Rashidi, A.; Miska, J.; Zhang, P.; Pituch, K.C.; Hou, D.; Xiao, T.; Fischietti, M.; Kang, S.J.; Appin, C.L.; et al. Myeloid-Derived Suppressive Cells Promote B cell-Mediated Immunosuppression via Transfer of PD-L1 in Glioblastoma. Cancer Immunol. Res. 2019, 7, 1928–1943. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Wei, K.C.; Chen, P.Y.; Lim, M.; Hwang, T.L. Roles of Neutrophils in Glioma and Brain Metastases. Front. Immunol. 2021, 12, 701383. [Google Scholar] [CrossRef] [PubMed]
- Magod, P.; Mastandrea, I.; Rousso-Noori, L.; Agemy, L.; Shapira, G.; Shomron, N.; Friedmann-Morvinski, D. Exploring the longitudinal glioma microenvironment landscape uncovers reprogrammed pro-tumorigenic neutrophils in the bone marrow. Cell Rep. 2021, 36, 109480. [Google Scholar] [CrossRef] [PubMed]
- Andzinski, L.; Kasnitz, N.; Stahnke, S.; Wu, C.F.; Gereke, M.; von Kockritz-Blickwede, M.; Schilling, B.; Brandau, S.; Weiss, S.; Jablonska, J. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 2016, 138, 1982–1993. [Google Scholar] [CrossRef]
- Scapini, P.; Lapinet-Vera, J.A.; Gasperini, S.; Calzetti, F.; Bazzoni, F.; Cassatella, M.A. The neutrophil as a cellular source of chemokines. Immunol. Rev. 2000, 177, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.V.; Fridlender, Z.G.; Granot, Z. The Multifaceted Roles Neutrophils Play in the Tumor Microenvironment. Cancer Microenviron. 2015, 8, 125–158. [Google Scholar] [CrossRef]
- Yee, P.P.; Wei, Y.; Kim, S.Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef]
- Kastenmuller, W.; Brandes, M.; Wang, Z.; Herz, J.; Egen, J.G.; Germain, R.N. Peripheral prepositioning and local CXCL9 chemokine-mediated guidance orchestrate rapid memory CD8+ T cell responses in the lymph node. Immunity 2013, 38, 502–513. [Google Scholar] [CrossRef]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef] [PubMed]
- Wendel, M.; Galani, I.E.; Suri-Payer, E.; Cerwenka, A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res. 2008, 68, 8437–8445. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Vijayan, D.; Putz, E.M.; Aguilera, A.R.; Markey, K.A.; Straube, J.; Kazakoff, S.; Nutt, S.L.; Takeda, K.; Hill, G.R.; et al. Interleukin-12 from CD103(+) Batf3-Dependent Dendritic Cells Required for NK-Cell Suppression of Metastasis. Cancer Immunol. Res. 2017, 5, 1098–1108. [Google Scholar] [CrossRef]
- Hochrein, H.; O’Keeffe, M.; Luft, T.; Vandenabeele, S.; Grumont, R.J.; Maraskovsky, E.; Shortman, K. Interleukin (IL)-4 is a major regulatory cytokine governing bioactive IL-12 production by mouse and human dendritic cells. J. Exp. Med. 2000, 192, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Tumor-derived factors modulating dendritic cell function. Cancer Immunol. Immunother. 2016, 65, 821–833. [Google Scholar] [CrossRef]
- Li, M.; Li, G.; Kiyokawa, J.; Tirmizi, Z.; Richardson, L.G.; Ning, J.; Das, S.; Martuza, R.L.; Stemmer-Rachamimov, A.; Rabkin, S.D.; et al. Characterization and oncolytic virus targeting of FAP-expressing tumor-associated pericytes in glioblastoma. Acta Neuropathol. Commun. 2020, 8, 221. [Google Scholar] [CrossRef]
- Kim, T.Y.; Srivastava, S.; Medikonda, R.; Pant, A.; Patel, K.K.; Rodriguez, F.; Lim, M. 347 Cancer-associated Fibroblasts Affect GBM Progression. Neurosurgery 2022, 68, 81. [Google Scholar] [CrossRef]
- Chen, Z.; Zhuo, S.; He, G.; Tang, J.; Hao, W.; Gao, W.Q.; Yang, K.; Xu, H. Prognosis and Immunotherapy Significances of a Cancer-Associated Fibroblasts-Related Gene Signature in Gliomas. Front. Cell Dev. Biol. 2021, 9, 721897. [Google Scholar] [CrossRef]
- Trylcova, J.; Busek, P.; Smetana, K., Jr.; Balaziova, E.; Dvorankova, B.; Mifkova, A.; Sedo, A. Effect of cancer-associated fibroblasts on the migration of glioma cells in vitro. Tumor Biol. 2015, 36, 5873–5879. [Google Scholar] [CrossRef]
- Mahata, B.; Biswas, S.; Rayman, P.; Chahlavi, A.; Ko, J.; Bhattacharjee, A.; Li, Y.T.; Li, Y.; Das, T.; Sa, G.; et al. GBM Derived Gangliosides Induce T Cell Apoptosis through Activation of the Caspase Cascade Involving Both the Extrinsic and the Intrinsic Pathway. PLoS ONE 2015, 10, e0134425. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutierrez-Vazquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Author Correction: Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 1533. [Google Scholar] [CrossRef] [PubMed]
- Book, A.A.; Fielding, K.E.; Kundu, N.; Wilson, M.A.; Fulton, A.M.; Laterra, J. IL-10 gene transfer to intracranial 9L glioma: Tumor inhibition and cooperation with IL-2. J. Neuroimmunol. 1998, 92, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, B.; Cyranowski, S. Recent Advances in Understanding Mechanisms of TGF Beta Signaling and Its Role in Glioma Pathogenesis. Adv. Exp. Med. Biol. 2020, 1202, 179–201. [Google Scholar] [CrossRef]
- Friese, M.A.; Wischhusen, J.; Wick, W.; Weiler, M.; Eisele, G.; Steinle, A.; Weller, M. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004, 64, 7596–7603. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Uhl, M.; Ma, J.Y.; Janssen, L.; Sriram, V.; Aulwurm, S.; Kerr, I.; Lam, A.; Webb, H.K.; Kapoun, A.M.; et al. Inhibiting TGF-beta signaling restores immune surveillance in the SMA-560 glioma model. Neuro Oncol. 2007, 9, 259–270. [Google Scholar] [CrossRef]
- Woroniecka, K.; Fecci, P.E. T-cell exhaustion in glioblastoma. Oncotarget 2018, 9, 35287–35288. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, H.; Xu, J.; Lu, Y.; Ji, X.; Yao, Y.; Chao, H.; Zhang, J.; Zhang, X.; Yao, S.; et al. Different T-cell subsets in glioblastoma multiforme and targeted immunotherapy. Cancer Lett. 2021, 496, 134–143. [Google Scholar] [CrossRef]
- Miska, J.; Lee-Chang, C.; Rashidi, A.; Muroski, M.E.; Chang, A.L.; Lopez-Rosas, A.; Zhang, P.; Panek, W.K.; Cordero, A.; Han, Y.; et al. HIF-1alpha Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep. 2019, 27, 226–237.e4. [Google Scholar] [CrossRef]
- Iwata, R.; Hyoung Lee, J.; Hayashi, M.; Dianzani, U.; Ofune, K.; Maruyama, M.; Oe, S.; Ito, T.; Hashiba, T.; Yoshimura, K.; et al. ICOSLG-mediated regulatory T-cell expansion and IL-10 production promote progression of glioblastoma. Neuro Oncol. 2020, 22, 333–344. [Google Scholar] [CrossRef]
- Cao, J.Y.; Guo, Q.; Guan, G.F.; Zhu, C.; Zou, C.Y.; Zhang, L.Y.; Cheng, W.; Wang, G.L.; Cheng, P.; Wu, A.H.; et al. Elevated lymphocyte specific protein 1 expression is involved in the regulation of leukocyte migration and immunosuppressive microenvironment in glioblastoma. Aging 2020, 12, 1656–1684. [Google Scholar] [CrossRef]
- Barsheshet, Y.; Wildbaum, G.; Levy, E.; Vitenshtein, A.; Akinseye, C.; Griggs, J.; Lira, S.A.; Karin, N. CCR8(+)FOXp3(+) Treg cells as master drivers of immune regulation. Proc. Natl. Acad. Sci. USA 2017, 114, 6086–6091. [Google Scholar] [CrossRef]
- Kidani, Y.; Nogami, W.; Yasumizu, Y.; Kawashima, A.; Tanaka, A.; Sonoda, Y.; Tona, Y.; Nashiki, K.; Matsumoto, R.; Hagiwara, M.; et al. CCR8-targeted specific depletion of clonally expanded Treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc. Natl. Acad. Sci. USA 2022, 119, e2114282119. [Google Scholar] [CrossRef]
- Lu, J.; Li, H.; Chen, Z.; Fan, L.; Feng, S.; Cai, X.; Wang, H. Identification of 3 subpopulations of tumor-infiltrating immune cells for malignant transformation of low-grade glioma. Cancer Cell Int. 2019, 19, 265. [Google Scholar] [CrossRef]
- Lee, S.J.; Kang, W.Y.; Yoon, Y.; Jin, J.Y.; Song, H.J.; Her, J.H.; Kang, S.M.; Hwang, Y.K.; Kang, K.J.; Joo, K.M.; et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer 2015, 15, 1011. [Google Scholar] [CrossRef]
- Baker, G.J.; Chockley, P.; Zamler, D.; Castro, M.G.; Lowenstein, P.R. Natural killer cells require monocytic Gr-1(+)/CD11b(+) myeloid cells to eradicate orthotopically engrafted glioma cells. Oncoimmunology 2016, 5, e1163461. [Google Scholar] [CrossRef]
- Mostafa, H.; Pala, A.; Hogel, J.; Hlavac, M.; Dietrich, E.; Westhoff, M.A.; Nonnenmacher, L.; Burster, T.; Georgieff, M.; Wirtz, C.R.; et al. Immune phenotypes predict survival in patients with glioblastoma multiforme. J. Hematol. Oncol. 2016, 9, 77. [Google Scholar] [CrossRef]
- Sharifzad, F.; Mardpour, S.; Mardpour, S.; Fakharian, E.; Taghikhani, A.; Sharifzad, A.; Kiani, S.; Heydarian, Y.; Los, M.J.; Azizi, Z.; et al. HSP70/IL-2 Treated NK Cells Effectively Cross the Blood Brain Barrier and Target Tumor Cells in a Rat Model of Induced Glioblastoma Multiforme (GBM). Int. J. Mol. Sci. 2020, 21, 2263. [Google Scholar] [CrossRef]
- De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2020, 10, 18. [Google Scholar] [CrossRef]
- Lin, Y.J.; Wu, C.Y.; Wu, J.Y.; Lim, M. The Role of Myeloid Cells in GBM Immunosuppression. Front. Immunol. 2022, 13, 887781. [Google Scholar] [CrossRef]
- Zhang, J.; Sarkar, S.; Cua, R.; Zhou, Y.; Hader, W.; Yong, V.W. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 2012, 33, 312–319. [Google Scholar] [CrossRef]
- Paulus, W.; Baur, I.; Huettner, C.; Schmausser, B.; Roggendorf, W.; Schlingensiepen, K.H.; Brysch, W. Effects of transforming growth factor-beta 1 on collagen synthesis, integrin expression, adhesion and invasion of glioma cells. J. Neuropathol. Exp. Neurol. 1995, 54, 236–244. [Google Scholar] [CrossRef]
- Zeiner, P.S.; Preusse, C.; Blank, A.E.; Zachskorn, C.; Baumgarten, P.; Caspary, L.; Braczynski, A.K.; Weissenberger, J.; Bratzke, H.; Reiss, S.; et al. MIF Receptor CD74 is Restricted to Microglia/Macrophages, Associated with a M1-Polarized Immune Milieu and Prolonged Patient Survival in Gliomas. Brain Pathol. 2015, 25, 491–504. [Google Scholar] [CrossRef]
- Gjorgjevski, M.; Hannen, R.; Carl, B.; Li, Y.; Landmann, E.; Buchholz, M.; Bartsch, J.W.; Nimsky, C. Molecular profiling of the tumor microenvironment in glioblastoma patients: Correlation of microglia/macrophage polarization state with metalloprotease expression profiles and survival. Biosci. Rep. 2019, 39, BSR20182361. [Google Scholar] [CrossRef]
- Wang, G.; Zhong, K.; Wang, Z.; Zhang, Z.; Tang, X.; Tong, A.; Zhou, L. Tumor-associated microglia and macrophages in glioblastoma: From basic insights to therapeutic opportunities. Front. Immunol. 2022, 13, 964898. [Google Scholar] [CrossRef]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef]
- Ma, J.; Chen, C.C.; Li, M. Macrophages/Microglia in the Glioblastoma Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 5775. [Google Scholar] [CrossRef]
- Brandenburg, S.; Blank, A.; Bungert, A.D.; Vajkoczy, P. Distinction of Microglia and Macrophages in Glioblastoma: Close Relatives, Different Tasks? Int. J. Mol. Sci. 2020, 22, 194. [Google Scholar] [CrossRef]
- Vakilian, A.; Khorramdelazad, H.; Heidari, P.; Sheikh Rezaei, Z.; Hassanshahi, G. CCL2/CCR2 signaling pathway in glioblastoma multiforme. Neurochem. Int. 2017, 103, 1–7. [Google Scholar] [CrossRef]
- Li, F.; Lv, B.; Liu, Y.; Hua, T.; Han, J.; Sun, C.; Xu, L.; Zhang, Z.; Feng, Z.; Cai, Y.; et al. Blocking the CD47-SIRPalpha axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology 2018, 7, e1391973. [Google Scholar] [CrossRef]
- Hu, J.; Dong, F.; He, Y.; Xia, X.; Cheng, F.; Chen, S.; Hou, X.; Zhang, P.; Liu, G.; Li, Y.; et al. LRIG2 promotes glioblastoma progression by modulating innate antitumor immunity through macrophage infiltration and polarization. J. Immunother. Cancer 2022, 10, e004452. [Google Scholar] [CrossRef]
- Dumas, A.A.; Pomella, N.; Rosser, G.; Guglielmi, L.; Vinel, C.; Millner, T.O.; Rees, J.; Aley, N.; Sheer, D.; Wei, J.; et al. Microglia promote glioblastoma via mTOR-mediated immunosuppression of the tumour microenvironment. EMBO J. 2020, 39, e103790. [Google Scholar] [CrossRef]
- Maas, S.L.N.; Abels, E.R.; Van De Haar, L.L.; Zhang, X.; Morsett, L.; Sil, S.; Guedes, J.; Sen, P.; Prabhakar, S.; Hickman, S.E.; et al. Glioblastoma hijacks microglial gene expression to support tumor growth. J. Neuroinflamm. 2020, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Di Ianni, N.; Musio, S.; Pellegatta, S. Altered Metabolism in Glioblastoma: Myeloid-Derived Suppressor Cell (MDSC) Fitness and Tumor-Infiltrating Lymphocyte (TIL) Dysfunction. Int. J. Mol. Sci. 2021, 22, 4460. [Google Scholar] [CrossRef] [PubMed]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 2018, 3, e122264. [Google Scholar] [CrossRef]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.; Ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. 2016, 18, 1253–1264. [Google Scholar] [CrossRef]
- Kohanbash, G.; Okada, H. Myeloid-derived suppressor cells (MDSCs) in gliomas and glioma-development. Immunol. Investig. 2012, 41, 658–679. [Google Scholar] [CrossRef]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef]
- Brandau, S.; Trellakis, S.; Bruderek, K.; Schmaltz, D.; Steller, G.; Elian, M.; Suttmann, H.; Schenck, M.; Welling, J.; Zabel, P.; et al. Myeloid-derived suppressor cells in the peripheral blood of cancer patients contain a subset of immature neutrophils with impaired migratory properties. J. Leukoc. Biol. 2011, 89, 311–317. [Google Scholar] [CrossRef]
- Lang, S.; Bruderek, K.; Kaspar, C.; Hoing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B.; et al. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell Subsets. Clin. Cancer Res. 2018, 24, 4834–4844. [Google Scholar] [CrossRef]
- Ley, K.; Hoffman, H.M.; Kubes, P.; Cassatella, M.A.; Zychlinsky, A.; Hedrick, C.C.; Catz, S.D. Neutrophils: New insights and open questions. Sci. Immunol. 2018, 3, eaat4579. [Google Scholar] [CrossRef]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349–353. [Google Scholar] [CrossRef]
- Mahiddine, K.; Blaisdell, A.; Ma, S.; Crequer-Grandhomme, A.; Lowell, C.A.; Erlebacher, A. Relief of tumor hypoxia unleashes the tumoricidal potential of neutrophils. J. Clin. Investig. 2020, 130, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Morad, H.; Luqman, S.; Tan, C.H.; Swann, V.; McNaughton, P.A. TRPM2 ion channels steer neutrophils towards a source of hydrogen peroxide. Sci. Rep. 2021, 11, 9339. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Alieva, M.; van der Most, T.; Klazen, J.A.Z.; Vollmann-Zwerenz, A.; Hau, P.; Vrisekoop, N. Neutrophils Promote Glioblastoma Tumor Cell Migration after Biopsy. Cells 2022, 11, 2196. [Google Scholar] [CrossRef]
- Lei, Y.Y.; Li, Y.T.; Hu, Q.L.; Wang, J.; Sui, A.X. Prognostic impact of neutrophil-to-lymphocyte ratio in gliomas: A systematic review and meta-analysis. World J. Surg. Oncol. 2019, 17, 152. [Google Scholar] [CrossRef]
- D’Agostino, P.M.; Gottfried-Blackmore, A.; Anandasabapathy, N.; Bulloch, K. Brain dendritic cells: Biology and pathology. Acta Neuropathol. 2012, 124, 599–614. [Google Scholar] [CrossRef]
- Flores, C.T.; Wildes, T.J.; Drake, J.A.; Moore, G.L.; Dean, B.D.; Abraham, R.S.; Mitchell, D.A. Lin(−)CCR2(+) hematopoietic stem and progenitor cells overcome resistance to PD-1 blockade. Nat. Commun. 2018, 9, 4313. [Google Scholar] [CrossRef]
- Miao, X.; Chen, Y.; Hao, K.; Zheng, M.; Chen, B.; Li, K.; Wang, Y.; Zhang, W.; Zhang, Y.; Mou, X.; et al. CD103(+) Cell Growth Factor Flt3L Enhances the Efficacy of Immune Checkpoint Blockades in Murine Glioblastoma Model. Oncol. Res. 2018, 26, 173–182. [Google Scholar] [CrossRef]
- Garzon-Muvdi, T.; Theodros, D.; Luksik, A.S.; Maxwell, R.; Kim, E.; Jackson, C.M.; Belcaid, Z.; Ganguly, S.; Tyler, B.; Brem, H.; et al. Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget 2018, 9, 20681–20697. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- De Smedt, T.; Van Mechelen, M.; De Becker, G.; Urbain, J.; Leo, O.; Moser, M. Effect of interleukin-10 on dendritic cell maturation and function. Eur. J. Immunol. 1997, 27, 1229–1235. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Tsumura, H.; Miwa, M.; Inaba, K. Contrasting effects of TGF-beta 1 and TNF-alpha on the development of dendritic cells from progenitors in mouse bone marrow. Stem Cells 1997, 15, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Wick, W.; Weller, M. Malignant glioma biology: Role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc. Res. Tech. 2001, 52, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Hishii, M.; Sato, K.; Okumura, K. Selective expression of interleukin-10 gene within glioblastoma multiforme. Brain Res. 1994, 649, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wu, A.; Kong, L.Y.; Wang, Y.; Fuller, G.; Fokt, I.; Melillo, G.; Priebe, W.; Heimberger, A.B. Hypoxia potentiates glioma-mediated immunosuppression. PLoS ONE 2011, 6, e16195. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.A.; Tomaszewski, W.H.; Haskell-Mendoza, A.P.; Hotchkiss, K.M.; Singh, K.; Reedy, J.L.; Fecci, P.E.; Sampson, J.H.; Khasraw, M. Targeting Immunometabolism in Glioblastoma. Front. Oncol. 2021, 11, 696402. [Google Scholar] [CrossRef]
- Ding, X.C.; Wang, L.L.; Zhang, X.D.; Xu, J.L.; Li, P.F.; Liang, H.; Zhang, X.B.; Xie, L.; Zhou, Z.H.; Yang, J.; et al. The relationship between expression of PD-L1 and HIF-1alpha in glioma cells under hypoxia. J. Hematol. Oncol. 2021, 14, 92. [Google Scholar] [CrossRef]
- Rolhion, C.; Penault-Llorca, F.; Kemeny, J.L.; Lemaire, J.J.; Jullien, C.; Labit-Bouvier, C.; Finat-Duclos, F.; Verrelle, P. Interleukin-6 overexpression as a marker of malignancy in human gliomas. J. Neurosurg. 2001, 94, 97–101. [Google Scholar] [CrossRef]
- Brat, D.J.; Bellail, A.C.; Van Meir, E.G. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol. 2005, 7, 122–133. [Google Scholar] [CrossRef]
- Won, W.J.; Deshane, J.S.; Leavenworth, J.W.; Oliva, C.R.; Griguer, C.E. Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress 2019, 3, 47–65. [Google Scholar] [CrossRef]
- Siska, P.J.; Rathmell, J.C. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015, 36, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; van der Windt, G.J.; Kishton, R.J.; Cohen, S.; Eisner, W.; MacIver, N.J.; Kater, A.P.; Weinberg, J.B.; Rathmell, J.C. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J. Immunol. 2016, 197, 2532–2540. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Kesarwani, P.; Prabhu, A.; Kant, S.; Kumar, P.; Graham, S.F.; Buelow, K.L.; Wilson, G.D.; Miller, C.R.; Chinnaiyan, P. Tryptophan Metabolism Contributes to Radiation-Induced Immune Checkpoint Reactivation in Glioblastoma. Clin. Cancer Res. 2018, 24, 3632–3643. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Chen, S.; Zhang, P.; Guo, D.; Wang, B. Targeted Arginine Metabolism Therapy: A Dilemma in Glioma Treatment. Front. Oncol. 2022, 12, 938847. [Google Scholar] [CrossRef]
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef]
- Hajji, N.; Garcia-Revilla, J.; Soto, M.S.; Perryman, R.; Symington, J.; Quarles, C.C.; Healey, D.R.; Guo, Y.; Orta-Vazquez, M.L.; Mateos-Cordero, S.; et al. Arginine deprivation alters microglial polarity and synergizes with radiation to eradicate non-arginine-auxotrophic glioblastoma tumors. J. Clin. Investig. 2022, 132, e142137. [Google Scholar] [CrossRef]
- Ma, Q.; Schlegel, F.; Bachmann, S.B.; Schneider, H.; Decker, Y.; Rudin, M.; Weller, M.; Proulx, S.T.; Detmar, M. Lymphatic outflow of cerebrospinal fluid is reduced in glioma. Sci. Rep. 2019, 9, 14815. [Google Scholar] [CrossRef]
- Toh, C.H.; Siow, T.Y. Factors Associated With Dysfunction of Glymphatic System in Patients With Glioma. Front. Oncol. 2021, 11, 744318. [Google Scholar] [CrossRef]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef] [PubMed]
- Ayasoufi, K.; Pfaller, C.K.; Evgin, L.; Khadka, R.H.; Tritz, Z.P.; Goddery, E.N.; Fain, C.E.; Yokanovich, L.T.; Himes, B.T.; Jin, F.; et al. Brain cancer induces systemic immunosuppression through release of non-steroid soluble mediators. Brain 2020, 143, 3629–3652. [Google Scholar] [CrossRef] [PubMed]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, M.P.; Lin, Y.; New, K.C.; Bulur, P.A.; O’Neill, B.P.; Gastineau, D.A.; Dietz, A.B. Systemic immune suppression in glioblastoma: The interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010, 12, 631–644. [Google Scholar] [CrossRef]
- Haddad, A.F.; Young, J.S.; Oh, J.Y.; Okada, H.; Aghi, M.K. The immunology of low-grade gliomas. Neurosurg. Focus 2022, 52, E2. [Google Scholar] [CrossRef]
- Yang, Y.; Tian, Y.; Li, Q.; Jiang, R.; Zhang, J. Uncovering the Immune Cell Infiltration Landscape in Low-Grade Glioma for Aiding Immunotherapy. J. Oncol. 2022, 2022, 3370727. [Google Scholar] [CrossRef]
- Weenink, B.; Draaisma, K.; Ooi, H.Z.; Kros, J.M.; Sillevis Smitt, P.A.E.; Debets, R.; French, P.J. Low-grade glioma harbors few CD8 T cells, which is accompanied by decreased expression of chemo-attractants, not immunogenic antigens. Sci. Rep. 2019, 9, 14643. [Google Scholar] [CrossRef]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wohrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015, 17, 1064–1075. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Preusser, M. In search of a target: PD-1 and PD-L1 profiling across glioma types. Neuro Oncol. 2016, 18, 1331–1332. [Google Scholar] [CrossRef]
- Garber, S.T.; Hashimoto, Y.; Weathers, S.P.; Xiu, J.; Gatalica, Z.; Verhaak, R.G.; Zhou, S.; Fuller, G.N.; Khasraw, M.; de Groot, J.; et al. Immune checkpoint blockade as a potential therapeutic target: Surveying CNS malignancies. Neuro Oncol. 2016, 18, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Zhao, Q.; Huang, L.; Zheng, Y.; Li, L.; He, Q.; Zhang, C.; Li, F.; Maimela, N.R.; Sun, Z.; et al. The R132H mutation in IDH1 promotes the recruitment of NK cells through CX3CL1/CX3CR1 chemotaxis and is correlated with a better prognosis in gliomas. Immunol. Cell Biol. 2019, 97, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Shi, F.D.; Jung, S.; Pien, G.C.; Wang, J.; Salazar-Mather, T.P.; He, T.T.; Weaver, J.T.; Ljunggren, H.G.; Biron, C.A.; et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006, 20, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Vidyarthi, A.; Agnihotri, T.; Khan, N.; Singh, S.; Tewari, M.K.; Radotra, B.D.; Chatterjee, D.; Agrewala, J.N. Predominance of M2 macrophages in gliomas leads to the suppression of local and systemic immunity. Cancer Immunol. Immunother. 2019, 68, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [PubMed]
- Buerki, R.A.; Horbinski, C.M.; Kruser, T.; Horowitz, P.M.; James, C.D.; Lukas, R.V. An overview of meningiomas. Future Oncol. 2018, 14, 2161–2177. [Google Scholar] [CrossRef]
- Borch, J.S.; Haslund-Vinding, J.; Vilhardt, F.; Maier, A.D.; Mathiesen, T. Meningioma-Brain Crosstalk: A Scoping Review. Cancers 2021, 13, 4267. [Google Scholar] [CrossRef]
- Materi, J.; Mampre, D.; Ehresman, J.; Rincon-Torroella, J.; Chaichana, K.L. Predictors of recurrence and high growth rate of residual meningiomas after subtotal resection. J. Neurosurg. 2020, 134, 410–416. [Google Scholar] [CrossRef]
- Garzon-Muvdi, T.; Bailey, D.D.; Pernik, M.N.; Pan, E. Basis for Immunotherapy for Treatment of Meningiomas. Front. Neurol. 2020, 11, 945. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Kannapadi, N.V.; Shah, P.P.; Mathios, D.; Jackson, C.M. Synthesizing Molecular and Immune Characteristics to Move Beyond WHO Grade in Meningiomas: A Focused Review. Front. Oncol. 2022, 12, 892004. [Google Scholar] [CrossRef] [PubMed]
- Rapp, C.; Dettling, S.; Liu, F.; Ull, A.T.; Warta, R.; Jungk, C.; Roesch, S.; Mock, A.; Sahm, F.; Schmidt, M.; et al. Cytotoxic T Cells and their Activation Status are Independent Prognostic Markers in Meningiomas. Clin. Cancer Res. 2019, 25, 5260–5270. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kresl, P.; Rajky, O.; Widhalm, G.; Ricken, G.; Hainfellner, J.A.; Marosi, C.; Birner, P.; Preusser, M. Analysis of the inflammatory tumor microenvironment in meningeal neoplasms. Clin. Neuropathol. 2020, 39, 256–262. [Google Scholar] [CrossRef]
- Fang, L.; Lowther, D.E.; Meizlish, M.L.; Anderson, R.C.; Bruce, J.N.; Devine, L.; Huttner, A.J.; Kleinstein, S.H.; Lee, J.Y.; Stern, J.N.; et al. The immune cell infiltrate populating meningiomas is composed of mature, antigen-experienced T and B cells. Neuro Oncol. 2013, 15, 1479–1490. [Google Scholar] [CrossRef]
- Zador, Z.; Landry, A.P.; Balas, M.; Cusimano, M.D. Landscape of immune cell gene expression is unique in predominantly WHO grade 1 skull base meningiomas when compared to convexity. Sci. Rep. 2020, 10, 9065. [Google Scholar] [CrossRef] [PubMed]
- Proctor, D.T.; Huang, J.; Lama, S.; Albakr, A.; Van Marle, G.; Sutherland, G.R. Tumor-associated macrophage infiltration in meningioma. Neurooncol. Adv. 2019, 1, vdz018. [Google Scholar] [CrossRef]
- Domingues, P.H.; Teodosio, C.; Otero, A.; Sousa, P.; Ortiz, J.; Macias Mdel, C.; Goncalves, J.M.; Nieto, A.B.; Lopes, M.C.; de Oliveira, C.; et al. Association between inflammatory infiltrates and isolated monosomy 22/del(22q) in meningiomas. PLoS ONE 2013, 8, e74798. [Google Scholar] [CrossRef]
- Morantz, R.A.; Wood, G.W.; Foster, M.; Clark, M.; Gollahon, K. Macrophages in experimental and human brain tumors. Part 2: Studies of the macrophage content of human brain tumors. J. Neurosurg. 1979, 50, 305–311. [Google Scholar] [CrossRef]
- Asai, J.; Suzuki, R.; Fujimoto, T.; Suzuki, T.; Nakagawa, N.; Nagashima, G.; Miyo, T.; Hokaku, H.; Takei, A. Fluorescence automatic cell sorter and immunohistochemical investigation of CD68-positive cells in meningioma. Clin. Neurol. Neurosurg. 1999, 101, 229–234. [Google Scholar] [CrossRef]
- Presta, I.; Guadagno, E.; Di Vito, A.; Malara, N.; Mignogna, C.; Maisano, D.; Donato, A.; Cardillo, G.; Del Basso De Caro, M.L.; Donato, G. Innate immunity may play a role in growth and relapse of chordoid meningioma. Int. J. Immunopathol. Pharmacol. 2017, 30, 429–433. [Google Scholar] [CrossRef]
- Grund, S.; Schittenhelm, J.; Roser, F.; Tatagiba, M.; Mawrin, C.; Kim, Y.J.; Bornemann, A. The microglial/macrophagic response at the tumour-brain border of invasive meningiomas. Neuropathol. Appl. Neurobiol. 2009, 35, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.D.; Veliceasa, D.; Lamano, J.B.; Lamano, J.B.; Kaur, G.; Biyashev, D.; Horbinski, C.M.; Kruser, T.J.; Bloch, O. Systemic and local immunosuppression in patients with high-grade meningiomas. Cancer Immunol. Immunother. 2019, 68, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Pinton, L.; Solito, S.; Masetto, E.; Vettore, M.; Cane, S.; Puppa, A.D.; Mandruzzato, S. Immunosuppressive activity of tumor-infiltrating myeloid cells in patients with meningioma. Oncoimmunology 2018, 7, e1440931. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tian, F.; Lun, P.; Feng, Y. Profiles of immune infiltration and its relevance to survival outcome in meningiomas. Biosci. Rep. 2020, 40, BSR20200538. [Google Scholar] [CrossRef]
- Reszec, J.; Hermanowicz, A.; Kochanowicz, J.; Turek, G.; Mariak, Z.; Chyczewski, L. Mast cells evaluation in meningioma of various grades. Folia Histochem. Cytobiol. 2012, 50, 542–546. [Google Scholar] [CrossRef]
- Reszec, J.; Hermanowicz, A.; Rutkowski, R.; Bernaczyk, P.; Mariak, Z.; Chyczewski, L. Evaluation of mast cells and hypoxia inducible factor-1 expression in meningiomas of various grades in correlation with peritumoral brain edema. J. Neurooncol. 2013, 115, 119–125. [Google Scholar] [CrossRef]
- Schober, R.; Himuro, H.; Wechsler, W. Cystic changes and vascular permeability in meningiomas. Clin. Neuropathol. 1988, 7, 16–21. [Google Scholar]
- Li, G.; Hattermann, K.; Mentlein, R.; Mehdorn, H.M.; Held-Feindt, J. The transmembrane chemokines CXCL16 and CX3CL1 and their receptors are expressed in human meningiomas. Oncol. Rep. 2013, 29, 563–570. [Google Scholar] [CrossRef]
- Wang, S.; Liechty, B.; Patel, S.; Weber, J.S.; Hollmann, T.J.; Snuderl, M.; Karajannis, M.A. Programmed death ligand 1 expression and tumor infiltrating lymphocytes in neurofibromatosis type 1 and 2 associated tumors. J. Neurooncol. 2018, 138, 183–190. [Google Scholar] [CrossRef]
- Kosugi, K.; Tamura, R.; Ohara, K.; Morimoto, Y.; Kuranari, Y.; Oishi, Y.; Yoshida, K.; Toda, M. Immunological and vascular characteristics in cavernous sinus meningioma. J. Clin. Neurosci. 2019, 67, 198–203. [Google Scholar] [CrossRef]
- Sato, K.; Kuratsu, J.; Takeshima, H.; Yoshimura, T.; Ushio, Y. Expression of monocyte chemoattractant protein-1 in meningioma. J. Neurosurg. 1995, 82, 874–878. [Google Scholar] [CrossRef]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Jabini, R.; Moradi, A.; Afsharnezhad, S.; Ayatollahi, H.; Behravan, J.; Raziee, H.R.; Mosaffa, F. Pathodiagnostic parameters and evaluation of O(6)- methyl guanine methyl transferase gene promoter methylation in meningiomas. Gene 2014, 538, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Polyzoidis, S.; Koletsa, T.; Panagiotidou, S.; Ashkan, K.; Theoharides, T.C. Mast cells in meningiomas and brain inflammation. J. Neuroinflamm. 2015, 12, 170. [Google Scholar] [CrossRef]
- Tirakotai, W.; Mennel, H.D.; Celik, I.; Hellwig, D.; Bertalanffy, H.; Riegel, T. Secretory meningioma: Immunohistochemical findings and evaluation of mast cell infiltration. Neurosurg. Rev. 2006, 29, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Tirakotai, W.; Mennel, H.D.; Lapanich, S.; Sure, U.; Bertalanffy, H.; Celik, I. The Possible Role of Mast Cells and VEGF in Peritumoural Oedema of Secretory Meningioma. J. Med. Assoc. Thail. 2016, 99 (Suppl. 3), S8–S115. [Google Scholar]
- Park, K.J.; Kang, S.H.; Chae, Y.S.; Yu, M.O.; Cho, T.H.; Suh, J.K.; Lee, H.K.; Chung, Y.G. Influence of interleukin-6 on the development of peritumoral brain edema in meningiomas. J. Neurosurg. 2010, 112, 73–80. [Google Scholar] [CrossRef]
- Toh, C.H.; Siow, T.Y.; Castillo, M. Peritumoral Brain Edema in Meningiomas May Be Related to Glymphatic Dysfunction. Front. Neurosci. 2021, 15, 674898. [Google Scholar] [CrossRef]
- George, B.; Bresson, D.; Herman, P.; Froelich, S. Chordomas: A Review. Neurosurg. Clin. N. Am. 2015, 26, 437–452. [Google Scholar] [CrossRef]
- Dridi, M.; Krebs-Drouot, L.; Meyronet, D.; Dumollard, J.M.; Vassal, F.; Jouanneau, E.; Jacquesson, T.; Barrey, C.; Grange, S.; Boutonnat, J.; et al. The Immune Microenvironment of Chordomas: An Immunohistochemical Analysis. Cancers 2021, 13, 3335. [Google Scholar] [CrossRef] [PubMed]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology 2020, 9, 1747340. [Google Scholar] [CrossRef] [PubMed]
- Mathios, D.; Ruzevick, J.; Jackson, C.M.; Xu, H.; Shah, S.R.; Taube, J.M.; Burger, P.C.; McCarthy, E.F.; Quinones-Hinojosa, A.; Pardoll, D.M.; et al. PD-1, PD-L1, PD-L2 expression in the chordoma microenvironment. J. Neurooncol. 2015, 121, 251–259. [Google Scholar] [CrossRef]
- Zou, M.X.; Peng, A.B.; Lv, G.H.; Wang, X.B.; Li, J.; She, X.L.; Jiang, Y. Expression of programmed death-1 ligand (PD-L1) in tumor-infiltrating lymphocytes is associated with favorable spinal chordoma prognosis. Am. J. Transl. Res. 2016, 8, 3274–3287. [Google Scholar] [PubMed]
- Feng, Y.; Shen, J.; Gao, Y.; Liao, Y.; Cote, G.; Choy, E.; Chebib, I.; Mankin, H.; Hornicek, F.; Duan, Z. Expression of programmed cell death ligand 1 (PD-L1) and prevalence of tumor-infiltrating lymphocytes (TILs) in chordoma. Oncotarget 2015, 6, 11139–11149. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.Q.; Adam, B.; Jack, A.S.; Lam, A.; Broad, R.W.; Chik, C.L. Immune Cell Infiltrates in Pituitary Adenomas: More Macrophages in Larger Adenomas and More T Cells in Growth Hormone Adenomas. Endocr. Pathol. 2015, 26, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Yagnik, G.; Rutowski, M.J.; Shah, S.S.; Aghi, M.K. Stratifying nonfunctional pituitary adenomas into two groups distinguished by macrophage subtypes. Oncotarget 2019, 10, 2212–2223. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Jacob, J.B.; Jacob, M.K.; Parajuli, P. Review of immune checkpoint inhibitors in immuno-oncology. Adv. Pharmacol. 2021, 91, 111–139. [Google Scholar] [CrossRef]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef]
- Huang, B.Y.; Zhan, Y.P.; Zong, W.J.; Yu, C.J.; Li, J.F.; Qu, Y.M.; Han, S. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS ONE 2015, 10, e0134715. [Google Scholar] [CrossRef]
- Mathios, D.; Kim, J.E.; Mangraviti, A.; Phallen, J.; Park, C.K.; Jackson, C.M.; Garzon-Muvdi, T.; Kim, E.; Theodros, D.; Polanczyk, M.; et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 2016, 8, 370ra180. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Sun, L.; Mochizuki, A.Y.; Reynoso, J.G.; Orpilla, J.; Chow, F.; Kienzler, J.C.; Everson, R.G.; Nathanson, D.A.; Bensinger, S.J.; et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat. Commun. 2021, 12, 6938. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Omuro, A.; Reardon, D.A.; Sampson, J.H.; Baehring, J.; Sahebjam, S.; Cloughesy, T.F.; Chalamandaris, A.G.; Potter, V.; Butowski, N.; Lim, M. Nivolumab plus radiotherapy with or without temozolomide in newly diagnosed glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neurooncol. Adv. 2022, 4, vdac025. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase 3 Trial of Chemoradiotherapy With Temozolomide Plus Nivolumab or Placebo for Newly Diagnosed Glioblastoma With Methylated MGMT Promoter. Neuro Oncol. 2022, 24, 1395–1949. [Google Scholar] [CrossRef] [PubMed]
- Riviere, P.; Goodman, A.M.; Okamura, R.; Barkauskas, D.A.; Whitchurch, T.J.; Lee, S.; Khalid, N.; Collier, R.; Mareboina, M.; Frampton, G.M.; et al. High Tumor Mutational Burden Correlates with Longer Survival in Immunotherapy-Naive Patients with Diverse Cancers. Mol. Cancer Ther. 2020, 19, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021, 184, 596–614.e14. [Google Scholar] [CrossRef]
- Takamatsu, S.; Hamanishi, J.; Brown, J.B.; Yamaguchi, K.; Yamanoi, K.; Murakami, K.; Gotoh, O.; Mori, S.; Mandai, M.; Matsumura, N. Mutation burden-orthogonal tumor genomic subtypes delineate responses to immune checkpoint therapy. J. Immunother. Cancer 2022, 10, e004831. [Google Scholar] [CrossRef]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef]
- Dunn, I.F.; Du, Z.; Touat, M.; Sisti, M.B.; Wen, P.Y.; Umeton, R.; Dubuc, A.M.; Ducar, M.; Canoll, P.D.; Severson, E.; et al. Mismatch repair deficiency in high-grade meningioma: A rare but recurrent event associated with dramatic immune activation and clinical response to PD-1 blockade. JCO Precis. Oncol. 2018, 2018, PO.18.00190. [Google Scholar] [CrossRef] [PubMed]
- Johanns, T.M.; Miller, C.A.; Dorward, I.G.; Tsien, C.; Chang, E.; Perry, A.; Uppaluri, R.; Ferguson, C.; Schmidt, R.E.; Dahiya, S.; et al. Immunogenomics of Hypermutated Glioblastoma: A Patient with Germline POLE Deficiency Treated with Checkpoint Blockade Immunotherapy. Cancer Discov. 2016, 6, 1230–1236. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Fecci, P.E.; Ochiai, H.; Mitchell, D.A.; Grossi, P.M.; Sweeney, A.E.; Archer, G.E.; Cummings, T.; Allison, J.P.; Bigner, D.D.; Sampson, J.H. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin. Cancer Res. 2007, 13, 2158–2167. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef]
- Mair, M.J.; Kiesel, B.; Feldmann, K.; Widhalm, G.; Dieckmann, K.; Wohrer, A.; Mullauer, L.; Preusser, M.; Berghoff, A.S. LAG-3 expression in the inflammatory microenvironment of glioma. J. Neurooncol. 2021, 152, 533–539. [Google Scholar] [CrossRef]
- Ladomersky, E.; Zhai, L.; Lenzen, A.; Lauing, K.L.; Qian, J.; Scholtens, D.M.; Gritsina, G.; Sun, X.; Liu, Y.; Yu, F.; et al. IDO1 Inhibition Synergizes with Radiation and PD-1 Blockade to Durably Increase Survival Against Advanced Glioblastoma. Clin. Cancer Res. 2018, 24, 2559–2573. [Google Scholar] [CrossRef]
- Ghouzlani, A.; Kandoussi, S.; Tall, M.; Reddy, K.P.; Rafii, S.; Badou, A. Immune Checkpoint Inhibitors in Human Glioma Microenvironment. Front. Immunol. 2021, 12, 679425. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, L.; Ye, M.; Kang, C.; You, H. Treatment Progress of Immune Checkpoint Blockade Therapy for Glioblastoma. Front. Immunol. 2020, 11, 592612. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Rhodin, K.E.; Dechant, C.; Cui, X.; Chongsathidkiet, P.; Wilkinson, D.; Waibl-Polania, J.; Sanchez-Perez, L.; Fecci, P.E. 4-1BB Agonism Averts TIL Exhaustion and Licenses PD-1 Blockade in Glioblastoma and Other Intracranial Cancers. Clin. Cancer Res. 2020, 26, 1349–1358. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Belcaid, Z.; Phallen, J.A.; Zeng, J.; See, A.P.; Mathios, D.; Gottschalk, C.; Nicholas, S.; Kellett, M.; Ruzevick, J.; Jackson, C.; et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLoS ONE 2014, 9, e101764. [Google Scholar] [CrossRef]
- Giles, A.J.; Hao, S.; Padget, M.; Song, H.; Zhang, W.; Lynes, J.; Sanchez, V.; Liu, Y.; Jung, J.; Cao, X.; et al. Efficient ADCC killing of meningioma by avelumab and a high-affinity natural killer cell line, haNK. JCI Insight 2019, 4, e130688. [Google Scholar] [CrossRef] [PubMed]
- Gelerstein, E.; Berger, A.; Jonas-Kimchi, T.; Strauss, I.; Kanner, A.A.; Blumenthal, D.T.; Gottfried, M.; Margalit, N.; Ram, Z.; Shahar, T. Regression of intracranial meningioma following treatment with nivolumab: Case report and review of the literature. J. Clin. Neurosci. 2017, 37, 51–53. [Google Scholar] [CrossRef]
- Nidamanuri, P.; Drappatz, J. Immune checkpoint inhibitor therapy for recurrent meningiomas: A retrospective chart review. J. Neurooncol. 2022, 157, 271–276. [Google Scholar] [CrossRef]
- Bi, W.L.; Nayak, L.; Meredith, D.M.; Driver, J.; Du, Z.; Hoffman, S.; Li, Y.; Lee, E.Q.; Beroukhim, R.; Rinne, M.; et al. Activity of PD-1 blockade with nivolumab among patients with recurrent atypical/anaplastic meningioma: Phase II trial results. Neuro Oncol. 2022, 24, 101–113. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Zhao, T.; Li, C.; Ge, H.; Lin, Y.; Kang, D. Glioblastoma vaccine tumor therapy research progress. Chin. Neurosurg. J. 2022, 8, 2. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; O’Rourke, D.M.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; et al. Rindopepimut with Bevacizumab for Patients with Relapsed EGFRvIII-Expressing Glioblastoma (ReACT): Results of a Double-Blind Randomized Phase II Trial. Clin. Cancer Res. 2020, 26, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Cao, T.Q.; Wainwright, D.A.; Lee-Chang, C.; Miska, J.; Sonabend, A.M.; Heimberger, A.B.; Lukas, R.V. Next Steps for Immunotherapy in Glioblastoma. Cancers 2022, 14, 4023. [Google Scholar] [CrossRef]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807. [Google Scholar] [CrossRef]
- Narita, Y.; Okita, Y.; Arakawa, Y. Evaluation of the efficacy and safety of TAS0313 in adults with recurrent glioblastoma. Cancer Immunol. Immunother. 2022, 71, 2703–2715. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Dutoit, V.; Allard, M.; Grandjean Hallez, N.; Marinari, E.; Widmer, V.; Philippin, G.; Corlazzoli, F.; Gustave, R.; Kreutzfeldt, M.; et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro Oncol. 2019, 21, 923–933. [Google Scholar] [CrossRef]
- Rampling, R.; Peoples, S.; Mulholland, P.J.; James, A.; Al-Salihi, O.; Twelves, C.J.; McBain, C.; Jefferies, S.; Jackson, A.; Stewart, W.; et al. A Cancer Research UK First Time in Human Phase I Trial of IMA950 (Novel Multipeptide Therapeutic Vaccine) in Patients with Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2016, 22, 4776–4785. [Google Scholar] [CrossRef]
- Boydell, E.; Marinari, E.; Migliorini, D.; Dietrich, P.Y.; Patrikidou, A.; Dutoit, V. Exploratory Study of the Effect of IMA950/Poly-ICLC Vaccination on Response to Bevacizumab in Relapsing High-Grade Glioma Patients. Cancers 2019, 11, 464. [Google Scholar] [CrossRef]
- Ogino, H.; Taylor, J.W.; Nejo, T.; Gibson, D.; Watchmaker, P.B.; Okada, K.; Saijo, A.; Tedesco, M.R.; Shai, A.; Wong, C.M.; et al. Randomized trial of neoadjuvant vaccination with tumor-cell lysate induces T cell response in low-grade gliomas. J. Clin. Investig. 2022, 132, e151239. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F.; et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin. Cancer Res. 2005, 11, 5515–5525. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosci. 2021, 15, 662064. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Tang, X.; Liu, F.; Liu, Z.; Cao, Y.; Zhang, Z.; Wang, Y.; Huang, J.; Fan, S.; Zhao, S.; Chen, Y.; et al. Bioactivity and safety of B7-H3-targeted chimeric antigen receptor T cells against anaplastic meningioma. Clin. Transl. Immunol. 2020, 9, e1137. [Google Scholar] [CrossRef]
- Long, C.; Li, G.; Zhang, C.; Jiang, T.; Li, Y.; Duan, X.; Zhong, G. B7-H3 as a Target for CAR-T Cell Therapy in Skull Base Chordoma. Front. Oncol. 2021, 11, 659662. [Google Scholar] [CrossRef] [PubMed]
- Danilova, L.; Anagnostou, V.; Caushi, J.X.; Sidhom, J.W.; Guo, H.; Chan, H.Y.; Suri, P.; Tam, A.; Zhang, J.; Asmar, M.E.; et al. The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Cardon, T.; Aboulouard, S.; Hajjaji, N.; Kobeissy, F.; Duhamel, M.; Fournier, I.; Salzet, M. Surfaceome Proteomic of Glioblastoma Revealed Potential Targets for Immunotherapy. Front. Immunol. 2021, 12, 746168. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.H.B.; Leon, A.J.; Hui, W.; Lee, S.C.; Batruch, I.; Faust, K.; Klekner, A.; Hutoczki, G.; Koritzinsky, M.; Richer, M.; et al. Topographic mapping of the glioblastoma proteome reveals a triple-axis model of intra-tumoral heterogeneity. Nat. Commun. 2022, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Stafford, J.H.; Hirai, T.; Deng, L.; Chernikova, S.B.; Urata, K.; West, B.L.; Brown, J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. 2016, 18, 797–806. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016, 18, 557–564. [Google Scholar] [CrossRef]
- Gyori, D.; Lim, E.L.; Grant, F.M.; Spensberger, D.; Roychoudhuri, R.; Shuttleworth, S.J.; Okkenhaug, K.; Stephens, L.R.; Hawkins, P.T. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight 2018, 3, e120631. [Google Scholar] [CrossRef]
- Barca, C.; Foray, C.; Hermann, S.; Herrlinger, U.; Remory, I.; Laoui, D.; Schafers, M.; Grauer, O.M.; Zinnhardt, B.; Jacobs, A.H. The Colony Stimulating Factor-1 Receptor (CSF-1R)-Mediated Regulation of Microglia/Macrophages as a Target for Neurological Disorders (Glioma, Stroke). Front. Immunol. 2021, 12, 787307. [Google Scholar] [CrossRef]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668. [Google Scholar] [CrossRef]
- Przystal, J.M.; Becker, H.; Canjuga, D.; Tsiami, F.; Anderle, N.; Keller, A.L.; Pohl, A.; Ries, C.H.; Schmittnaegel, M.; Korinetska, N.; et al. Targeting CSF1R Alone or in Combination with PD1 in Experimental Glioma. Cancers 2021, 13, 2400. [Google Scholar] [CrossRef]
- Ding, A.S.; Routkevitch, D.; Jackson, C.; Lim, M. Targeting Myeloid Cells in Combination Treatments for Glioma and Other Tumors. Front. Immunol. 2019, 10, 1715. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ding, H.; Li, Z.; Peng, Y.; Tan, H.; Wang, C.; Huang, G.; Li, W.; Ma, G.; Wei, W. Exploration and functionalization of M1-macrophage extracellular vesicles for effective accumulation in glioblastoma and strong synergistic therapeutic effects. Signal Transduct. Target. Ther. 2022, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.; Yaghoobi, V.; Miyagishima, D.; Vesely, M.D.; Zhang, T.; Badri, T.; Nassar, A.; Han, X.; Sanmamed, M.F.; Youngblood, M.; et al. Targeting the CSF1/CSF1R axis is a potential treatment strategy for malignant meningiomas. Neuro Oncol. 2021, 23, 1922–1935. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).