
Glycoproteomics in Cerebrospinal Fluid Reveals Brain-Specific Glycosylation Changes

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
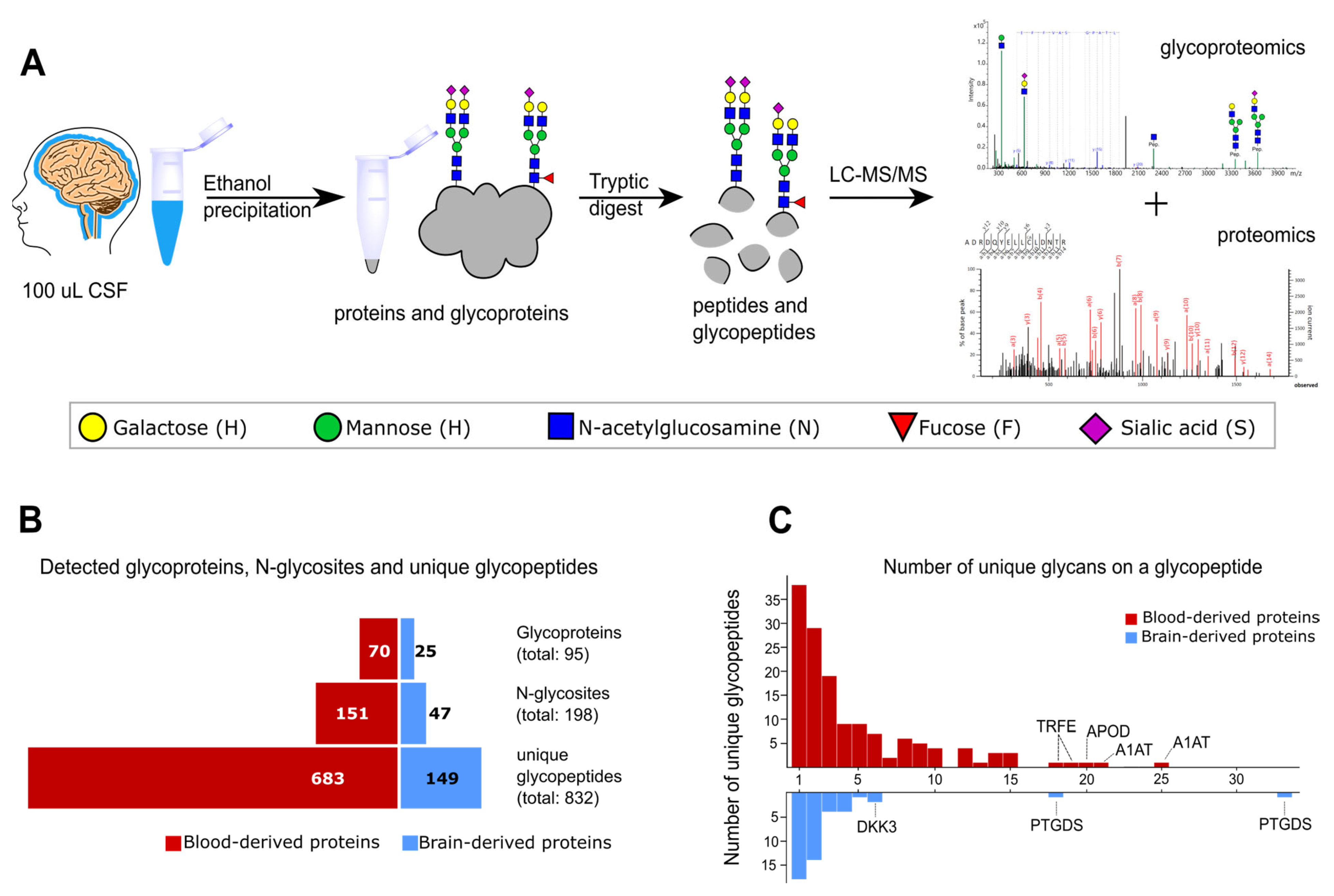
2.1. Glycoproteomics Identification in Non-Enriched CSF Samples
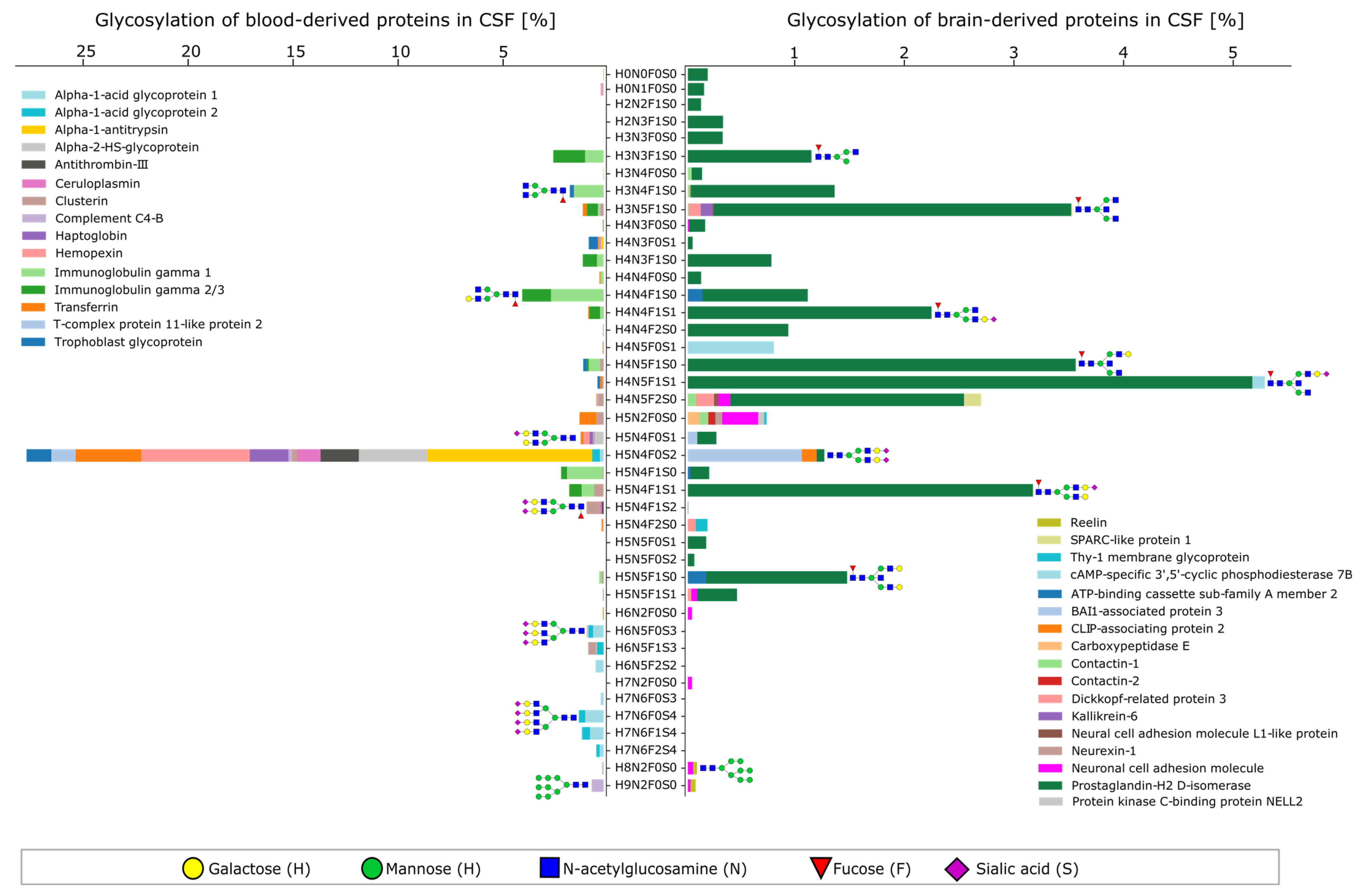
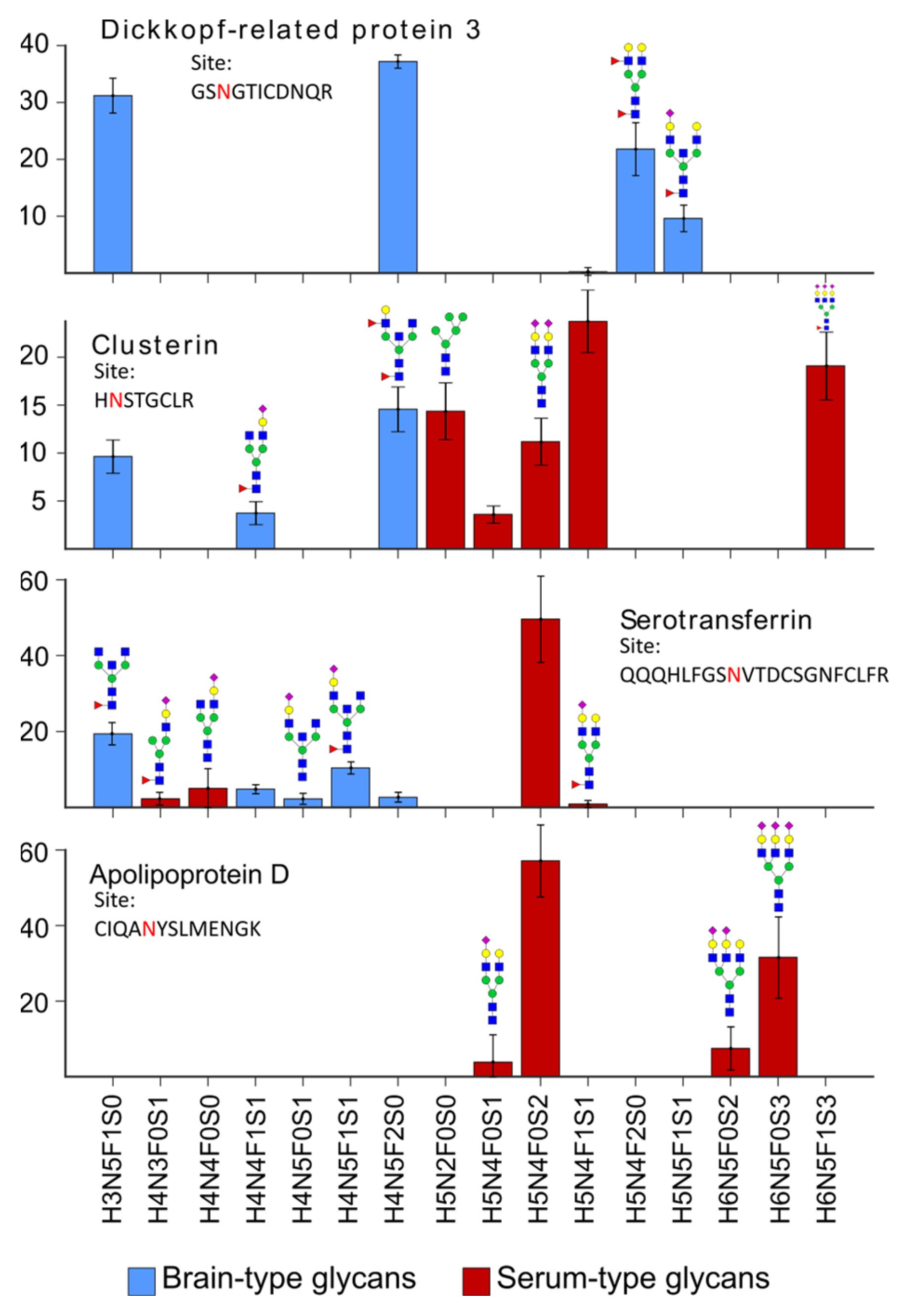
2.2. Characteristics of the CSF Glycoproteome in Healthy Individuals
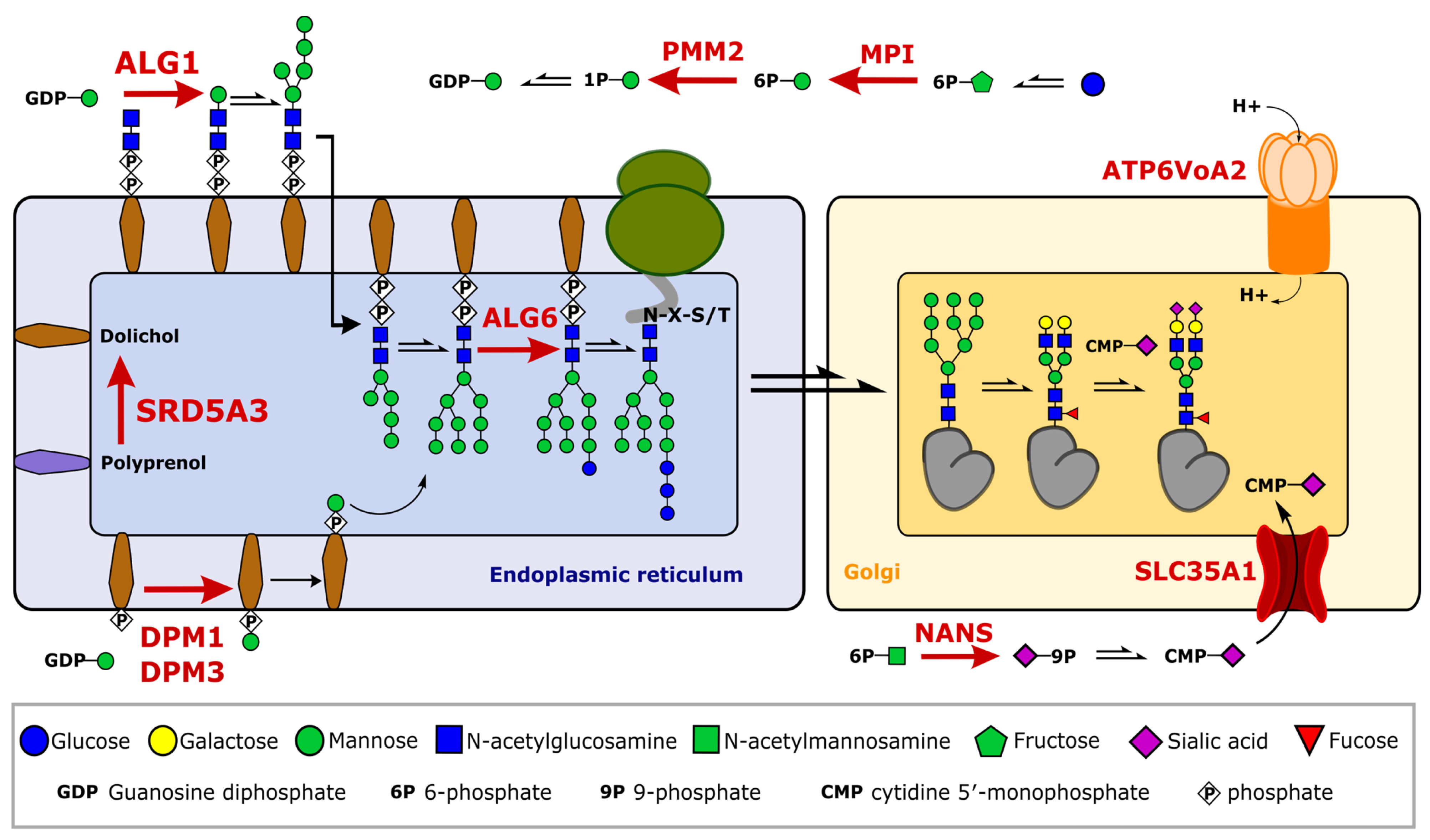
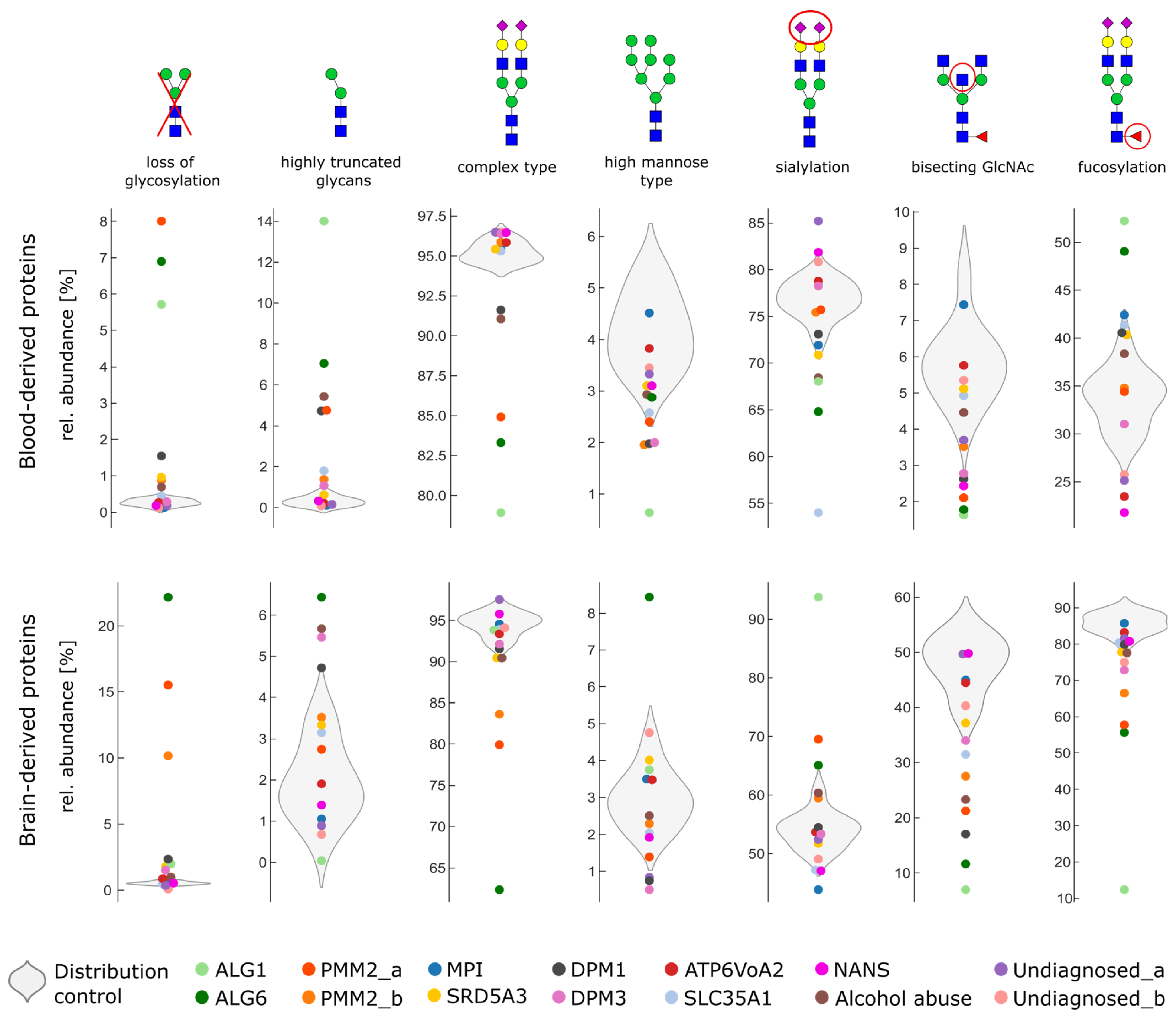
2.3. Glycosylation Changes in CDG Patients
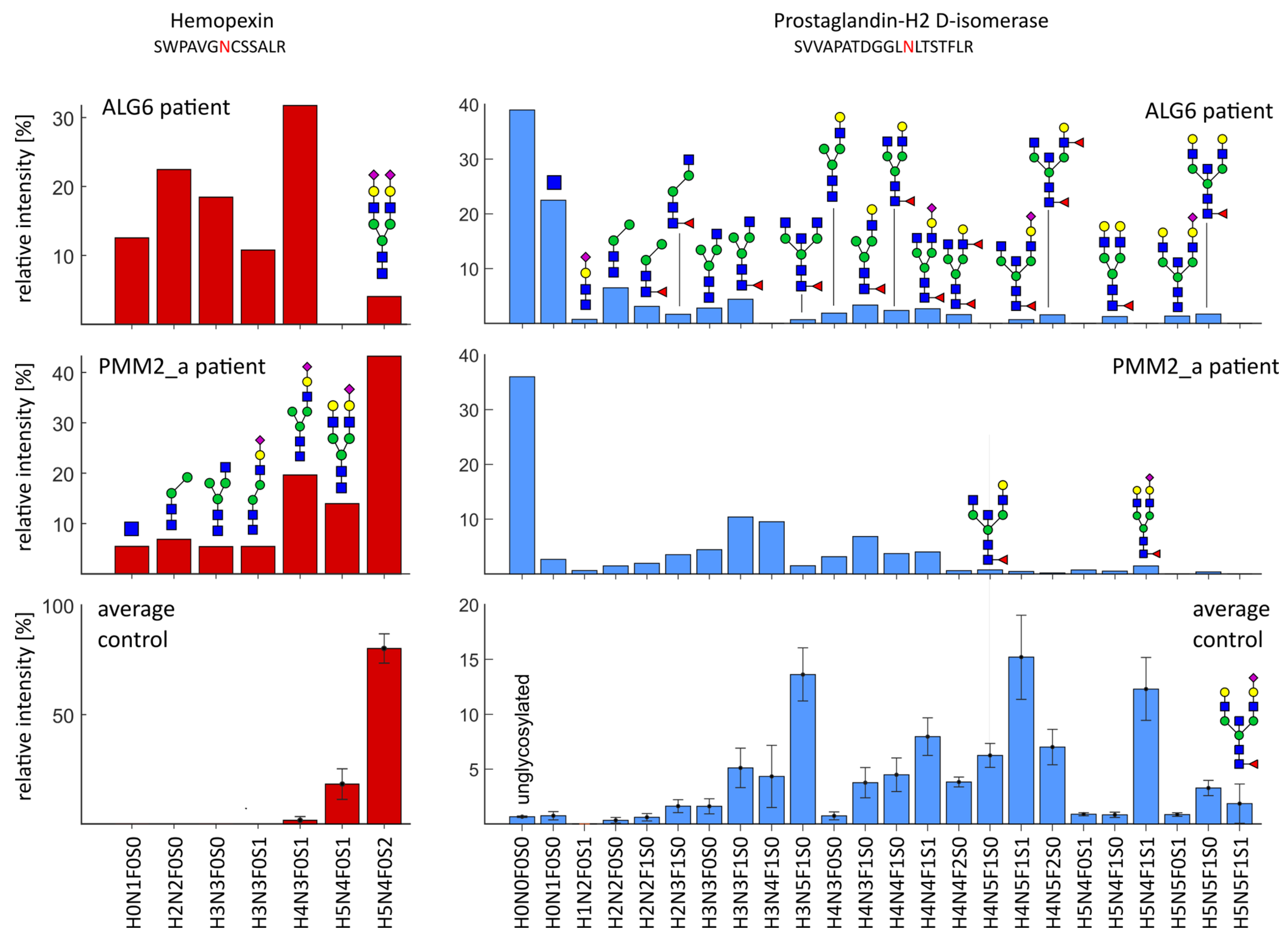
2.4. Severe Changes in Glycosylation for Patient ALG6 and PMM2
3. Discussion
4. Materials and Methods
4.1. CSF Samples
4.2. Digestion of Proteins and Glycoproteins from CSF
4.3. LC-MS/MS Measurements
4.4. Identification of Glycopeptide Spectra and Target List Generation
4.5. Target Extraction and Relative Quantification of Glycopeptides
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoffmann, A.; Nimtz, M.; Wurster, U.; Conradt, H.S. Carbohydrate Structures of β-Trace Protein from Human Cerebrospinal Fluid: Evidence for “Brain-Type”N-Glycosylation. J. Neurochem. 1994, 63, 2185–2196. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, H.; Dityatev, A. Polysialic Acid in Brain Development and Synaptic Plasticity. In SialoGlyco Chemistry and Biology I: Biosynthesis, Structural Diversity and Sialoglycopathologies; Springer: Berlin/Heidelberg, Germany, 2015; pp. 55–96. [Google Scholar]
- Schnaar, R.L.; Gerardy-Schahn, R.; Hildebrandt, H. Sialic Acids in the Brain: Gangliosides and Polysialic Acid in Nervous System Development, Stability, Disease, and Regeneration. Physiol. Rev. 2014, 94, 461–518. [Google Scholar] [CrossRef] [PubMed]
- Regan, P.; McClean, P.L.; Smyth, T.; Doherty, M. Early Stage Glycosylation Biomarkers in Alzheimer’s Disease. Medicines 2019, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Jin, H.; Wu, Z.; Han, Y.; Chen, J.; Mao, C.; Hao, P.; Zhang, X.; Liu, C.-F.; Yang, S. Mass Spectrometry-Based Analysis of Serum N-Glycosylation Changes in Patients with Parkinson’s Disease. ACS Chem. Neurosci. 2022, 13, 1719–1726. [Google Scholar] [CrossRef]
- Palmigiano, A.; Barone, R.; Sturiale, L.; Sanfilippo, C.; Bua, R.O.; Romeo, D.A.; Messina, A.; Capuana, M.L.; Maci, T.; Le Pira, F.; et al. CSF N-glycoproteomics for early diagnosis in Alzheimer’s disease. J. Proteom. 2016, 131, 29–37. [Google Scholar] [CrossRef]
- Murakami, Y.; Saito, K.; Ito, H.; Hashimoto, Y. Transferrin isoforms in cerebrospinal fluid and their relation to neurological diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 198–210. [Google Scholar] [CrossRef]
- Hoshi, K.; Matsumoto, Y.; Ito, H.; Saito, K.; Honda, T.; Yamaguchi, Y.; Hashimoto, Y. A unique glycan-isoform of transferrin in cerebrospinal fluid: A potential diagnostic marker for neurological diseases. Biochim. Biophys. Acta BBA—Gen. Subj. 2017, 1861, 2473–2478. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, Q.; Yu, Q.; Johnson, J.; Shipman, R.; Zhong, X.; Huang, J.; Asthana, S.; Carlsson, C.; Okonkwo, O.; et al. In-depth Site-specific Analysis of N-glycoproteome in Human Cerebrospinal Fluid and Glycosylation Landscape Changes in Alzheimer’s Disease. Mol. Cell. Proteom. 2021, 20, 100081. [Google Scholar] [CrossRef]
- Reiber, H. Dynamics of brain-derived proteins in cerebrospinal fluid. Clin. Chim. Acta Int. J. Clin. Chem. 2001, 310, 173–186. [Google Scholar] [CrossRef]
- Meier, F.; Park, M.A.; Mann, M. Trapped Ion Mobility Spectrometry and Parallel Accumulation—Serial Fragmentation in Proteomics. Mol. Cell. Proteom. 2021, 20, 100138. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.-D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation—Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef]
- Polasky, D.A.; Yu, F.; Teo, G.C.; Nesvizhskii, A.I. Fast and comprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco. Nat. Methods 2020, 17, 1125–1132. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: www.proteinatlas.org (accessed on 1 October 2021).
- Margolis, R.U.; Margolis, R.K. Neurobiology of Glycoconjugates; Springer: Boston, MA, USA, 1998. [Google Scholar]
- Clerc, F.; Reiding, K.R.; Jansen, B.C.; Kammeijer, G.S.M.; Bondt, A.; Wuhrer, M. Human plasma protein N-glycosylation. Glycoconj. J. 2016, 33, 309–343. [Google Scholar] [CrossRef]
- Hildebrandt, H.; Mühlenhoff, M.; Weinhold, B.; Gerardy-Schahn, R. Dissecting polysialic acid and NCAM functions in brain development. J. Neurochem. 2007, 103 (Suppl. S1), 56–64. [Google Scholar] [CrossRef]
- Oltmann-Norden, I.; Galuska, S.P.; Hildebrandt, H.; Geyer, R.; Gerardy-Schahn, R.; Geyer, H.; Mühlenhoff, M. Impact of the Polysialyltransferases ST8SiaII and ST8SiaIV on Polysialic Acid Synthesis during Postnatal Mouse Brain Development. J. Biol. Chem. 2008, 283, 1463–1471. [Google Scholar] [CrossRef]
- Hu, Y.; Shah, P.; Clark, D.J.; Ao, M.; Zhang, H. Reanalysis of Global Proteomic and Phosphoproteomic Data Identified a Large Number of Glycopeptides. Anal. Chem. 2018, 90, 8065–8071. [Google Scholar] [CrossRef]
- von der Ohe, M.; Wheeler, S.F.; Wuhrer, M.; Harvey, D.J.; Liedtke, S.; Mühlenhoff, M.; Gerardy-Schahn, R.; Geyer, H.; Dwek, R.A.; Geyer, R.; et al. Localization and characterization of polysialic acid–containing N-linked glycans from bovine NCAM. Glycobiology 2002, 12, 47–63. [Google Scholar] [CrossRef]
- Hoffmann, A.; Nimtz, M.; Getzlaff, R.; Conradt, H.S. ‘Brain-type’ N-glycosylation of asialo-transferrin from human cerebrospinal fluid. FEBS Lett. 1995, 359, 164–168. [Google Scholar] [CrossRef]
- Murakami, Y.; Takahashi, K.; Hoshi, K.; Ito, H.; Kanno, M.; Saito, K.; Nollet, K.; Yamaguchi, Y.; Miyajima, M.; Arai, H.; et al. Spontaneous intracranial hypotension is diagnosed by a combination of lipocalin-type prostaglandin D synthase and brain-type transferrin in cerebrospinal fluid. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, K.; Ito, H.; Abe, E.; Fuwa, T.J.; Kanno, M.; Murakami, Y.; Abe, M.; Murakami, T.; Yoshihara, A.; Ugawa, Y.; et al. Transferrin Biosynthesized in the Brain Is a Novel Biomarker for Alzheimer’s Disease. Metabolites 2021, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Vanderver, A.; Hathout, Y.; Maletkovic, J.; Gordon, E.S.; Mintz, M.; Timmons, M.; Hoffman, E.P.; Horzinski, L.; Niel, F.; Fogli, A.; et al. Sensitivity and specificity of decreased CSF asialotransferrin for eIF2B-related disorder. Neurology 2008, 70, 2226–2232. [Google Scholar] [CrossRef] [PubMed]
- Wopereis, S.; Morava, É.; Grünewald, S.; Adamowicz, M.; Huijben, K.M.L.C.; Lefeber, D.J.; Wevers, R.A. Patients with unsolved congenital disorders of glycosylation type II can be subdivided in six distinct biochemical groups. Glycobiology 2005, 15, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Stibler, H.; Borg, S.; Allgulander, C. Clinical Significance of Abnormal Heterogeneity of Transferrin in Relation to Alcohol Consumption. Acta Med. Scand. 1979, 206, 275–281. [Google Scholar] [CrossRef]
- Suzuki, T.; Eguchi, A.; Shigefuku, R.; Nagao, S.; Morikawa, M.; Sugimoto, K.; Iwasa, M.; Takei, Y. Accuracy of carbohydrate-deficient transferrin as a biomarker of chronic alcohol abuse during treatment for alcoholism. Hepatol. Res. 2022, 52, 120–127. [Google Scholar] [CrossRef]
- van Scherpenzeel, M.; Steenbergen, G.; Morava, E.; Wevers, R.A.; Lefeber, D.J. High-resolution mass spectrometry glycoprofiling of intact transferrin for diagnosis and subtype identification in the congenital disorders of glycosylation. Transl. Res. 2015, 166, 639–649. [Google Scholar] [CrossRef]
- Barone, R.; Sturiale, L.; Sofia, V.; Ignoto, A.; Fiumara, A.; Sorge, G.; Garozzo, D.; Zappia, M. Clinical phenotype correlates to glycoprotein phenotype in a sib pair with CDG-Ia. Am. J. Med. Genet. Part A 2008, 146A, 2103–2108. [Google Scholar] [CrossRef]
- Witters, P.; Cassiman, D.; Morava, E. Nutritional Therapies in Congenital Disorders of Glycosylation (CDG). Nutrients 2017, 9, 1222. [Google Scholar] [CrossRef]
- Grünewald, S.; Schollen, E.; Van Schaftingen, E.; Jaeken, J.; Matthijs, G. High Residual Activity of PMM2 in Patients’ Fibroblasts: Possible Pitfall in the Diagnosis of CDG-Ia (Phosphomannomutase Deficiency). Am. J. Hum. Genet. 2001, 68, 347–354. [Google Scholar] [CrossRef]
- Callewaert, N.; Schollen, E.; Vanhecke, A.; Jaeken, J.; Matthijs, G.; Contreras, R. Increased fucosylation and reduced branching of serum glycoprotein N-glycans in all known subtypes of congenital disorder of glycosylation I. Glycobiology 2003, 13, 367–375. [Google Scholar] [CrossRef]
- Hipgrave Ederveen, A.L.; de Haan, N.; Baerenfaenger, M.; Lefeber, D.J.; Wuhrer, M. Dissecting Total Plasma and Protein-Specific Glycosylation Profiles in Congenital Disorders of Glycosylation. Int. J. Mol. Sci. 2020, 21, 7635. [Google Scholar] [CrossRef]
- Wessels, H.J.; Kulkarni, P.; van Dael, M.; Suppers, A.; Willems, E.; Zijlstra, F.; Kragt, E.; Gloerich, J.; Schmit, P.-O.; Pengelley, S.; et al. Plasma glycoproteomics delivers high-specificity disease biomarkers by detecting site-specific glycosylation abnormalities. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lieber, C.S. Carbohydrate Deficient Transferrin in Alcoholic Liver Disease: Mechanisms and Clinical Implications. Alcohol 1999, 19, 249–254. [Google Scholar] [CrossRef]
- Gong, M.; Castillo, L.; Redman, R.S.; Garige, M.; Hirsch, K.; Azuine, M.; Amdur, R.L.; Seth, D.; Haber, P.S.; Lakshman, M.R. Down-regulation of liver Galβ1, 4GlcNAc α2, 6-sialyltransferase gene by ethanol significantly correlates with alcoholic steatosis in humans. Metabolism 2008, 57, 1663–1668. [Google Scholar] [CrossRef]
- Ghosh, P.; Liu, Q.-H.; Lakshman, M.R. Long-term ethanol exposure impairs glycosylation of both N- and O-glycosylated proteins in rat liver. Metabolism 1995, 44, 890–898. [Google Scholar] [CrossRef]
- Searashi, Y.; Yamauchi, M.; Sakamoto, K.; Ohata, M.; Asakura, T.; Ohkawa, K. Acetaldehyde-Induced Growth Retardation and Micro-Heterogeneity of the Sugar Chain in Transferrin Synthesized by HepG2 Cells. Alcohol. Clin. Exp. Res. 2002, 26, 32s–37s. [Google Scholar] [CrossRef]
- Zhang, W.; James, P.M.; Ng, B.G.; Li, X.; Xia, B.; Rong, J.; Asif, G.; Raymond, K.; Jones, M.A.; Hegde, M.; et al. A Novel N-Tetrasaccharide in Patients with Congenital Disorders of Glycosylation, Including Asparagine-Linked Glycosylation Protein 1, Phosphomannomutase 2, and Mannose Phosphate Isomerase Deficiencies. Clin. Chem. 2016, 62, 208–217. [Google Scholar] [CrossRef]
- Bakar, N.A.; Ashikov, A.; Brum, J.M.; Smeets, R.; Kersten, M.; Huijben, K.; Keng, W.T.; Speck-Martins, C.E.; de Carvalho, D.R.; de Rizzo, I.M.P.O.; et al. Synergistic use of glycomics and single molecule Molecular Inversion Probes (smMIPs) for identification of congenital disorders of glycosylation type-1. J. Inherit. Metab. Dis. 2022, 45, 769. [Google Scholar] [CrossRef]
- Stavenhagen, K.; Hinneburg, H.; Thaysen-Andersen, M.; Hartmann, L.; Varón Silva, D.; Fuchser, J.; Kaspar, S.; Rapp, E.; Seeberger, P.H.; Kolarich, D. Quantitative mapping of glycoprotein micro-heterogeneity and macro-heterogeneity: An evaluation of mass spectrometry signal strengths using synthetic peptides and glycopeptides. J. Mass Spectrom. 2013, 48, 627–639. [Google Scholar] [CrossRef]
- de Loos, F.; Huijben, K.M.; van der Kar, N.C.; Monnens, L.A.; van den Heuvel, L.P.; Groener, J.E.; de Moor, R.A.; Wevers, R.A. Hemolytic Uremic Syndrome Attributable to Streptococcus pneumoniae Infection: A Novel Cause for Secondary Protein N-Glycan Abnormalities. Clin. Chem. 2002, 48, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Pronicka, E.; Adamowicz, M.; Kowalik, A.; Płoski, R.; Radomyska, B.; Rogaszewska, M.; Rokicki, D.; Sykut-Cegielska, J. Elevated Carbohydrate-Deficient Transferrin (CDT) and Its Normalization on Dietary Treatment as a Useful Biochemical Test for Hereditary Fructose Intolerance and Galactosemia. Pediatr. Res. 2007, 62, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Hansson, S.F.; Andréasson, U.; Wall, M.; Skoog, I.; Andreasen, N.; Wallin, A.; Zetterberg, H.; Blennow, K. Reduced levels of amyloid-beta-binding proteins in cerebrospinal fluid from Alzheimer’s disease patients. J. Alzheimer’s Dis. 2009, 16, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Puchades, M.; Hansson, S.F.; Nilsson, C.L.; Andreasen, N.; Blennow, K.; Davidsson, P. Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer’s disease. Mol. Brain Res. 2003, 118, 140–146. [Google Scholar] [CrossRef]
- Martins-De-Souza, D.; Wobrock, T.; Zerr, I.; Schmitt, A.; Gawinecka, J.; Schneider-Axmann, T.; Falkai, P.; Turck, C.W. Different apolipoprotein E, apolipoprotein A1 and prostaglandin-H2 D-isomerase levels in cerebrospinal fluid of schizophrenia patients and healthy controls. World J. Biol. Psychiatry 2010, 11, 719–728. [Google Scholar] [CrossRef]
- Salazar, I.L.; Lourenço, A.S.T.; Manadas, B.; Baldeiras, I.; Ferreira, C.; Teixeira, A.C.; Mendes, V.M.; Novo, A.M.; Machado, R.; Batista, S.; et al. Posttranslational modifications of proteins are key features in the identification of CSF biomarkers of multiple sclerosis. J. Neuroinflamm. 2022, 19, 44. [Google Scholar] [CrossRef]
- Foster, E.M.; Fernandes, M.; Dangla-Valls, A.; Hublitz, P.; Pangalos, M.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Glycosylated clusterin species facilitate Aβ toxicity in human neurons. Sci. Rep. 2022, 12, 18639. [Google Scholar] [CrossRef]
- Alagesan, K.; Kolarich, D. To enrich or not to enrich: Enhancing (glyco)peptide ionization using the CaptiveSpray nanoBooster™. bioRxiv 2019. [Google Scholar] [CrossRef]
- Momčilović, A.; de Haan, N.; Hipgrave Ederveen, A.L.; Bondt, A.; Koeleman, C.A.M.; Falck, D.; de Neef, L.A.; Mesker, W.E.; Tollenaar, R.; de Ru, A.; et al. Simultaneous Immunoglobulin A and G Glycopeptide Profiling for High-Throughput Applications. Anal. Chem. 2020, 92, 4518–4526. [Google Scholar] [CrossRef]
- Abu Bakar, N.; Lefeber, D.J.; van Scherpenzeel, M. Clinical glycomics for the diagnosis of congenital disorders of glycosylation. J. Inherit. Metab. Dis. 2018, 41, 499–513. [Google Scholar] [CrossRef]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baerenfaenger, M.; Post, M.A.; Langerhorst, P.; Huijben, K.; Zijlstra, F.; Jacobs, J.F.M.; Verbeek, M.M.; Wessels, H.J.C.T.; Lefeber, D.J. Glycoproteomics in Cerebrospinal Fluid Reveals Brain-Specific Glycosylation Changes. Int. J. Mol. Sci. 2023, 24, 1937. https://doi.org/10.3390/ijms24031937
Baerenfaenger M, Post MA, Langerhorst P, Huijben K, Zijlstra F, Jacobs JFM, Verbeek MM, Wessels HJCT, Lefeber DJ. Glycoproteomics in Cerebrospinal Fluid Reveals Brain-Specific Glycosylation Changes. International Journal of Molecular Sciences. 2023; 24(3):1937. https://doi.org/10.3390/ijms24031937
Chicago/Turabian StyleBaerenfaenger, Melissa, Merel A. Post, Pieter Langerhorst, Karin Huijben, Fokje Zijlstra, Joannes F. M. Jacobs, Marcel M. Verbeek, Hans J. C. T. Wessels, and Dirk J. Lefeber. 2023. "Glycoproteomics in Cerebrospinal Fluid Reveals Brain-Specific Glycosylation Changes" International Journal of Molecular Sciences 24, no. 3: 1937. https://doi.org/10.3390/ijms24031937
APA StyleBaerenfaenger, M., Post, M. A., Langerhorst, P., Huijben, K., Zijlstra, F., Jacobs, J. F. M., Verbeek, M. M., Wessels, H. J. C. T., & Lefeber, D. J. (2023). Glycoproteomics in Cerebrospinal Fluid Reveals Brain-Specific Glycosylation Changes. International Journal of Molecular Sciences, 24(3), 1937. https://doi.org/10.3390/ijms24031937