
A New Plasmacytoid Dendritic Cell-Based Vaccine in Combination with Anti-PD-1 Expands the Tumor-Specific CD8+ T Cells of Lung Cancer Patients

 ,
,  , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Results
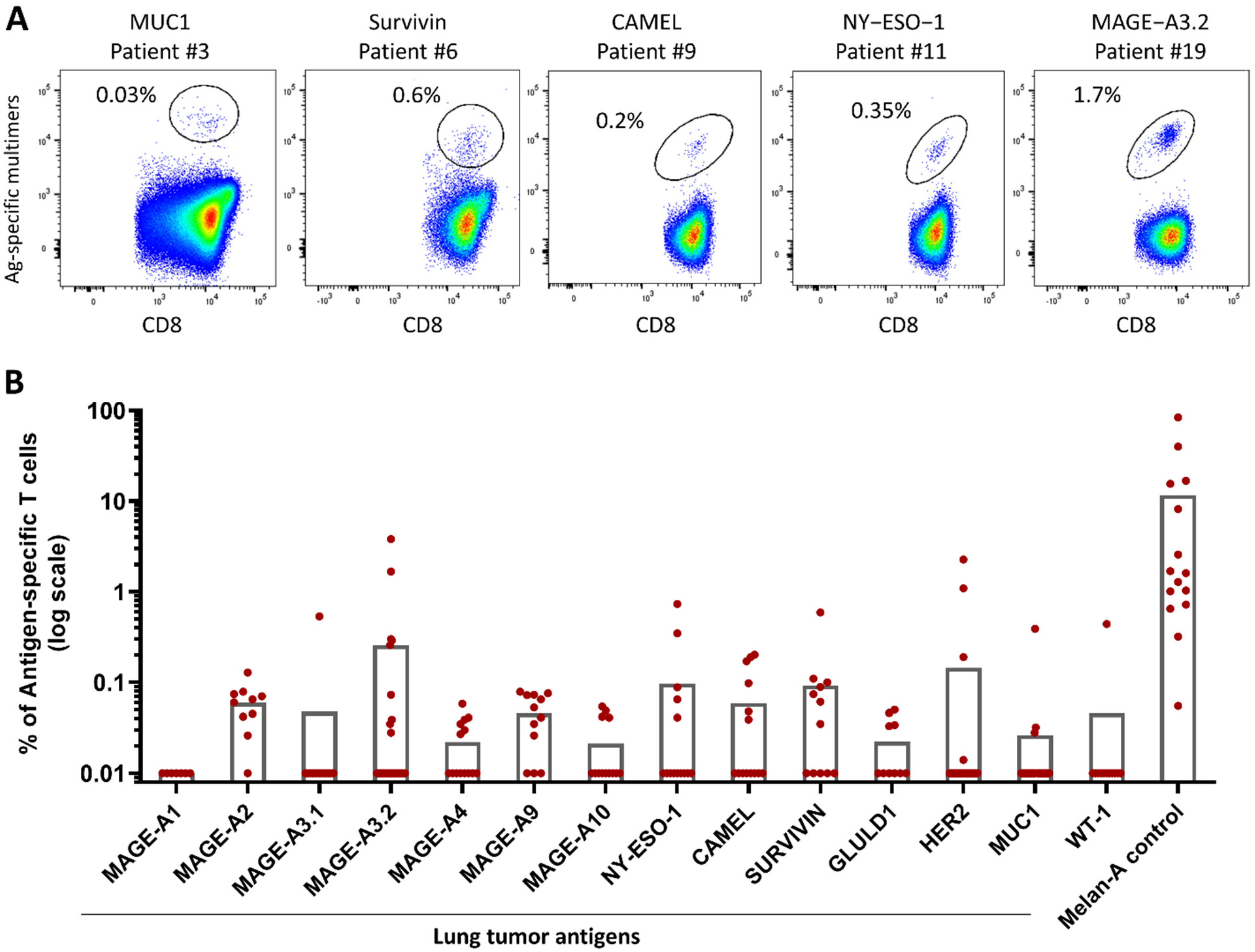
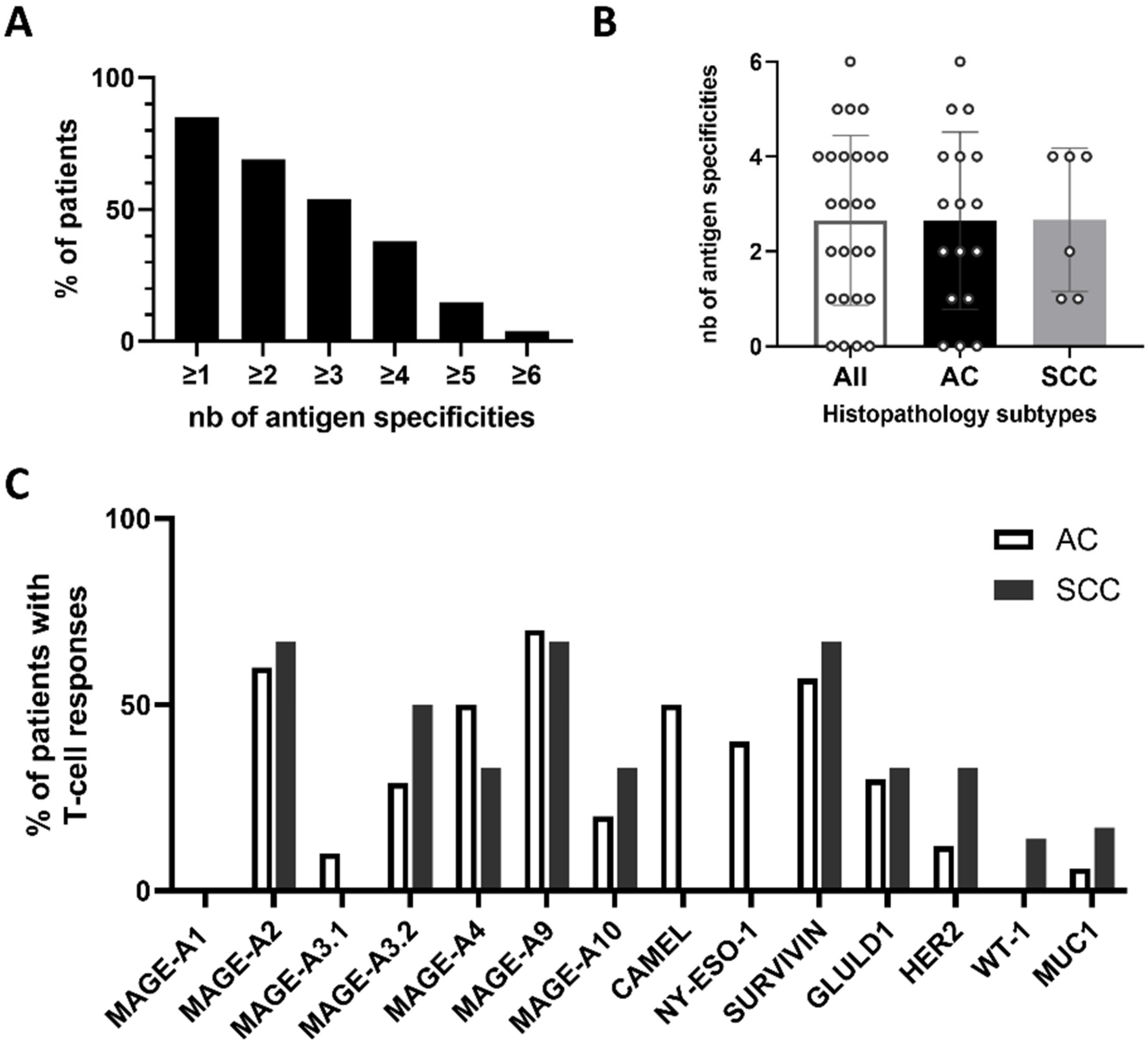
2.1. PDC*Line Cells Induce a Broad Antitumor Response in Lung Cancer Patients
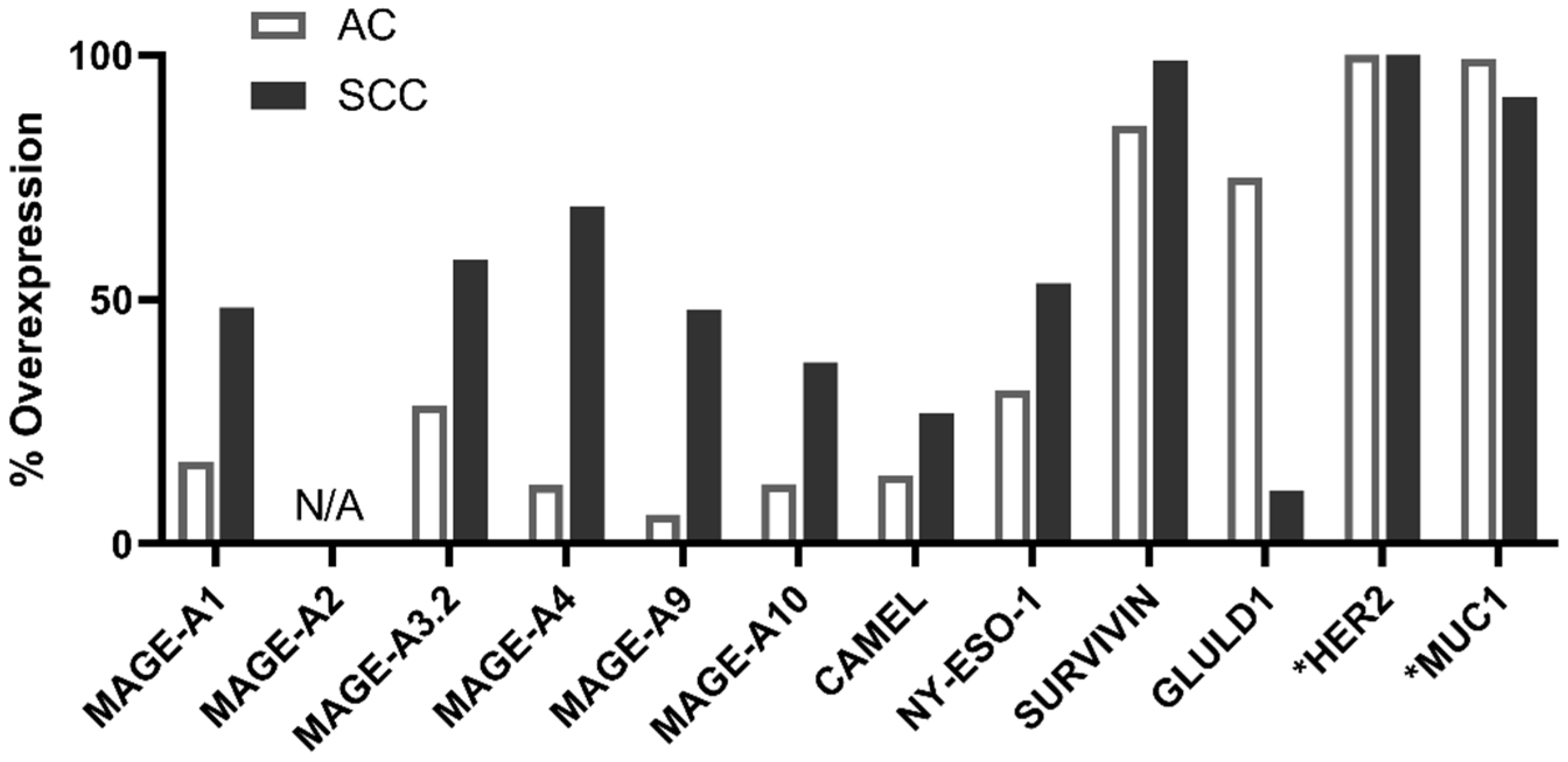
2.2. PDC*Line Cells Induce Antitumor Response in Both Subtypes of NSCLC
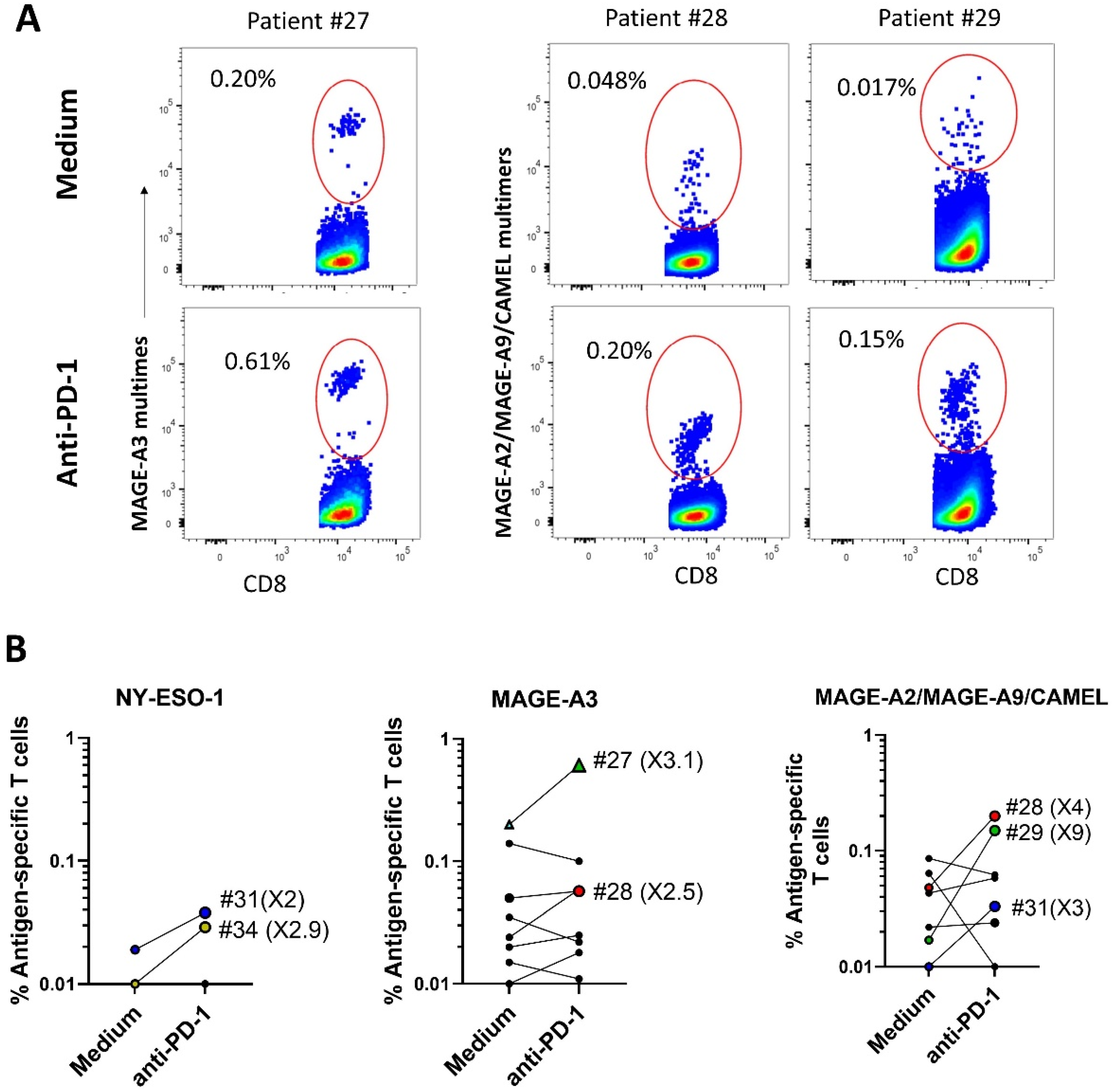
2.3. ICI Synergizes with Peptide-Loaded PDC*Line Cells to Expand Antitumor CD8+ T Cells
3. Discussion
4. Materials and Methods
4.1. Patients and Peripheral Blood Mononuclear Cells
4.2. Preparation of Tumor-Peptide-Loaded PDC*Line
4.3. Patients’ PBMC Stimulation with Loaded PDC*Line Cells
4.4. Detection of Antigen-Specific CD8+ T Cells
4.5. Transcriptomic Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Mithoowani, H.; Febbraro, M. Non-Small-Cell Lung Cancer in 2022: A Review for General Practitioners in Oncology. Current Oncology 2022, 29, 1828–1839. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, C.J.; Adderley, H.; Ortega-Franco, A.; Khan, A.; Reck, M.; Califano, R. First-Line Immune Checkpoint Inhibition for Advanced Non-Small-Cell Lung Cancer: State of the Art and Future Directions. Drugs 2020, 80, 1783–1797. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant Anti-PD-1 Immunotherapy Promotes a Survival Benefit with Intratumoral and Systemic Immune Responses in Recurrent Glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Pardoll, D.M. Neoadjuvant Checkpoint Blockade for Cancer Immunotherapy. Science 2020, 367, eaax0182. [Google Scholar] [CrossRef]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic Cancer Vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Plumas, J. Harnessing Dendritic Cells for Innovative Therapeutic Cancer Vaccines. Curr. Opin. Oncol. 2022, 34, 161–168. [Google Scholar] [CrossRef]
- Chaperot, L.; Blum, A.; Manches, O.; Lui, G.; Angel, J.; Molens, J.-P.; Plumas, J. Virus or TLR Agonists Induce TRAIL-Mediated Cytotoxic Activity of Plasmacytoid Dendritic Cells. J. Immunol. 2006, 176, 248–255. [Google Scholar] [CrossRef]
- Lui, G.; Manches, O.; Angel, J.; Molens, J.-P.; Chaperot, L.; Plumas, J. Plasmacytoid Dendritic Cells Capture and Cross-Present Viral Antigens from Influenza-Virus Exposed Cells. PLoS ONE 2009, 4, e7111. [Google Scholar] [CrossRef]
- van der Sluis, R.M.; Egedal, J.H.; Jakobsen, M.R. Plasmacytoid Dendritic Cells as Cell-Based Therapeutics: A Novel Immunotherapy to Treat Human Immunodeficiency Virus Infection? Front. Cell. Infect. Microbiol. 2020, 10, 249. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. The Multifaceted Biology of Plasmacytoid Dendritic Cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Hernández, S.S.; Jakobsen, M.R.; Bak, R.O. Plasmacytoid Dendritic Cells as a Novel Cell-Based Cancer Immunotherapy. Int. J. Mol. Sci. 2022, 23, 11397. [Google Scholar] [CrossRef] [PubMed]
- Monti, M.; Consoli, F.; Vescovi, R.; Bugatti, M.; Vermi, W. Human Plasmacytoid Dendritic Cells and Cutaneous Melanoma. Cells 2020, 9, 417. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Aarntzen, E.H.J.G.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.G.; van Rossum, M.; et al. Natural Human Plasmacytoid Dendritic Cells Induce Antigen-Specific T-Cell Responses in Melanoma Patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef]
- Aspord, C.; Charles, J.; Leccia, M.-T.; Laurin, D.; Richard, M.-J.; Chaperot, L.; Plumas, J. A Novel Cancer Vaccine Strategy Based on HLA-A*0201 Matched Allogeneic Plasmacytoid Dendritic Cells. PLoS ONE 2010, 5, e10458. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Leccia, M.-T.; Salameire, D.; Laurin, D.; Chaperot, L.; Charles, J.; Plumas, J. HLA-A*0201 + Plasmacytoid Dendritic Cells Provide a Cell-Based Immunotherapy for Melanoma Patients. J. Investig. Dermatol. 2012, 132, 2395–2406. [Google Scholar] [CrossRef]
- Charles, J.; Chaperot, L.; Hannani, D.; Costa, J.B.; Templier, I.; Trabelsi, S.; Gil, H.; Moisan, A.; Persoons, V.; Hegelhofer, H.; et al. An Innovative Plasmacytoid Dendritic Cell Line-Based Cancer Vaccine Primes and Expands Antitumor T-Cells in Melanoma Patients in a First-in-Human Trial. OncoImmunology 2020, 9, 1738812. [Google Scholar] [CrossRef]
- Vigneron, N.; Stroobant, V.; Van den Eynde, B.J.; van der Bruggen, P. Database of T Cell-Defined Human Tumor Antigens: The 2013 Update. Cancer Immun 2013, 13, 15. [Google Scholar]
- Oka, Y.; Elisseeva, O.A.; Tsuboi, A.; Ogawa, H.; Tamaki, H.; Li, H.; Oji, Y.; Kim, E.H.; Soma, T.; Asada, M.; et al. Human Cytotoxic T-Lymphocyte Responses Specific for Peptides of the Wild-Type Wilms’ Tumor Gene (WT1) Product. Immunogenetics 2000, 51, 99–107. [Google Scholar] [CrossRef]
- Stevens, D.; Ingels, J.; Van Lint, S.; Vandekerckhove, B.; Vermaelen, K. Dendritic Cell-Based Immunotherapy in Lung Cancer. Front. Immunol. 2021, 11, 620374. [Google Scholar] [CrossRef]
- Ottaviani, S.; Zhang, Y.; Boon, T.; van der Bruggen, P. A MAGE-1 Antigenic Peptide Recognized by Human Cytolytic T Lymphocytes on HLA-A2 Tumor Cells. Cancer Immunol. Immunother 2005, 54, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, S.; Schirle, M.; Gückel, B.; Dumrese, T.; Stumm, S.; Kayser, S.; Moris, A.; Wallwiener, D.; Rammensee, H.-G.; Stevanovic, S. A MAGE-A1 HLA-A*0201 Epitope Identified by Mass Spectrometry. Cancer Res. 2001, 61, 4072–4077. [Google Scholar] [PubMed]
- Kawashima, I.; Hudson, S.J.; Tsai, V.; Southwood, S.; Takesako, K.; Appella, E.; Sette, A.; Celis, E. The Multi-Epitope Approach for Immunotherapy for Cancer: Identification of Several CTL Epitopes from Various Tumor-Associated Antigens Expressed on Solid Epithelial Tumors. Hum. Immunol. 1998, 59, 1–14. [Google Scholar] [CrossRef] [PubMed]
- van der Bruggen, P.; Bastin, J.; Gajewski, T.; Coulie, P.G.; Boël, P.; Smet, C.D.; Traversari, C.; Townsend, A.; Boon, T. A Peptide Encoded by Human Gene MAGE-3 and Presented by HLA-A2 Induces Cytolytic T Lymphocytes That Recognize Tumor Cells Expressing MAGE-3. Eur. J. Immunol. 1994, 24, 3038–3043. [Google Scholar] [CrossRef] [PubMed]
- Duffour, M.-T.; Chaux, P.; Lurquin, C.; Cornelis, G.; Boon, T.; Bruggen, P.V. der A MAGE-A4 Peptide Presented by HLA-A2 Is Recognized by Cytolytic T Lymphocytes. Eur. J. Immunol. 1999, 29, 3329–3337. [Google Scholar] [CrossRef]
- Oehlrich, N.; Devitt, G.; Linnebacher, M.; Schwitalle, Y.; Grosskinski, S.; Stevanovic, S.; Zöller, M. Generation of RAGE-1 and MAGE-9 Peptide-Specific Cytotoxic T-Lymphocyte Lines for Transfer in Patients with Renal Cell Carcinoma. Int. J. Cancer 2005, 117, 256–264. [Google Scholar] [CrossRef]
- Huang, L.-Q.; Brasseur, F.; Serrano, A.; Plaen, E.D.; van der Bruggen, P.; Boon, T.; Pel, A.V. Cytolytic T Lymphocytes Recognize an Antigen Encoded by MAGE-A10 on a Human Melanoma. J. Immunol. 1999, 162, 6849–6854. [Google Scholar] [CrossRef]
- Chen, J.-L.; Dunbar, P.R.; Gileadi, U.; Jager, E.; Gnjatic, S.; Nagata, Y.; Stockert, E.; Panicali, D.L.; Chen, Y.-T.; Knuth, A.; et al. Identification of NY-ESO-1 Peptide Analogues Capable of Improved Stimulation of Tumor-Reactive CTL. J. Immunol. 2000, 165, 948–955. [Google Scholar] [CrossRef]
- Jäger, E.; Chen, Y.-T.; Drijfhout, J.W.; Karbach, J.; Ringhoffer, M.; Jäger, D.; Arand, M.; Wada, H.; Noguchi, Y.; Stockert, E.; et al. Simultaneous Humoral and Cellular Immune Response against Cancer–Testis Antigen NY-ESO-1: Definition of Human Histocompatibility Leukocyte Antigen (HLA)-A2–Binding Peptide Epitopes. J. Exp. Med. 1998, 187, 265–270. [Google Scholar] [CrossRef]
- Valmori, D.; Dutoit, V.; Liénard, D.; Rimoldi, D.; Pittet, M.J.; Champagne, P.; Ellefsen, K.; Sahin, U.; Speiser, D.; Lejeune, F.; et al. Naturally Occurring Human Lymphocyte Antigen-A2 Restricted CD8+ T-Cell Response to the Cancer Testis Antigen NY-ESO-1 in Melanoma Patients. Cancer Res. 2000, 60, 4499–4506. [Google Scholar]
- Aarnoudse, C.A.; van den Doel, P.B.; Heemskerk, B.; Schrier, P.I. Interleukin-2-Induced, Melanoma-Specific T Cells Recognize Camel, an Unexpected Translation Product of LAGE-1. Int. J. Cancer 1999, 82, 442–448. [Google Scholar] [CrossRef]
- Nakatsugawa, M.; Horie, K.; Yoshikawa, T.; Shimomura, M.; Kikuchi, Y.; Sakemura, N.; Suzuki, S.; Nobuoka, D.; Hirohashi, Y.; Torigoe, T.; et al. Identification of an HLA-A*0201-Restricted Cytotoxic T Lymphocyte Epitope from the Lung Carcinoma Antigen, Lengsin. Int. J. Oncol. 2011, 39, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Diestelkoetter, P.; Weigle, B.; Schmachtenberg, F.; Stevanovic, S.; Ockert, D.; Rammensee, H.G.; Rieber, E.P. Generation of Survivin-Specific CD8+ T Effector Cells by Dendritic Cells Pulsed with Protein or Selected Peptides. Cancer Res. 2000, 60, 4845–4849. [Google Scholar] [PubMed]
- Schmidt, S.M.; Schag, K.; Müller, M.R.; Weck, M.M.; Appel, S.; Kanz, L.; Grünebach, F.; Brossart, P. Survivin Is a Shared Tumor-Associated Antigen Expressed in a Broad Variety of Malignancies and Recognized by Specific Cytotoxic T Cells. Blood 2003, 102, 571–576. [Google Scholar] [CrossRef]
- Fisk, B.; Blevins, T.L.; Wharton, J.T.; Ioannides, C.G. Identification of an Immunodominant Peptide of HER-2/Neu Protooncogene Recognized by Ovarian Tumor-Specific Cytotoxic T Lymphocyte Lines. J. Exp. Med. 1995, 181, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Brossart, P.; Heinrich, K.S.; Stuhler, G.; Behnke, L.; Reichardt, V.L.; Stevanovic, S.; Muhm, A.; Rammensee, H.G.; Kanz, L.; Brugger, W. Identification of HLA-A2-Restricted T-Cell Epitopes Derived from the MUC1 Tumor Antigen for Broadly Applicable Vaccine Therapies. Blood 1999, 93, 4309–4317. [Google Scholar] [CrossRef] [PubMed]
- Boon, T.; Coulie, P.G.; Van den Eynde, B.J.; Bruggen, P. van der Human T Cell Responses Against Melanoma. Annu. Rev. Immunol. 2006, 24, 175–208. [Google Scholar] [CrossRef]
- Germeau, C.; Ma, W.; Schiavetti, F.; Lurquin, C.; Henry, E.; Vigneron, N.; Brasseur, F.; Lethé, B.; Plaen, E.D.; Velu, T.; et al. High Frequency of Antitumor T Cells in the Blood of Melanoma Patients before and after Vaccination with Tumor Antigens. J. Exp. Med. 2005, 201, 241–248. [Google Scholar] [CrossRef]
- Lenogue, K.; Walencik, A.; Laulagnier, K.; Molens, J.-P.; Benlalam, H.; Dreno, B.; Coulie, P.; Pule, M.; Chaperot, L.; Plumas, J. Engineering a Human Plasmacytoid Dendritic Cell-Based Vaccine to Prime and Expand Multispecific Viral and Tumor Antigen-Specific T-Cells. Vaccines 2021, 9, 141. [Google Scholar] [CrossRef]
- Palata, O.; Podzimkova Hradilova, N.; Mysiková, D.; Kutna, B.; Mrazkova, H.; Lischke, R.; Spisek, R.; Adkins, I. Detection of Tumor Antigens and Tumor-Antigen Specific T Cells in NSCLC Patients: Correlation of the Quality of T Cell Responses with NSCLC Subtype. Immunol. Lett. 2020, 219, 46–53. [Google Scholar] [CrossRef]
- Groeper, C.; Gambazzi, F.; Zajac, P.; Bubendorf, L.; Adamina, M.; Rosenthal, R.; Zerkowski, H.-R.; Heberer, M.; Spagnoli, G.C. Cancer/Testis Antigen Expression and Specific Cytotoxic T Lymphocyte Responses in Non Small Cell Lung Cancer. Int. J. Cancer 2007, 120, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Cetin, K.; Ettinger, D.S.; Hei, Y.; O’Malley, C.D. Survival by Histologic Subtype in Stage IV Nonsmall Cell Lung Cancer Based on Data from the Surveillance, Epidemiology and End Results Program. CLEP 2011, 3, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, S.; Debernardi, A.; Jacquiau, B.; Vitte, A.-L.; Vesin, A.; Nagy-Mignotte, H.; Moro-Sibilot, D.; Brichon, P.-Y.; Lantuejoul, S.; Hainaut, P.; et al. Ectopic Activation of Germline and Placental Genes Identifies Aggressive Metastasis-Prone Lung Cancers. Sci. Transl. Med. 2013, 5, 186ra66. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen Type | Antigen | Peptide Sequence (HLA-A*02:01) | References |
|---|---|---|---|
| Cancer-germline Antigens | MAGE-A1 | 278KVLEYVIKV286 | [21,22] |
| MAGE-A2 | 157YLQLVFGIEV166 | [23] | |
| MAGE-A3.1 | 112KVAELVHFL120 | [23] | |
| MAGE-A3.2 | 271FLWGPRALV279 | [24] | |
| MAGE-A4 | 230GVYDGREHTV239 | [25] | |
| MAGE-A9 | 223ALSVMGVYV231 | [26] | |
| MAGE-A10 | 254GLYDGMEHL262 | [27] | |
| NY-ESO-1 (CTAG1B) | 157SLLMWITQC165 | [28,29,30] | |
| CAMEL (CTAG2) | 152MLMAQEALAFL162 | [31] | |
| Overexpressed Antigens | GLULD1 (LGSN) | 270FLPEFGISSA279 | [32] |
| SURVIVIN (BIRC5) | 95ELTLGEFLKL104 | [33,34] | |
| HER2 (ERBB2) | 369KIFGSLAFL377 | [35] | |
| WT-1 | 126RMFPNAPYL134 | [19] | |
| Post-translational | MUC-1 | 12LLLLTVLTV20 | [36] |
| MAGE-A1 | MAGE-A2 | MAGE-A3.1 | MAGE-A3.2 | MAGE-A4 | MAGE-A9 | MAGE-A10 | NY-ESO-1 | CAMEL | GLULD1 | SURV | HER2 | MUC-1 | WT-1 | At Least One Ag |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0/14 | 9/14 | 1/14 | 9/26 | 6/14 | 9/14 | 4/14 | 5/14 | 6/14 | 4/14 | 7/12 | 4/26 | 3/26 | 1/12 | 22/26 |
| 0% | 64% | 7.1% | 34.6% | 43% | 64% | 29% | 36% | 43% | 29% | 58% | 15% | 11.5% | 8.3% | 84.6% |
| Patient Number | Histology | Stage | Smoking Status |
|---|---|---|---|
| 1 | SCC | IIB | Active |
| 2 | AC | IIIA | Active |
| 3 | SCC | IA | Active |
| 4 | AC | IA | Former |
| 5 | AC | IIA | Non-smoker |
| 6 | TC | IB | Former |
| 7 | SCC | IIA | Active |
| 8 | SCC | IB | Active |
| 9 | AC | IIIA | Active |
| 10 | AC | IIB | Former |
| 11 | AC | IIIA | Active |
| 12 | AC | IIIA | Active |
| 13 | AC | IA | Active |
| 14 | AC | IV | Active |
| 15 | LCNC | IIIA | Active |
| 16 | AC | IIB | Active |
| 17 | AC | IA | Active |
| 18 | AC | IB | Active |
| 19 | SCC | IIB | Active |
| 20 | AC | IB | Former |
| 21 | AC | IIA | Active |
| 22 | AC | IA | Active |
| 23 | TC | IA | Active |
| 24 | AC | nd | Former |
| 25 | SCC | nd | Unknown |
| 26 | AC | II | Former |
| 27 | SCC | IIB | Active |
| 28 | SCC | IIIA | Active |
| 29 | AC | IIb | Former |
| 30 | AC | IIB | Former |
| 31 | AC | IIA | Former |
| 32 | AC | IB | Former |
| 33 | AC | IIB | Non-smoker |
| 34 | SCC | IV | Former |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hannani, D.; Leplus, E.; Laurin, D.; Caulier, B.; Aspord, C.; Madelon, N.; Bourova-Flin, E.; Brambilla, C.; Brambilla, E.; Toffart, A.-C.; et al. A New Plasmacytoid Dendritic Cell-Based Vaccine in Combination with Anti-PD-1 Expands the Tumor-Specific CD8+ T Cells of Lung Cancer Patients. Int. J. Mol. Sci. 2023, 24, 1897. https://doi.org/10.3390/ijms24031897
Hannani D, Leplus E, Laurin D, Caulier B, Aspord C, Madelon N, Bourova-Flin E, Brambilla C, Brambilla E, Toffart A-C, et al. A New Plasmacytoid Dendritic Cell-Based Vaccine in Combination with Anti-PD-1 Expands the Tumor-Specific CD8+ T Cells of Lung Cancer Patients. International Journal of Molecular Sciences. 2023; 24(3):1897. https://doi.org/10.3390/ijms24031897
Chicago/Turabian StyleHannani, Dalil, Estelle Leplus, David Laurin, Benjamin Caulier, Caroline Aspord, Natacha Madelon, Ekaterina Bourova-Flin, Christian Brambilla, Elisabeth Brambilla, Anne-Claire Toffart, and et al. 2023. "A New Plasmacytoid Dendritic Cell-Based Vaccine in Combination with Anti-PD-1 Expands the Tumor-Specific CD8+ T Cells of Lung Cancer Patients" International Journal of Molecular Sciences 24, no. 3: 1897. https://doi.org/10.3390/ijms24031897
APA StyleHannani, D., Leplus, E., Laurin, D., Caulier, B., Aspord, C., Madelon, N., Bourova-Flin, E., Brambilla, C., Brambilla, E., Toffart, A.-C., Laulagnier, K., Chaperot, L., & Plumas, J. (2023). A New Plasmacytoid Dendritic Cell-Based Vaccine in Combination with Anti-PD-1 Expands the Tumor-Specific CD8+ T Cells of Lung Cancer Patients. International Journal of Molecular Sciences, 24(3), 1897. https://doi.org/10.3390/ijms24031897