
KRAS and BRAF Mutation Rates and Survival Outcomes in Colorectal Cancer in an Ethnically Diverse Patient Cohort

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics and Clinicopathological Variables
2.2. Tumor Characterisitcs
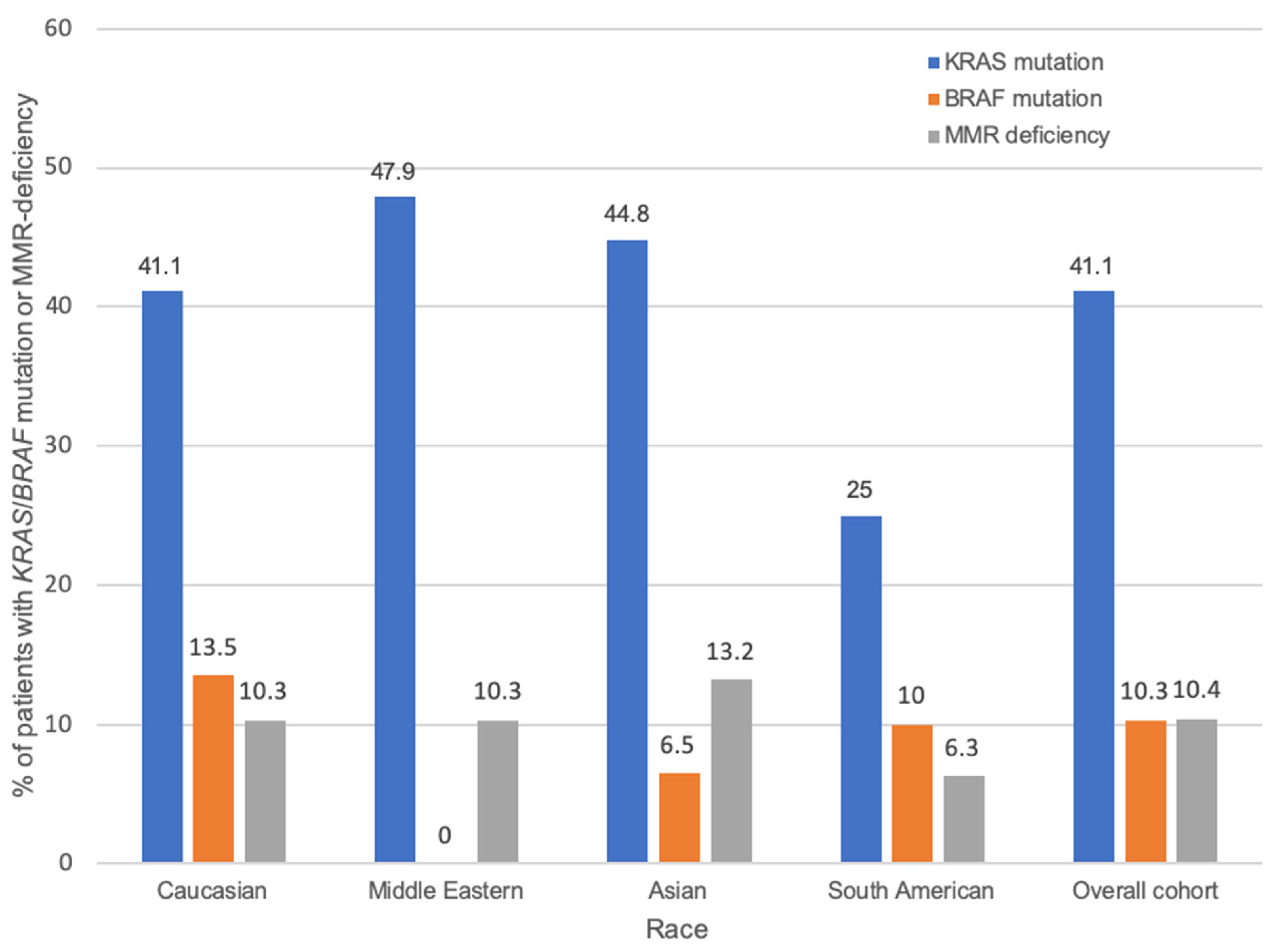
2.3. Race and KRAS/BRAF
2.4. Survival Outcomes
2.4.1. Race
2.4.2. KRAS/BRAF/MMR Status
2.4.3. Multivariable Analysis
2.5. Six Ethnic Groups
3. Discussion
4. Materials and Methods
4.1. Study Population and Clinicopathological Data
4.2. Survival Outcomes
4.3. Somatic Mutation Analysis
4.4. Determination of MMR Status
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Worldwide Cancer Data [Internet]. London: World Cancer Research Fund International. 2018. Available online: https://www.wcrf.org/dietandcancer/worldwide-cancer-data/ (accessed on 3 August 2021).
- Tan, C.; Du, X. KRAS mutation testing in metastatic colorectal cancer. World J. Gastroenterol. 2012, 18, 5171–5180. [Google Scholar]
- Kafatos, G.; Niepel, D.; Lowe, K.; Jenkins-Anderson, S.; Westhead, H.; Garawin, T.; Traugottová, Z.; Bilalis, A.; Molnar, E.; Timar, J.; et al. RAS mutation prevalence among patients with metastatic colorectal cancer: A meta-analysis of real-world data. Biomark. Med. 2017, 11, 751–760. [Google Scholar] [CrossRef]
- Al-Sohaily, S.; Biankin, A.; Leong, R.; Kohonen-Corish, M.; Warusavitarne, J. Molecular pathways in colorectal cancer. J. Gastroenterol. Hepatol. 2012, 27, 1423–1431. [Google Scholar] [CrossRef]
- László, L.; Kurilla, A.; Takács, T.; Kudlik, G.; Koprivanacz, K.; Buday, L.; Vas, V. Recent Updates on the Significance of KRAS Mutations in Colorectal Cancer Biology. Cells 2021, 10, 667. [Google Scholar] [CrossRef]
- Li, W.; Li, H.; Liu, R.; Yang, X.; Gao, Y.; Niu, Y.; Geng, J.; Xue, Y.; Jin, X.; You, Q.; et al. Comprehensive Analysis of the Relationship Between RAS and RAF Mutations and MSI Status of Colorectal Cancer in Northeastern China. Cell. Physiol. Biochem. 2018, 50, 1496–1509. [Google Scholar] [CrossRef]
- Al-Shamsi, H.O.; Jones, J.; Fahmawi, Y.; Dahbour, I.; Tabash, A.; Abdel-Wahab, R.; Abousamra, A.O.S.; Shaw, K.R.; Xiao, L.; Hassan, M.M.; et al. Molecular spectrum of KRAS, NRAS, BRAF, PIK3CA, TP53, and APC somatic gene mutations in Arab patients with colorectal cancer: Determination of frequency and distribution pattern. J. Gastrointest. Oncol. 2016, 7, 882–902. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Hussain, S.; Mehrotra, R.; Kumar, R.S.; Kumar, K.; Pande, P.; Doval, D.C.; Basir, S.F.; Bharadwaj, M. Kras Gene Mutation and RASSF1A, FHIT and MGMT Gene Promoter Hypermethylation: Indicators of Tumor Staging and Metastasis in Adenocarcinomatous Sporadic Colorectal Cancer in Indian Population. PLoS ONE 2013, 8, e60142. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, B.N.; Bibi, A.; Rashid, M.U.; Khan, Y.I.; Chaudri, M.S.; Shakoori, A.R. Spectrum of K ras mutations in Pakistani colorectal cancer patients. Braz. J. Med. Biol. Res. 2014, 47, 35–41. [Google Scholar] [CrossRef]
- Natsume, S.; Yamaguchi, T.; Takao, M.; Iijima, T.; Wakaume, R.; Takahashi, K.; Matsumoto, H.; Nakano, D.; Horiguchi, S.-I.; Koizumi, K.; et al. Clinicopathological and molecular differences between right-sided and left-sided colorectal cancer in Japanese patients. Jpn. J. Clin. Oncol. 2018, 48, 609–618. [Google Scholar] [CrossRef]
- Hoffman, S.J.; Wu, M.L.-C. Phenotypic and genotypic differences in colorectal carcinoma among Caucasians, Asians, and Hispanics lack statistical significance. Pathol. Res. Pract. 2018, 214, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Huang, D.; Poston, G.; Ma, X.; Wang, R.; Sheng, W.; Zhou, X.; Zhu, X.; Cai, S. The molecular heterogeneity of sporadic colorectal cancer with different tumor sites in Chinese patients. Oncotarget 2017, 8, 49076–49083. [Google Scholar] [CrossRef]
- Levi, M.; Prayogi, G.; Sastranagara, F.; Sudianto, E.; Widjajahakim, G.; Gani, W.; Mahanadi, A.; Agnes, J.; Khairunisa, B.H.; Utomo, A.R. Clinicopathological Associations of K-RAS and N-RAS Mutations in Indonesian Colorectal Cancer Cohort. J. Gastrointest. Cancer 2018, 49, 124–131. [Google Scholar] [CrossRef]
- Guo, F.; Gong, H.; Zhao, H.; Chen, J.; Zhang, Y.; Zhang, L.; Shi, X.; Zhang, A.; Jin, H.; Zhang, J.; et al. Mutation status and prognostic values of KRAS, NRAS, BRAF and PIK3CA in 353 Chinese colorectal cancer patients. Sci. Rep. 2018, 8, 6076. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Huang, Y.; Yu, X.; Li, X.; Zheng, S.; Ding, K.; Xu, J. Clinicopathologic distribution of KRAS and BRAF mutations in a Chinese population with colorectal cancer precursor lesions. Oncotarget 2016, 7, 17265–17274. [Google Scholar] [CrossRef]
- Sirisena, N.D.; Deen, K.; Mandawala, D.E.N.; Herath, P.; Dissanayake, V.H.W. The pattern of KRAS mutations in metastatic colorectal cancer: A retrospective audit from Sri Lanka. BMC Res. Notes 2017, 10, 392. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tang, L.; Yi, J.; Li, G.; Lu, Y.; Xu, Y.; Zhao, S.; Mao, R.; Li, X.; Ren, L.; et al. Unique characteristics of CpG island methylator phenotype (CIMP) in a Chinese population with colorectal cancer. BMC Gastroenterol. 2019, 19, 173. [Google Scholar] [CrossRef] [PubMed]
- Korphaisarn, K.; Pongpaibul, A.; Roothumnong, E.; Pongsuktavorn, K.; Thamlikitkul, L.; Anekpuritanang, T.; Poungvarin, N.; Thongnoppakhun, W.; Pithukpakorn, M. High Frequency of KRAS Codon 146 and FBXW7 Mutations in Thai Patients with Stage II-III Colon Cancer. Asian Pac. J. Cancer Prev. 2019, 20, 2319–2326. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Z.; Zhang, Z.-C.; Wang, F.; Zhao, Q.; Zhang, D.-S.; Wang, F.-H.; Wang, Z.-Q.; Luo, H.-Y.; He, M.-M.; Wang, D.-S.; et al. Mutation profiling in chinese patients with metastatic colorectal cancer and its correlation with clinicopathological features and anti-EGFR treatment response. Oncotarget 2016, 7, 28356–28368. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Le, D.T.; Duong, Q.H.; Tatipamula, V.B.; Nguyen, B. High frequency of microsatellite instability and its substantial co-existence with KRAS and BRAF mutations in Vietnamese patients with colorectal cancer. Oncol. Lett. 2020, 21, 41. [Google Scholar] [CrossRef]
- Guedes, J.G.; Veiga, I.; Rocha, P.; Pinto, P.; Pinto, C.; Pinheiro, M.; Peixoto, A.; Fragoso, M.; Raimundo, A.; Ferreira, P.; et al. High resolution melting analysis of KRAS, BRAF and PIK3CA in KRAS exon 2 wild-type metastatic colorectal cancer. BMC Cancer 2013, 13, 169. [Google Scholar] [CrossRef]
- Kosmidou, V.; Oikonomou, E.; Vlassi, M.; Avlonitis, S.; Katseli, A.; Tsipras, I.; Mourtzoukou, D.; Kontogeorgos, G.; Zografos, G.; Pintzas, A. Tumor heterogeneity revealed by KRAS, BRAF, and PIK3CA pyrosequencing: KRAS and PIK3CA intratumor mutation profile differences and their therapeutic implications. Hum. Mutat. 2014, 35, 329–340. [Google Scholar] [CrossRef]
- Vignot, S.; Lefebvre, C.; Frampton, G.M.; Meurice, G.; Yelensky, R.; Palmer, G.; Capron, F.; Lazar, V.; Hannoun, L.; Miller, V.A.; et al. Comparative analysis of primary tumour and matched metastases in colorectal cancer patients: Evaluation of concordance between genomic and transcriptional profiles. Eur. J. Cancer 2015, 51, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.S.; Bondy, M.L.; El-Badawy, S.A.; Mokhtar, N.; Eissa, S.; Bayoumy, S.; Seifeldin, I.A.; Houlihan, P.S.; Lukish, J.R.; Watanabe, T.; et al. Contrasting molecular pathology of colorectal carcinoma in Egyptian and Western patients. Br. J. Cancer 2001, 85, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Zahrani, A.; Kandil, M.; Badar, T.; Abdelsalam, M.; Al-Faiar, A.; Ismail, A. Clinico-pathological study of K-ras mutations in colorectal tumors in Saudi Arabia. Tumori 2014, 100, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.; Saer-Ghorra, C.; Trak-Smayra, V.; Nadiri, S.; Yazbeck, C.; Baz, M.; Kattane, J.G. Molecular characteristics of colorectal cancer in a Middle Eastern population in a single institution. Ann. Saudi Med. 2018, 38, 251–259. [Google Scholar] [CrossRef]
- Mohsen, N.; Ahmadreza, S.; Fatemeh, H.; Fatemeh, H.; Fariba, E.R. Frequency of K-RAS and N-RAS Gene Mutations in Colorectal Cancers in Southeastern Iran. Asian Pac. J. Cancer Prev. 2016, 17, 4511–4515. [Google Scholar] [PubMed]
- Koochak, A.; Rakhshani, N.; Niya, M.H.K.; Tameshkel, F.S.; Sohrabi, M.R.; Babaee, M.R.; Rezvani, H.; Bahar, B.; Imanzade, F.; Zamani, F.; et al. Mutation Analysis of KRAS and BRAF Genes in Metastatic Colorectal Cancer: A First Large Scale Study from Iran. Asian Pac. J. Cancer Prev. 2016, 17, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Amirifard, N.; Sadeghi, E.; Farshchian, N.; Haghparast, A.; Choubsaz, M. Evaluation of KRAS Gene Mutations in Metastatic Colo-rectal Cancer Patients in Kermanshah Province. Asian Pac. J. Cancer Prev. 2016, 17, 3085–3088. [Google Scholar]
- Karbalaie Niya, M.H.; Basi, A.; Koochak, A.; Safarnezhad Tameshkel, F.; Rakhshani, N.; Zamani, F.; Imanzade, F.; Rezvani, H.; Adib Sereshki, M.M.; Sohrabi, M.R. Sensitive High-Resolution Melting Analysis for Screening of KRAS and BRAF Mutations in Iranian Human Metastatic Colorectal Cancers. Asian Pac. J. Cancer Prev. 2016, 17, 5147–5152. [Google Scholar]
- Zekri, J.; Al-Shehri, A.; Mahrous, M.; Al-Rehaily, S.; Darwish, T.; Bassi, S.; El Taani, H.; Al Zahrani, A.; Elsamany, S.; Al-Maghrabi, J.; et al. Mutations in codons 12 and 13 of K-ras exon 2 in colorectal tumors of Saudi Arabian patients: Frequency, clincopathological associations, and clinical outcomes. Genet. Mol. Res. 2017, 16, 1–11. [Google Scholar] [CrossRef]
- Greenbaum, A.; Wiggins, C.; Meisner, A.L.; Rojo, M.; Kinney, A.Y.; Rajput, A. KRAS biomarker testing disparities in colorectal cancer patients in New Mexico. Heliyon 2017, 3, e00448. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, W.; Sobanski, T.; de Carvalho, A.C.; Evangelista, A.F.; Matsushita, M.; Berardinelli, G.N.; de Oliveira, M.A.; Reis, R.M.; Guimarães, D.P. Mutation profiling of cancer drivers in Brazilian colorectal cancer. Sci. Rep. 2019, 9, 13687. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, K.; Orellana, P.; Villarroel, C.; Contreras, L.; Kawachi, H.; Kobayashi, M.; Wielandt, A.M.; De la Fuente, M.; Triviño, J.C.; Kronberg, U.; et al. EGFR pathway subgroups in Chilean colorectal cancer patients, detected by mutational and expression profiles, associated to different clinicopathological features. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317724517. [Google Scholar] [CrossRef] [PubMed]
- Kamran, S.C.; Xie, J.; Cheung, A.T.M.; Mavura, M.Y.; Song, H.; Palapattu, E.L.; Madej, J.; Gusev, A.; Van Allen, E.M.; Huang, F.W. Tumor Mutations Across Racial Groups in a Re-al-World Data Registry. JCO Precis. Oncol. 2021, 5, 1654–1658. [Google Scholar] [CrossRef]
- Siraj, A.K.; Bu, R.; Prabhakaran, S.; Bavi, P.; Beg, S.; Al Hazmi, M.; Al-Rasheed, M.; Alobaisi, K.; Al-Dayel, F.; AlManea, H.; et al. A very low incidence of BRAF mutations in Middle Eastern colorectal carcinoma. Mol. Cancer 2014, 13, 168. [Google Scholar] [CrossRef]
- Ng, J.Y.; Lu, C.T.; Lam, A.K. BRAF mutation: Current and future clinical pathological applications in colorectal carcinoma. Histol. Histopathol. 2019, 34, 469–477. [Google Scholar] [PubMed]
- Huang, D.; Sun, W.; Zhou, Y.; Li, P.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef]
- Saxena, S.; Srinivas, V.; Deb, P.; Raman, D.K.; Jagani, R. A study of BRAF mutation in colorectal carcinoma in Indian population. J. Cancer Res. Ther. 2018, 14, 1403–1406. [Google Scholar] [CrossRef]
- Jang, M.H.; Kim, S.; Hwang, D.Y.; Kim, W.Y.; Lim, S.D.; Kim, W.S.; Hwang, T.S.; Han, H.S. BRAF-Mutated Colorectal Cancer Exhibits Distinct Clinico-pathological Features from Wild-Type BRAF-Expressing Cancer Independent of the Microsatellite Instability Status. J. Korean Med. Sci. 2017, 32, 38–46. [Google Scholar] [CrossRef]
- Cheng, H.-H.; Lin, J.-K.; Chen, W.-S.; Jiang, J.-K.; Yang, S.-H.; Chang, S.-C. Clinical significance of the BRAFV600E mutation in Asian patients with colorectal cancer. Int. J. Color. Dis. 2018, 33, 1173–1181. [Google Scholar] [CrossRef]
- Jiang, W.; Cai, M.; Li, S.; Bei, J.; Wang, F.; Hampel, H.; Ling, Y.; Frayling, I.M.; Sinicrope, F.A.; Rodriguez-Bigas, M.A.; et al. Universal screening for Lynch syndrome in a large consecutive cohort of Chinese colorectal cancer patients: High prevalence and unique molecular features. Int. J. Cancer 2019, 144, 2161–2168. [Google Scholar] [CrossRef]
- Sylvester, B.E.; Huo, D.; Khramtsov, A.; Zhang, J.; Smalling, R.V.; Olugbile, S.; Polite, B.N.; Olopade, O.I. Molecular analysis of colorectal tumors within a diverse patient cohort at a single institution. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 350–359. [Google Scholar] [CrossRef]
- Rimbert, J.; Tachon, G.; Junca, A.; Villalva, C.; Karayan-Tapon, L.; Tougeron, D. Association between clinicopathological character-istics and RAS mutation in colorectal cancer. Mod. Pathol. 2018, 31, 517–526. [Google Scholar] [CrossRef]
- Samara, M.; Kapatou, K.; Ioannou, M.; Kostopoulou, E.; Papamichali, R.; Papandreou, C.; Athanasiadis, A.; Koukoulis, G. Mutation profile of KRAS and BRAF genes in patients with colorectal cancer: Association with morphological and prognostic criteria. Genet. Mol. Res. 2015, 14, 16793–16802. [Google Scholar] [CrossRef] [PubMed]
- Kalady, M.F.M.; DeJulius, K.L.B.; Sanchez, J.A.M.; Jarrar, A.; Liu, X.; Manilich, E.; Skacel, M.M.; Church, J.M.M. BRAF Mutations in Colorectal Cancer Are Associated with Distinct Clinical Characteristics and Worse Prognosis. Dis. Colon Rectum 2012, 55, 128–133. [Google Scholar] [CrossRef]
- Waring, P.; Tie, J.; Maru, D.; Karapetis, C.S. RAS Mutations as Predictive Biomarkers in Clinical Management of Metastatic Colorectal Cancer. Clin. Color. Cancer 2016, 15, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Fujiyoshi, K.; Yamamoto, G.; Takahashi, A.; Arai, Y.; Yamada, M.; Kakuta, M.; Yamaguchi, K.; Akagi, Y.; Nishimura, Y.; Sakamoto, H.; et al. High concordance rate of KRAS/BRAF mutations and MSI-H between primary colorectal cancer and corresponding metastases. Oncol. Rep. 2017, 37, 785–792. [Google Scholar] [CrossRef]
- Yoon, H.H.; Shi, Q.; Alberts, S.R.; Goldberg, R.M.; Thibodeau, S.N.; Sargent, D.J.; Sinicrope, F.A. Racial Differences in BRAF/KRAS Mutation Rates and Survival in Stage III Colon Cancer Patients. J. Natl. Cancer Inst. 2015, 107, djv186. [Google Scholar] [CrossRef]
- Martínez, M.E.; Maltzman, T.; Marshall, J.R.; Einspahr, J.; Reid, M.E.; Sampliner, R.; Ahnen, D.J.; Hamilton, S.R.; Alberts, D.S. Risk factors for Ki-ras protooncogene mutation in sporadic colorectal adenomas. Cancer Res. 1999, 59, 5181–5185. [Google Scholar] [PubMed]
- Slattery, M.L.; Curtin, K.; Wolff, R.K.; Herrick, J.S.; Caan, B.J.; Samowitz, W. Diet, physical activity, and body size associations with rectal tumor mutations and epigenetic changes. Cancer Causes Control 2010, 21, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, J.; Prenen, H. Molecular genetics of colorectal cancer. Ann. Gastroenterol. 2014, 27, 9. [Google Scholar] [PubMed]
- Velho, S.; Moutinho, C.; Cirnes, L.; Albuquerque, C.; Hamelin, R.; Schmitt, F.; Carneiro, F.; Oliveira, C.; Seruca, R. BRAF, KRAS and PIK3CA mutations in colorectal serrated polyps and cancer: Primary or secondary genetic events in colorectal carcinogenesis? BMC Cancer 2008, 8, 255. [Google Scholar] [CrossRef]
- eviQ. Mismatch Repair (MMR) Genetic Testing: NSW Government. 2018 [Updated 29 November 2018]. Available online: https://www.eviq.org.au/cancer-genetics/adult/genetic-testing-for-heritable-pathogenic-variants/619-mismatch-repair-mmr-genetic-testing (accessed on 31 December 2020).
- Alqahtani, M.; Grieu, F.; Carrello, A.; Amanuel, B.; Mashour, M.; Alattas, R.; Al-Saleh, K.; Alsheikh, A.; Alqahtani, S.; Iacopetta, B. Screening for Lynch Syndrome in Young Colorectal Cancer Patients from Saudi Arabia Using Microsatellite Instability as the Initial Test. Asian Pac. J. Cancer Prev. 2016, 17, 1917–1923. [Google Scholar] [CrossRef] [PubMed]
- Marcella, S.; Miller, J.E. Racial differences in colorectal cancer mortality. The importance of stage and socioeconomic status. J. Clin. Epidemiol. 2001, 54, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Dimou, A.; Syrigos, K.N.; Saif, M.W. Disparities in colorectal cancer in African-Americans vs. Whites: Before and after diagnosis. World J. Gastroenterol. 2009, 15, 3734–3743. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.S.; Thomson, C.A.; Pettijohn, E.; Kato, I.; Rodabough, R.J.; Lane, D.; Hubbell, F.A.; O’Sullivan, M.J.; Adams-Campbell, L.; Mouton, C.P.; et al. Racial differences in colorectal cancer incidence and mortality in the Women’s Health Initiative. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Gomez, S.L.; O’Malley, C.D.; Stroup, A.; Shema, S.J.; Satariano, W.A. Longitudinal, population-based study of racial/ethnic differences in colorectal cancer survival: Impact of neighborhood socioeconomic status, treatment and comorbidity. BMC Cancer 2007, 7, 193. [Google Scholar] [CrossRef]
- Rui, Y.-Y.; Zhang, D.; Zhou, Z.-G.; Wang, C.; Yang, L.; Yu, Y.-Y.; Chen, H.-N. Can K-ras gene mutation be utilized as prognostic biomarker for colorectal cancer patients receiving chemotherapy? A me-ta-analysis and systematic review. PLoS ONE 2013, 8, e77901. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- Meng, M.; Zhong, K.; Jiang, T.; Liu, Z.; Kwan, H.Y.; Su, T. The current understanding on the impact of KRAS on colorectal cancer. Biomed. Pharmacother. 2021, 140, 111717. [Google Scholar] [CrossRef]
- Ogino, S.; Shima, K.; Meyerhardt, J.A.; McCleary, N.J.; Ng, K.; Hollis, D.; Saltz, L.B.; Mayer, R.J.; Schaefer, P.; Whittom, R.; et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: Results from intergroup trial CALGB 89803. Clin. Cancer Res. 2012, 18, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.F.; Shigeta, R.; Villoslada, P.; Selmi, C.; Tuomilehto, J.; Silva, G.; Belmont, J.W.; Klareskog, L.; Gregersen, P.K. European population substructure: Clustering of northern and southern populations. PLoS Genet. 2006, 2, e143. [Google Scholar] [CrossRef] [PubMed]
- Consortium APG. AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer—The stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Variable | Total N (%) | Race Unknown N | Caucasian N (%) | Middle Eastern N (%) | Asian N (%) | South American N (%) | p 1 |
|---|---|---|---|---|---|---|---|
| Median age at diagnosis (N = 413) | 66 | 69 | 63 | 56.5 | 70 | <0.001 | |
| Sex (N = 445) | 0.464 | ||||||
| Male | 250 (56.2) | 13 | 155 (53.8) | 27 (55.1) | 42 (61.8) | 13 (68.4) | |
| Female | 195 (43.8) | 8 | 133 (46.2) | 22 (44.9) | 26 (38.2) | 6 (31.6) | |
| Differentiation (N = 381) | 0.361 | ||||||
| Well | 12 (3.1) | 0 | 8 (3.3) | 2 (4.7) | 1 (1.6) | 1 (5.9) | |
| Moderate | 280 (73.5) | 10 | 183 (74.4) | 34 (79.1) | 43 (68.3) | 10 (58.8) | |
| Poor | 89 (23.4) | 2 | 55 (22.4) | 7 (16.3) | 19 (30.2) | 6 (35.3) | |
| AJCC Stage (N = 397) | 0.992 | ||||||
| 1 | 5 (1.3) | 0 | 4 (1.6) | 1 (2.1) | 0 (0) | 0 (0) | |
| 2 | 38 (9.6) | 0 | 26 (10.2) | 4 (8.3) | 7 (11.3) | 1 (5.6) | |
| 3 | 161 (40.6) | 4 | 101 (39.8) | 21 (43.8) | 27 (43.5) | 8 (44.4) | |
| 4 | 193 (48.6) | 11 | 123 (48.4) | 22 (45.8) | 28 (45.2) | 9 (50) | |
| Location (N = 390) | 0.126 | ||||||
| Right | 153 (39.2) | 4 | 101 (40.2) | 18 (40) | 19 (32.2) | 11 (64.7) | |
| Left | 237 (60.8) | 14 | 150 (59.8) | 27 (60) | 40 (67.8) | 6 (35.3) | |
| Chemotherapy 2 (N = 338) | 0.527 | ||||||
| Yes | 328 (97.0) | 15 | 207 (96.7) | 44 (100) | 51 (94.4) | 11 (100) | |
| No | 10 (3) | 0 | 7 (3.3) | 0 (0) | 3 (5.6) | 0 (0) | |
| KRAS status (N = 438) | 0.343 | ||||||
| WT | 258 (58.9) | 15 | 166 (58.9) | 25 (52.1) | 37 (55.2) | 15 (75) | |
| Mutant | 180 (41.1) | 6 | 116 (41.1) | 23 (47.9) | 30 (44.8) | 5 (25) | |
| BRAF status (N = 399) | 0.025 | ||||||
| WT | 358 (89.7) | 19 | 224 (86.5) | 39 (100) | 58 (93.5) | 18 (90) | |
| Mutant | 41 (10.3) | 0 | 35 (13.5) | 0 (0) | 4 (6.5) | 2 (10) | |
| MMR status (N = 388) | 0.882 | ||||||
| Proficient | 303 (89.6) | 15 | 192 (89.7) | 35 (89.7) | 46 (86.8) | 15 (93.8) | |
| Deficient | 35 (10.4) | 1 | 22 (10.3) | 4 (10.3) | 7 (13.2) | 1 (6.3) |
| KRAS | BRAF | MMR | |||||||
|---|---|---|---|---|---|---|---|---|---|
| WT N (%) | Mutant N (%) | p 1 | WT N (%) | Mutant N (%) | p 2 | Proficient N (%) | Deficient N (%) | p 3 | |
| Median age at diagnosis | 66 | 67 | 0.625 | 66 | 69 | 0.087 | 66 | 75 | 0.028 |
| Sex | 0.490 | 0.095 | 0.150 | ||||||
| Male | 140 (57.6) | 103 (42.4) | 205 (91.9) | 18 (8.1) | 172 (92) | 15 (8) | |||
| Female | 115 (61.2) | 73 (38.8) | 146 (86.4) | 23 (13.6) | 129 (86.6) | 20 (13.4) | |||
| Differentiation | 0.006 | 0.034 | 0.001 | ||||||
| Well | 7 (58.3) | 5 (41.7) | 9 (90) | 1 (10) | 10 (83.3) | 2 (16.7) | |||
| Moderate | 144 (53.1) | 127 (46.9) | 222 (92.9) | 17 (7.1) | 218 (93.6) | 15 (6.4) | |||
| Poor | 63 (72.4) | 24 (27.6) | 70 (83.3) | 14 (16.7) | 58 (78.4) | 16 (21.6) | |||
| T-stage | 0.168 | 0.136 | 0.297 | ||||||
| 1 | 2 (25) | 6 (75) | 4 (66.7) | 2 (33.3) | 5 (83.3) | 1 (16.7) | |||
| 2 | 9 (64.3) | 5 (35.7) | 10 (90.9) | 1 (9.1) | 6 (75) | 2 (25) | |||
| 3 | 99 (55.9) | 78 (44.1) | 147 (91.9) | 13 (8.1) | 148 (90.8) | 15 (9.2) | |||
| 4 | 80 (62.5) | 48 (37.5) | 104 (87.4) | 15 (12.6) | 109 (90.1) | 12 (9.9) | |||
| N-stage | 0.162 | 0.416 | 0.037 | ||||||
| 0 | 36 (53.7) | 31 (46.3) | 53 (86.9) | 8 (13.1) | 46 (88.5) | 6 (11.5) | |||
| 1 | 78 (54.9) | 64 (45.1) | 123 (92.5) | 10 (7.5) | 114 (85.7) | 19 (14.3) | |||
| 2 | 74 (65.5) | 39 (34.5) | 86 (88.7) | 11 (11.3) | 104 (95.4) | 5 (4.6) | |||
| AJCC Stage | 0.317 | 0.235 | 0.062 | ||||||
| 1 | 1 (20) | 4 (80) | 3 (75) | 1 (25) | 1 (100) | 0 (0) | |||
| 2 | 21 (55.3) | 17 (44.7) | 31 (86.1) | 5 (13.9) | 25 (86.2) | 4 (13.8) | |||
| 3 | 98 (60.9) | 63 (39.1) | 126 (91.3) | 12 (8.7) | 123 (86.6) | 19 (13.4) | |||
| 4 | 103 (56.6) | 79 (43.4) | 158 (92.9) | 12 (7.1) | 132 (95) | 7 (5) | |||
| Location | 0.243 | 0.001 | <0.001 | ||||||
| Right | 82 (54.7) | 68 (45.3) | 114 (83.8) | 22 (16.2) | 116 (82.3) | 25 (17.7) | |||
| Left | 140 (60.9) | 90 (39.1) | 193 (94.6) | 11 (5.4) | 172 (95.6) | 8 (4.4) | |||
| Vascular Invasion | 116 (51.4) | 64 (48.6) | 0.022 | 141 (87.6) | 20 (12.4) | 0.171 | 161 (90.4) | 17 (9.6) | 0.842 |
| Perineural Invasion | 48 (59.3) | 33 (40.7) | 1.0 | 64 (90.1) | 7 (9.9) | 1.0 | 75 (98.7) | 1 (1.3) | 0.002 |
| Circumferential tumor | 86 (66.2) | 44 (33.8) | 0.041 | 109 (88.6) | 14 (11.4) | 0.403 | 110 (88.7) | 14 (11.3) | 0.535 |
| Tumor-infiltrating lymphocytes | 5 (55.6) | 4 (44.4) | 1.0 | 7 (77.8) | 2 (22.2) | 0.232 | 4 (44.4) | 5 (55.6) | 0.001 |
| BRAF mutation | <0.001 | - | <0.001 | ||||||
| WT | 194 (55.9) | 153 (44.1) | - | - | - | 249 (93.3) | 18 (6.7) | ||
| Mutant | 35 (87.5) | 5 (12.5) | - | - | - | 18 (54.5) | 15 (45.5) | ||
| MMR | 0.002 | <0.001 | - | ||||||
| Proficient | 164 (56.0) | 129 (44.0) | 249 (93.3) | 18 (6.7) | - | - | |||
| Deficient | 29 (82.9) | 6 (17.1) | 18 (54.5) | 15 (45.5) | - | - | |||
| KRAS | BRAF | MMR | |||||||
|---|---|---|---|---|---|---|---|---|---|
| WT N (%) | Mutant N (%) | p 1 | WT N (%) | Mutant N (%) | p 2 | Proficient N (%) | Deficient N (%) | p 3 | |
| 0.343 | 0.025 | 0.882 | |||||||
| Caucasian | 166 (58.9) | 116 (41.1) | 224 (86.5) | 35 (13.5) | 192 (89.7) | 22 (10.3) | |||
| Middle Eastern | 25 (52.1) | 23 (47.9) | 39 (100) | 0 (0) | 35 (89.7) | 4 (10.3) | |||
| Asian | 37 (55.2) | 30 (44.8) | 58 (93.5) | 4 (6.5) | 46 (86.8) | 7 (13.2) | |||
| South American | 15 (75) | 5 (25) | 18 (90) | 2 (10) | 15 (93.8) | 1 (6.3) | |||
| N (%) | G12D | G12R | G12V | G12A | G12C | G12S | G13D |
|---|---|---|---|---|---|---|---|
| Caucasian | 40 (13.8) | 4 (1.4) | 41 (14.4) | 23 (7.9) | 19 (6.6) | 15 (5.2) | 17 (5.9) |
| Middle Eastern | 8 (16.7) | 1 (2.1) | 5 (10.4) | 3 (6.3) | 1 (2.1) | 3 (6.3) | 5 (10.4) |
| Asian | 11 (16.4) | 0 (0) | 9 (13.4) | 7 (10.4) | 6 (9.0) | 3 (4.5) | 5 (7.5) |
| South American | 1 (5.0) | 0 (0) | 1 (5) | 1 (5.0) | 2 (10.0) | 1 (5.0) | 2 (10.0) |
| Missing Race | 1 | 0 | 1 | 1 | 0 | 1 | 3 |
| p-value 1 | 0.61 | 0.68 | 0.75 | 0.86 | 0.37 | 0.95 | 0.46 |
| Variable in Cox Model | Hazard Ratio (95% CI) | p |
|---|---|---|
| Age | 1.022 (1.002–1.042) | 0.028 |
| Sex | ||
| Male | 1 (ref) | |
| Female | 1.098 (0.731–1.650) | 0.652 |
| Race | 0.775 | |
| Caucasian | 1 (ref) | |
| Middle Eastern | 1.389 (0.698–2.802) | 0.385 |
| Asian | 0.928 (0.51–1.69) | 0.808 |
| South American | 1.191 (0.473–2.998) | 0.710 |
| KRAS status | ||
| WT | 1 (ref) | |
| Mutant | 0.999 (0.654–1.527) | 0.996 |
| BRAF status | ||
| WT | 1 (ref) | |
| Mutant | 2.278 (1.099–4.722) | 0.027 |
| MMR status | ||
| Proficient | 1 (ref) | |
| Deficient | 0.332 (0.119–0.929) | 0.036 |
| AJCC stage | <0.001 | |
| 4 | 1 (ref) | |
| 3 | 0.302 (0.191–0.479) | <0.001 |
| 2 | 0.877 (0.445–1.727) | 0.704 |
| 1 | 0.000 2 | 0.967 |
| Differentiation | 0.009 | |
| Well | 1 (ref) | |
| Moderate | 3.009 (0.715–12.667) | 0.133 |
| Poor | 5.459 (1.243–23.975) | 0.025 |
| Location | ||
| Right | 1 (ref) | |
| Left | 0.728 (0.471–1.125) | 0.153 |
| Chemotherapy received 3 | ||
| No | 1 (ref) | |
| Yes | 0.281 (0.096–0.819) | 0.020 |
| KRAS | BRAF | MMR | |||||||
|---|---|---|---|---|---|---|---|---|---|
| WT N (%) | Mutant N (%) | p 1 | WT N (%) | Mutant N (%) | p 2 | Proficient N (%) | Deficient N (%) | p 3 | |
| 0.009 | 0.010 | 0.566 | |||||||
| Anglo-Saxon | 110 (64) | 62 (36) | 130 (81.8) | 27 (17.2) | 118 (86.8) | 18 (13.2) | |||
| Middle Eastern | 25 (52.1) | 23 (47.9) | 39 (100) | 0 (0) | 35 (89.7) | 4 (10.3) | |||
| Asian | 37 (55.2) | 30 (44.8) | 58 (93.5) | 4 (6.5) | 46 (86.8) | 7 (13.2) | |||
| South American | 15 (75) | 5 (25) | 18 (90) | 2 (10) | 15 (93.7) | 1 (6.3) | |||
| Southern European | 26 (68.4) | 12 (31.6) | 33 (89.2) | 4 (10.8) | 29 (93.5) | 2 (6.5) | |||
| Other European | 30 (41.7) | 42 (58.3) | 61 (90.2) | 4 (9.8) | 45 (95.7) | 2 (4.3) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habashy, P.; Lea, V.; Wilkinson, K.; Wang, B.; Wu, X.-J.; Roberts, T.L.; Ng, W.; Rutland, T.; Po, J.W.; Becker, T.; et al. KRAS and BRAF Mutation Rates and Survival Outcomes in Colorectal Cancer in an Ethnically Diverse Patient Cohort. Int. J. Mol. Sci. 2023, 24, 17509. https://doi.org/10.3390/ijms242417509
Habashy P, Lea V, Wilkinson K, Wang B, Wu X-J, Roberts TL, Ng W, Rutland T, Po JW, Becker T, et al. KRAS and BRAF Mutation Rates and Survival Outcomes in Colorectal Cancer in an Ethnically Diverse Patient Cohort. International Journal of Molecular Sciences. 2023; 24(24):17509. https://doi.org/10.3390/ijms242417509
Chicago/Turabian StyleHabashy, Paul, Vivienne Lea, Kate Wilkinson, Bin Wang, Xiao-Juan Wu, Tara Laurine Roberts, Weng Ng, Tristan Rutland, Joseph William Po, Therese Becker, and et al. 2023. "KRAS and BRAF Mutation Rates and Survival Outcomes in Colorectal Cancer in an Ethnically Diverse Patient Cohort" International Journal of Molecular Sciences 24, no. 24: 17509. https://doi.org/10.3390/ijms242417509
APA StyleHabashy, P., Lea, V., Wilkinson, K., Wang, B., Wu, X.-J., Roberts, T. L., Ng, W., Rutland, T., Po, J. W., Becker, T., Descallar, J., Lee, M., Mackenzie, S., Gupta, R., Cooper, W., Lim, S., Chua, W., & Lee, C. S. (2023). KRAS and BRAF Mutation Rates and Survival Outcomes in Colorectal Cancer in an Ethnically Diverse Patient Cohort. International Journal of Molecular Sciences, 24(24), 17509. https://doi.org/10.3390/ijms242417509