

Kernel Transcriptome Profiles of Susceptible Wheat Genotypes in Response to Wheat Dwarf Bunt


Abstract
:1. Introduction
2. Results
2.1. RNA Sequencing Analysis
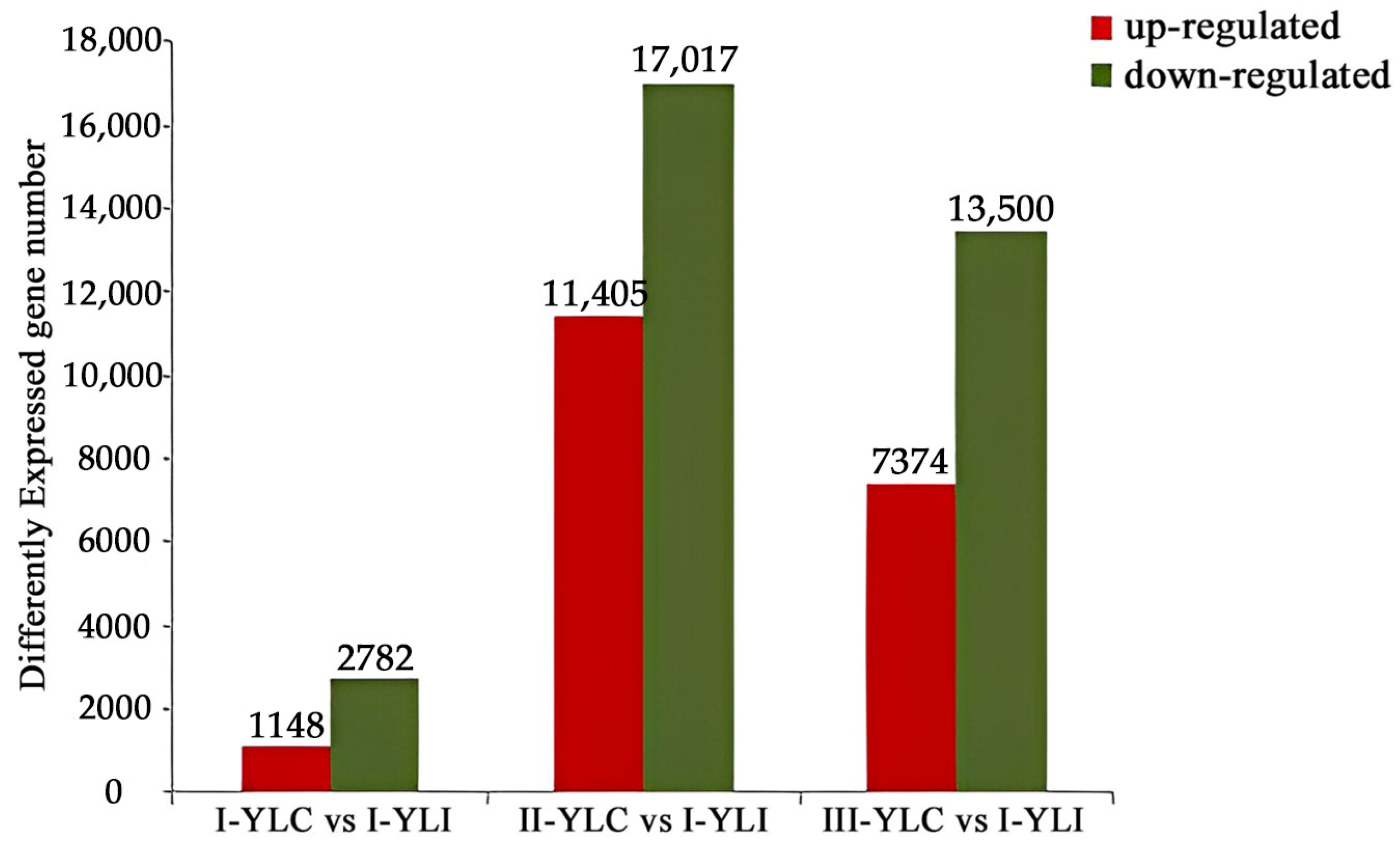
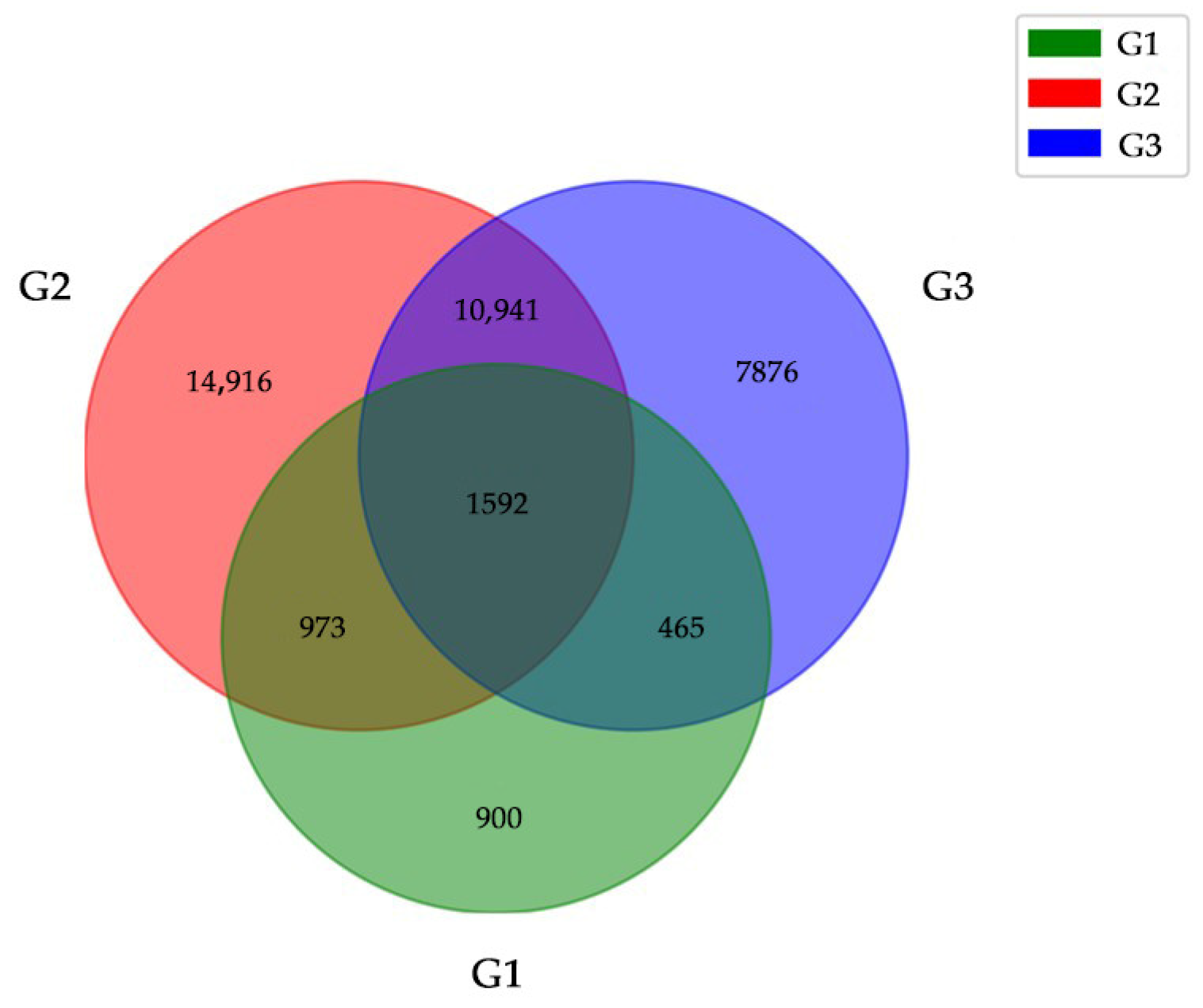
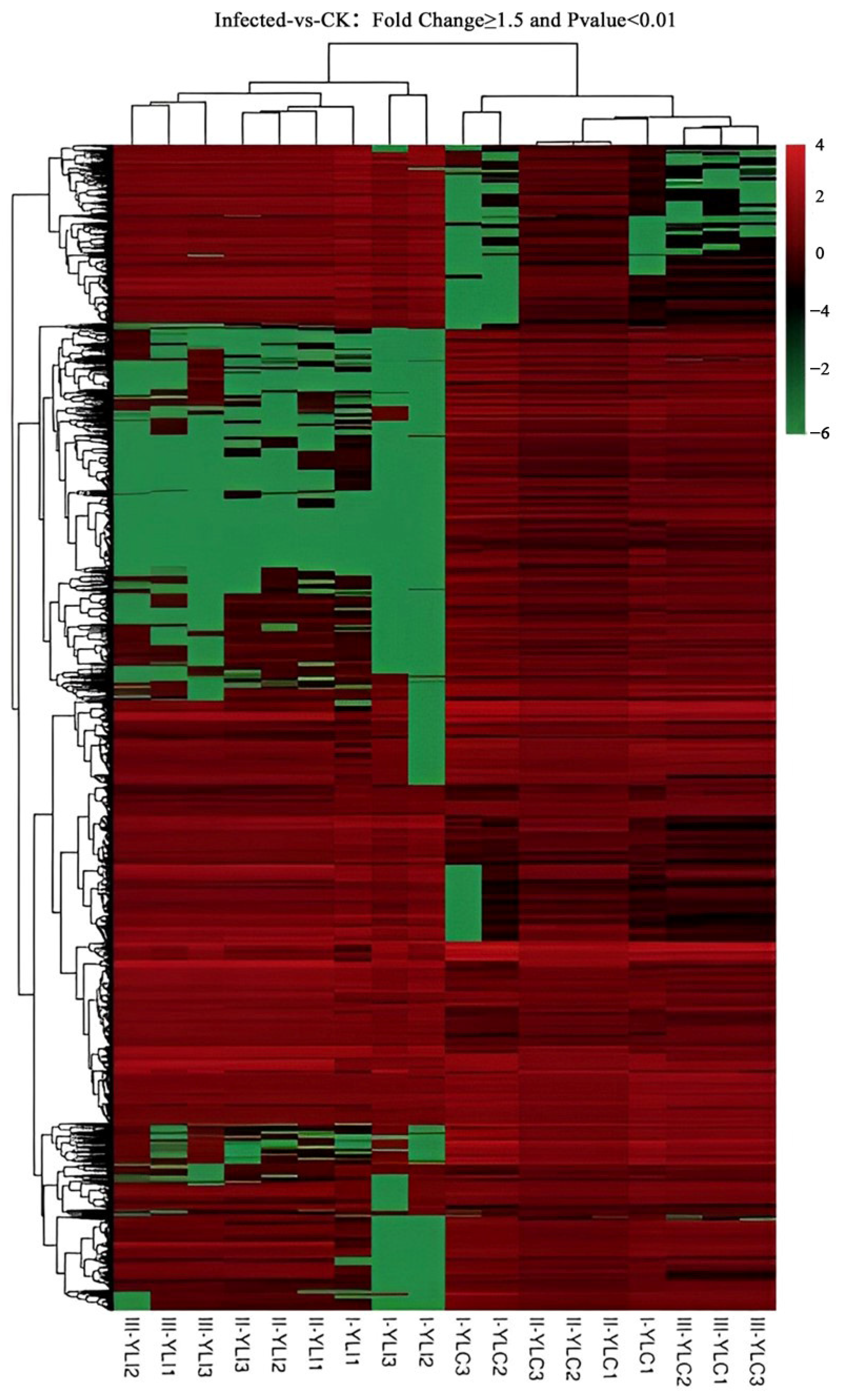
2.2. DEGs Identification
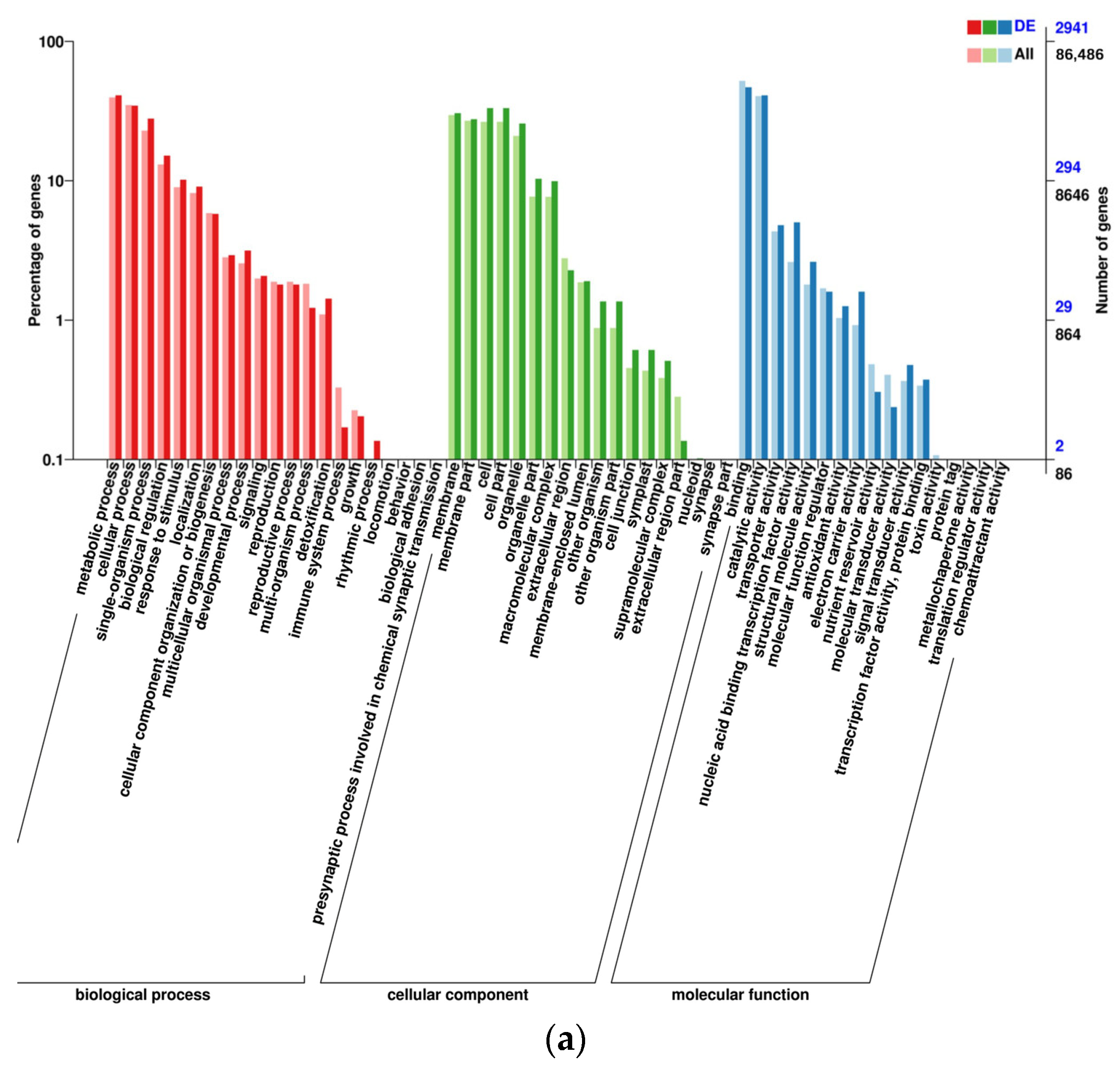
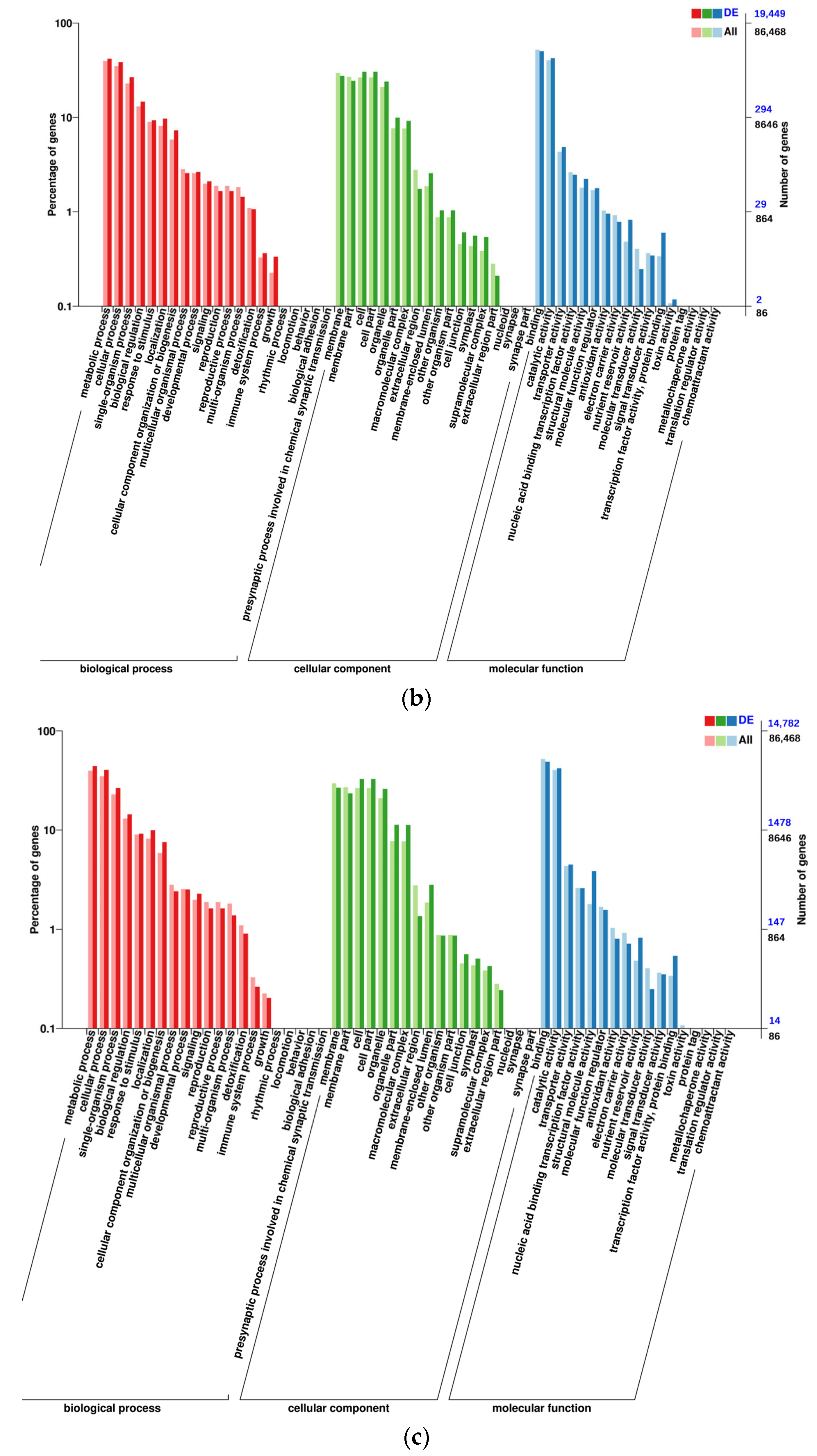
2.3. Gene Ontology Analysis of DEGs
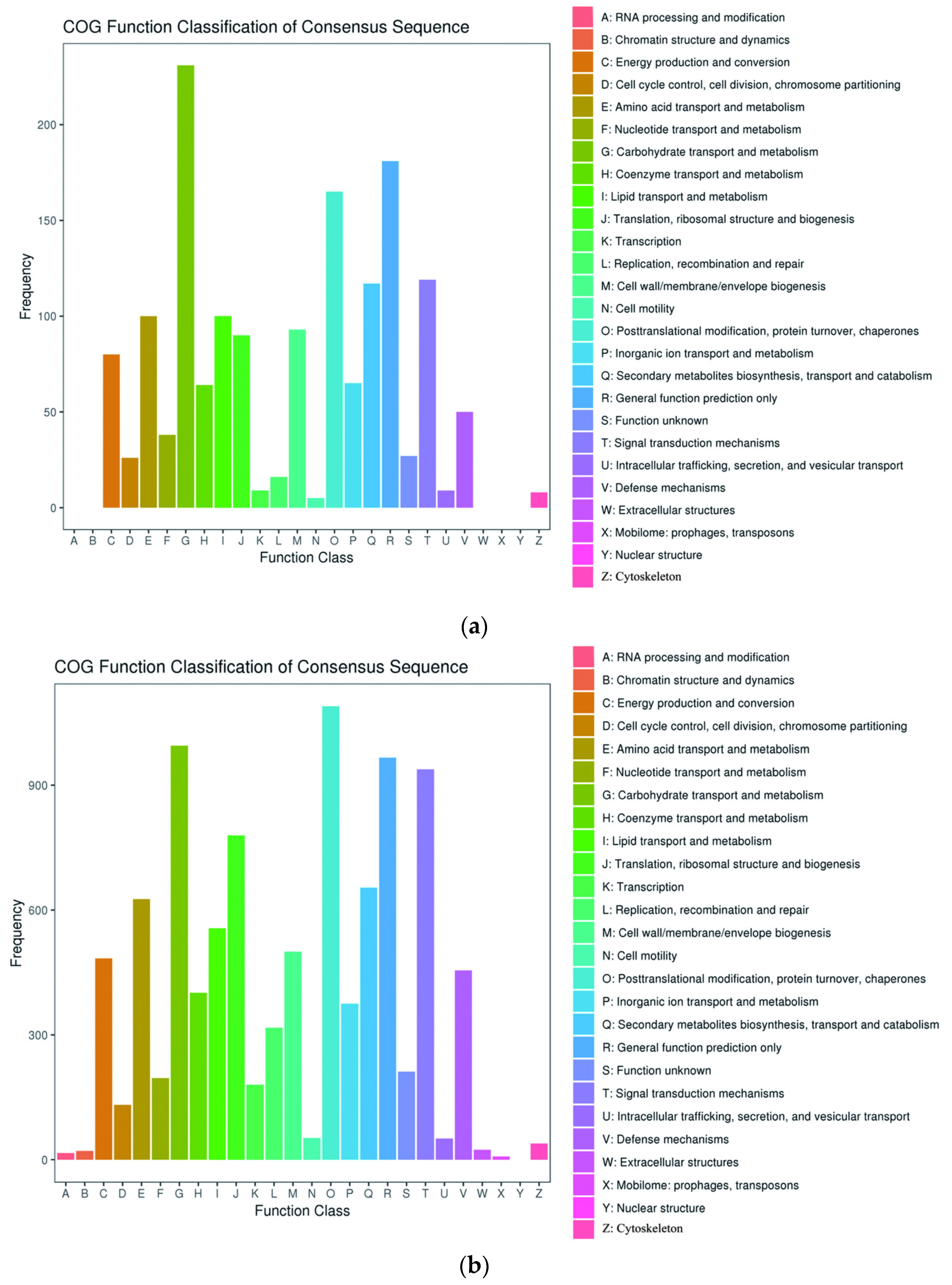
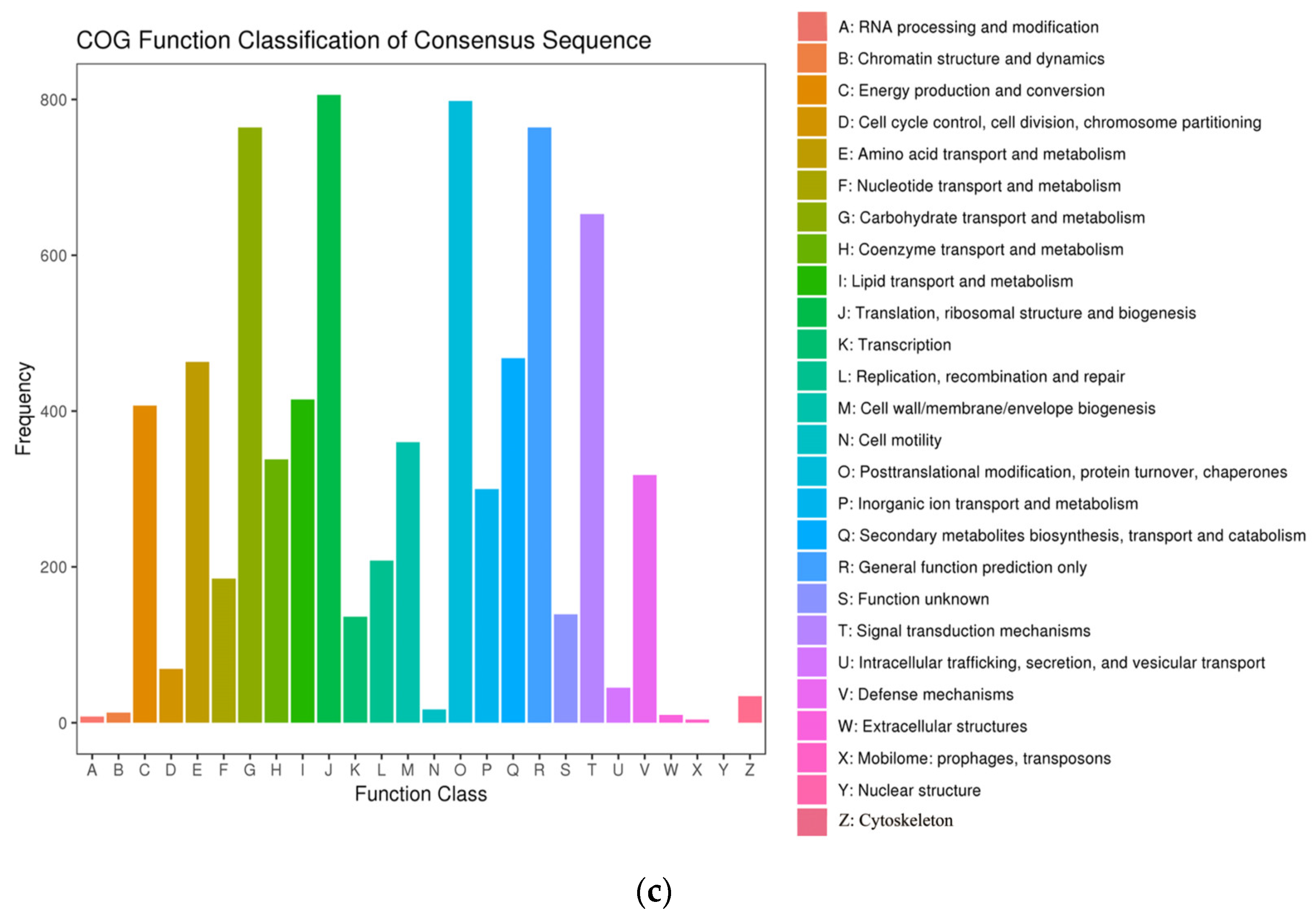
2.4. Cluster of Orthologous Groups Analysis of DEGs
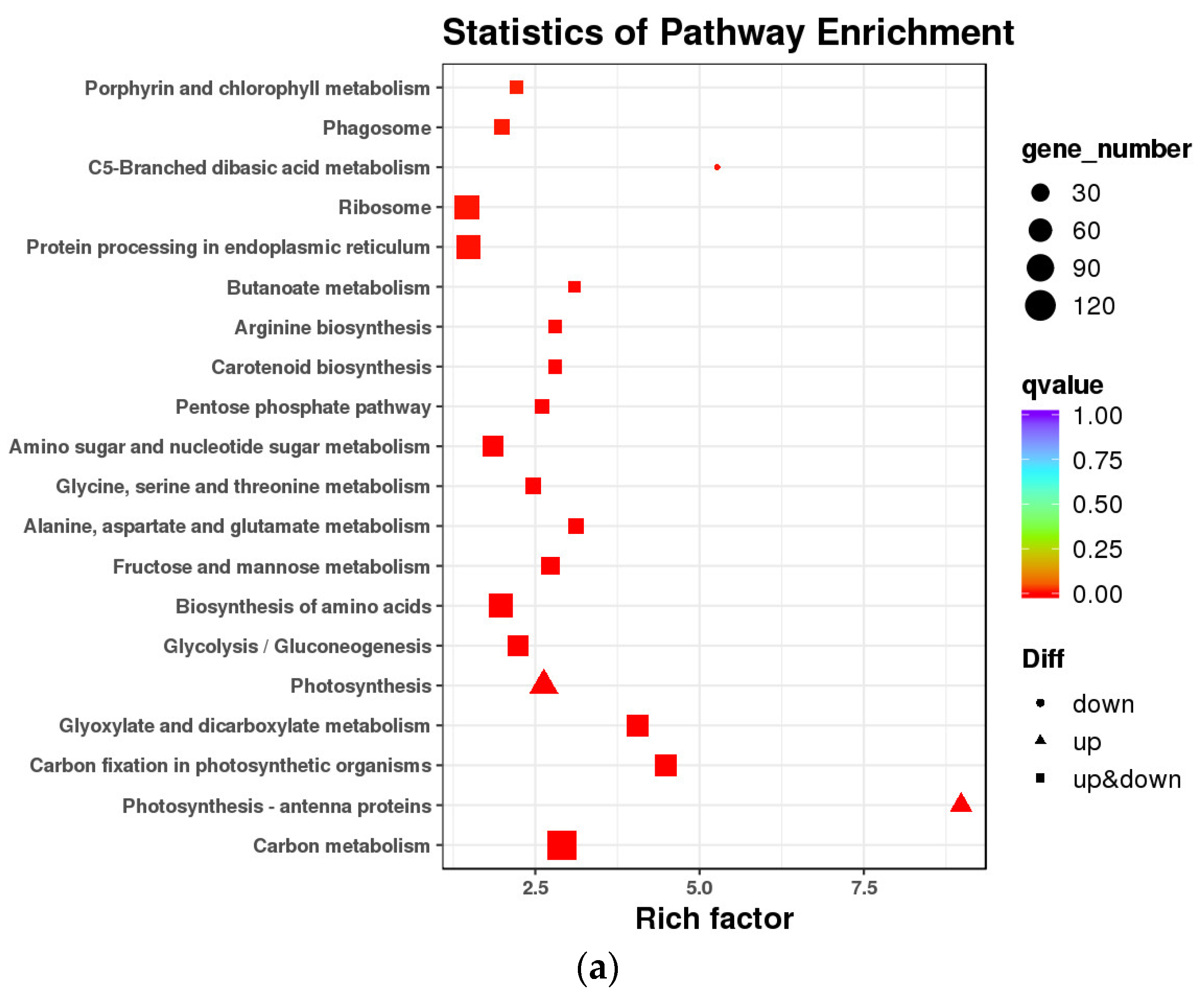
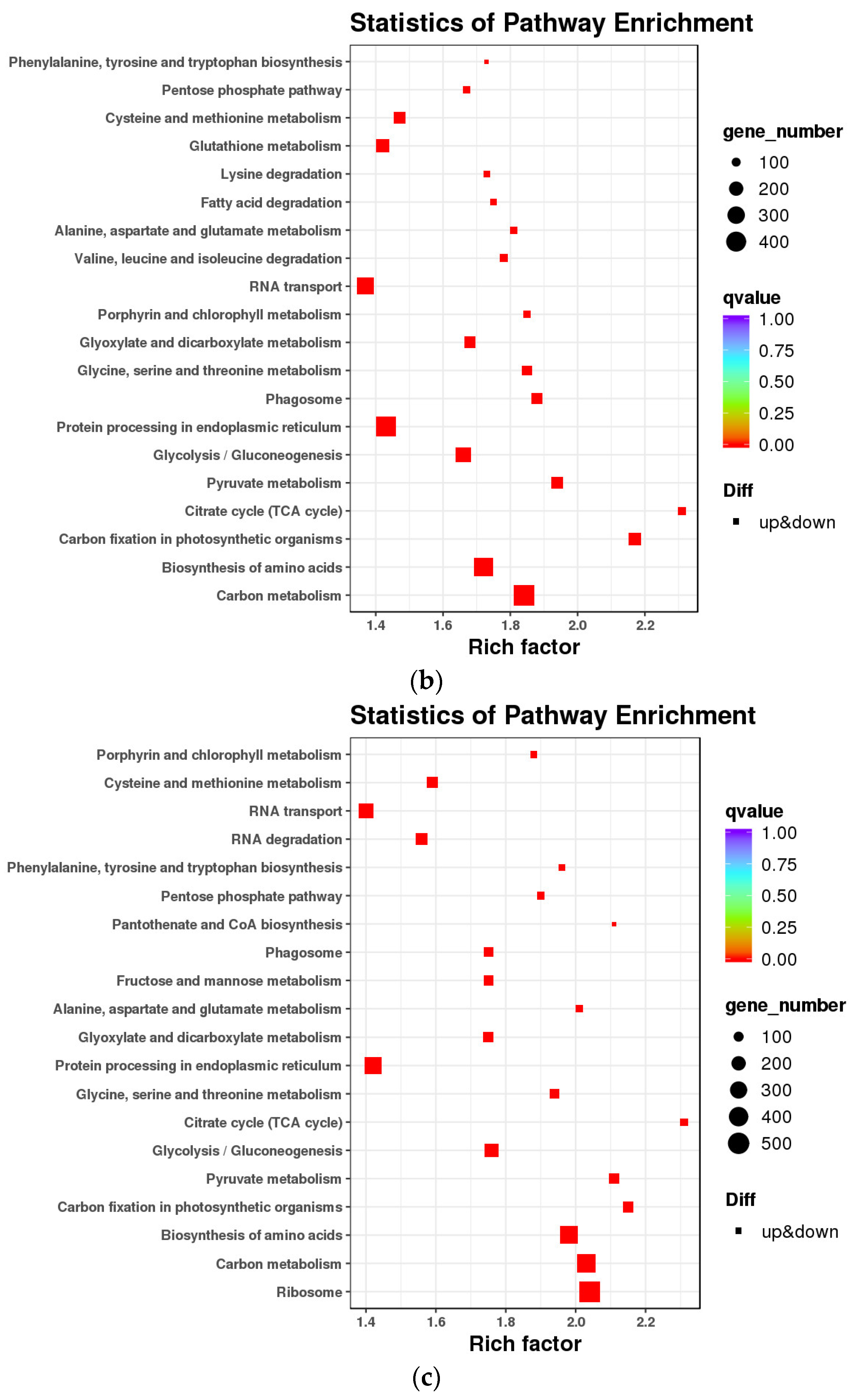
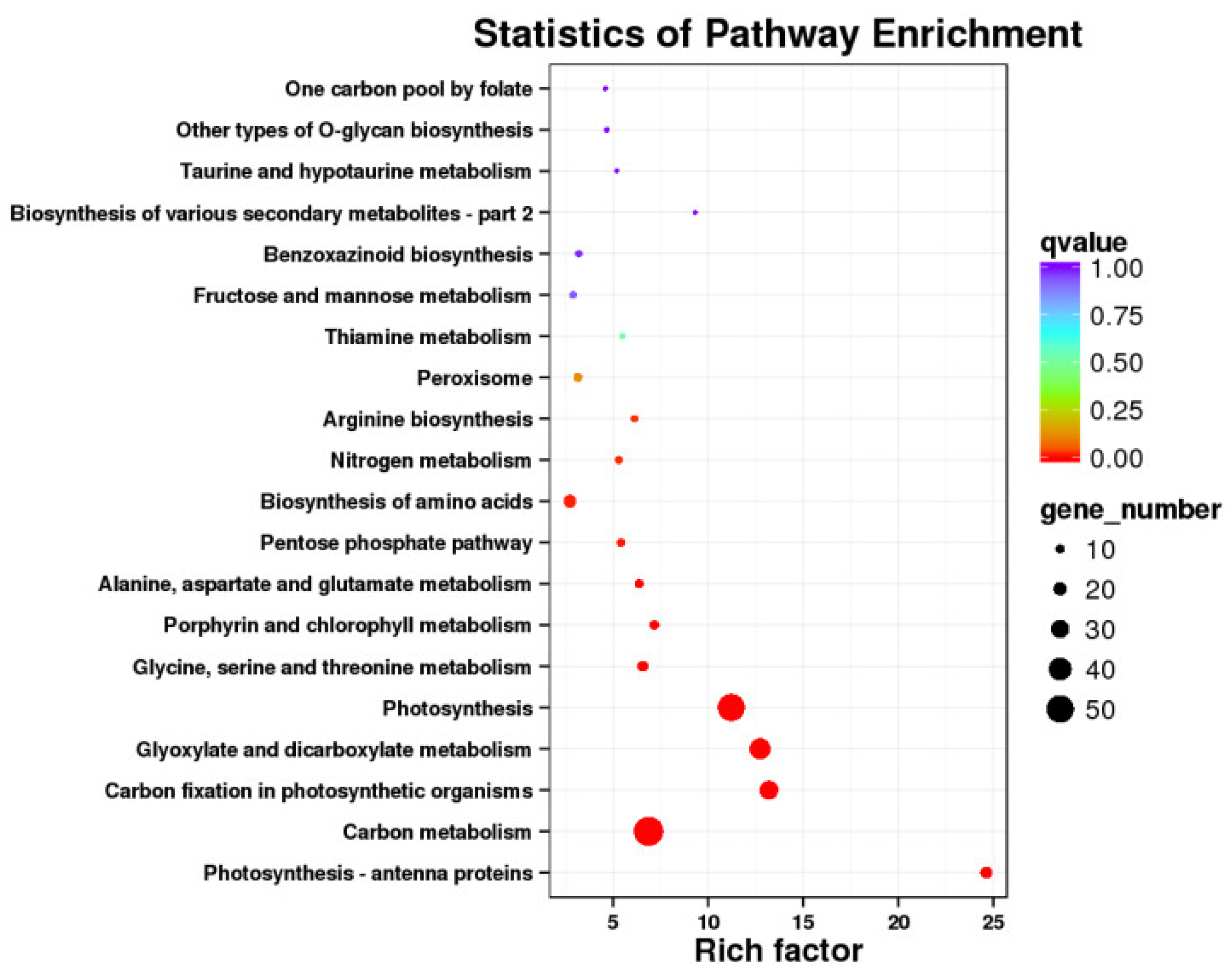
2.5. KEGG Pathway Enrichment Analysis of DEGs
2.6. Candidate Genes Related to Abnormal Kernels Development
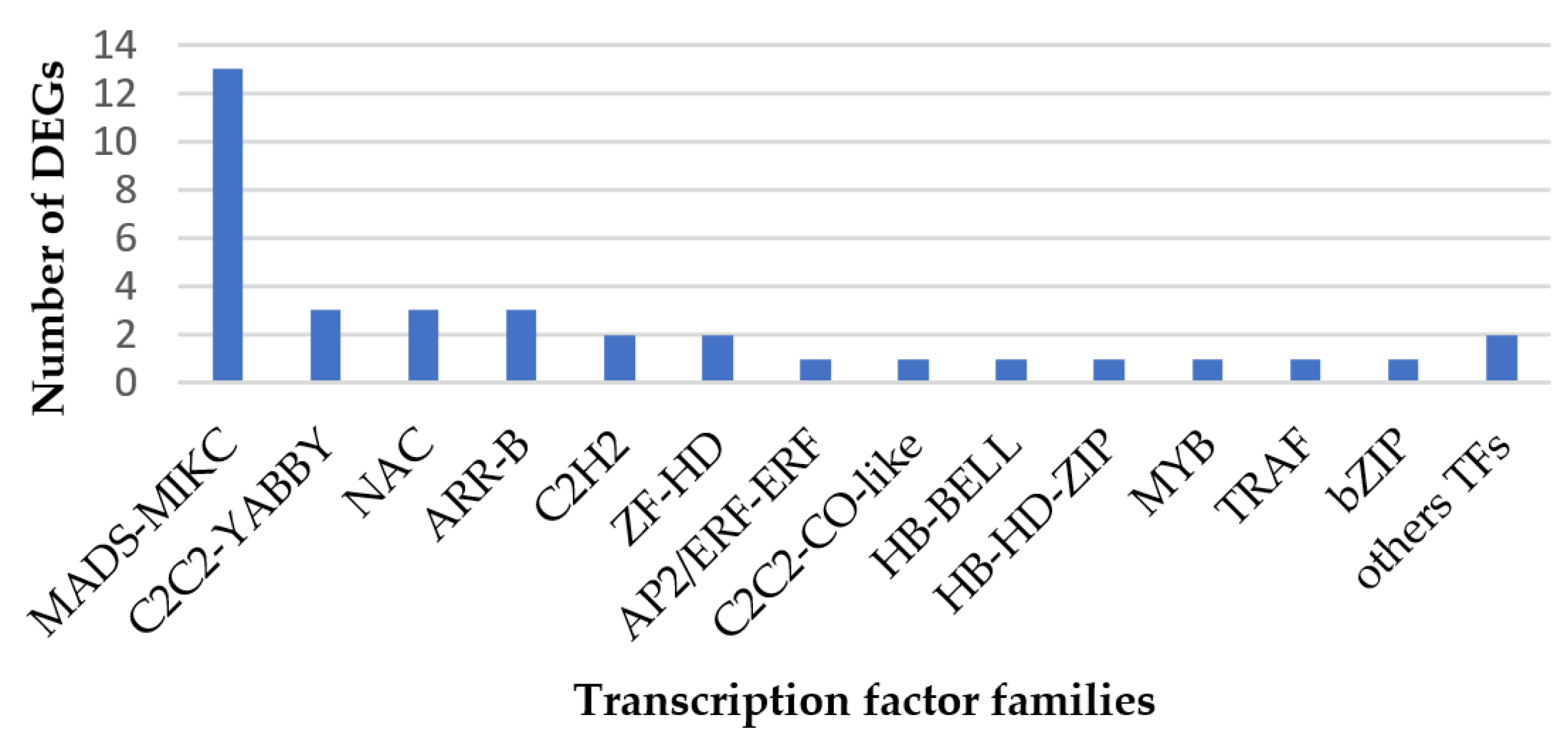
2.7. Transcription Factors Prediction
2.8. DEGs Related to Plant Hormone Biosynthesis
2.9. DEGs Involved in Response to Stimulus
2.10. qRT-PCR Analysis
3. Discussion
4. Materials and Methods
4.1. Fungal Material and Culture
4.2. Plants Material and Inoculation
4.3. RNA Extraction, cDNA Library Construction, and Sequencing
4.4. Quality Control, Mapping, and DEGs Screening
4.5. Functional Annotation of DEGs
4.6. Validation of RNA-Seq Results Using qRT-PCR
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jia, W.M.; Zhou, Y.L.; Duan, X.Y.; Yong, L.O.; Ding, S.L.; Cao, X.R.; Bruce, D.F. Assessment of risk of establishment of wheat dwarf bunt (Tilletia controversa) in China. J. Integr. Agric. 2013, 12, 87–94. [Google Scholar] [CrossRef]
- Shakoor, M.A.; Ahmad, M.; Ghazali, H.Z.; Ahmad, S.; Balouch, M.A.; Anjum, R.; Hussain, M. Chemotherapy of karnal bunt of wheat. Int. J. Adv. Res. Biol. Sci. 2014, 1, 163–188. [Google Scholar]
- Hoffmann, J.A. Bunt of wheat. Plant Dis. 1982, 66, 979–986. [Google Scholar] [CrossRef]
- Trione, E.J. Dwarf bunt of wheat and its importance in international wheat trade. Plant Dis. 1982, 66, 1083–1088. [Google Scholar] [CrossRef]
- Holton, C.S. Preliminary investigations of dwarf bunt of wheat. Phytopathology 1941, 31, 74–82. [Google Scholar]
- Wagner, F. Occurrence, spore germination, and infection of dwarf bunt on wheat. Z. Pflanzenkrankh. Pflanzenpathol. Pflanzensch. 1950, 1, 1–13. [Google Scholar]
- Slinkard, A.E.; Ellicitt, F.C. The effect of bunt incidence on the yield of wheat in eastern Washington. Agron. J. 1954, 46, 439–441. [Google Scholar] [CrossRef]
- Baylis, R.J. Studies of Tilletia controversa, the cause of dwarf bunt of winter. Can. J. Bot. 1958, 36, 17–32. [Google Scholar] [CrossRef]
- Tyler, L.J.; Jensen, N.F. Some factors that influence development of dwarf bunt in winter wheat. Phytopathology 1958, 48, 565–571. [Google Scholar]
- Purdy, L.H.; Kendrick, E.L.; Hoffmann, J.A.; Holton, C.S. Dwarf bunt of wheat. Annu. Rev. Microbiol. 1963, 17, 199–222. [Google Scholar] [CrossRef]
- Martens, J.; Seaman, W.W.; Atkinson, T.H. Diseases of Field Crops in Canada: An Illustrated Compendium; Canadian Phytopathological Society: Harrow, ON, USA, 1984; 160p. [Google Scholar]
- Muhae-Ud-Din, G.; Chen, D.; Liu, T.; Chen, W.; Gao, L. Characterization of the wheat cultivars against Tilletia controversa Kühn, causal agent of wheat dwarf bunt. Sci. Rep. 2020, 10, 9029. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Singh, D.V.; Srivastava, K.D. Studies on the ontogeny of teliospore ornamentation of Neovossia indica observed through scanning electron microscopy. Indian Phytopathol. 1999, 52, 417–419. [Google Scholar]
- Cohn, J.; Sessa, G.; Martin, G.B. Innate immunity in plants. Curr. Opin. Immunol. 2001, 13, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Gurjar, M.S.; Jain, S.; Aggarwal, R.; Saharan, M.S.; Kumar TP, J.; Kharbikar, L. Transcriptome analysis of wheat–Tilletia indica interaction provides defense and pathogenesis-related genes. Plants 2022, 11, 3061. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.; Schullehner, K.; Dick, R.; Fiesselmann, A.; Gierl, A. Benzoxazinoid biosynthesis, a model for evolution of secondary metabolic pathways in plants. Phytochemistry 2009, 70, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.S.; Qin, D.D.; Muhae-Ud-Din, G.; Liu, T.G.; Chen, W.Q.; Gao, L. Characterization of histological changes at the tillering stage (Z21) in resistant and susceptible wheat plants infected by Tilletia controversa Kühn. BMC Plant Biol. 2021, 21, 49. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Ren, Z.; Muhae-Ud-Din, G.; Guo, Q.; Liu, T.; Chen, W.; Gao, L. Transcriptomics Analysis of Wheat Tassel Response to Tilletia laevis Kühn, Which Causes Common Bunt of Wheat. Front. Plant Sci. 2022, 13, 823907. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, L.; Chen, Y.; Wei, L.; Hao, Y.; Yu, Z.; Wang, Z.; Zhang, H.; Zhang, X.; Li, M.; et al. The Regulatory Network of CMPG1-V in Wheat-Blumeria graminis f. sp. tritici interaction revealed by temporal profiling using RNA-Seq. Int. J. Mol. Sci. 2020, 21, 5967. [Google Scholar] [CrossRef]
- Kumar, J.; Gunapati, S.; Kianian, S.F.; Singh, S.P. Comparative analysis of transcriptome in two wheat genotypes with contrasting levels of drought tolerance. Protoplasma 2018, 255, 1487–1504. [Google Scholar] [CrossRef]
- Zou, H.; Tzarfati, R.; Hübner, S.; Krugman, T.; Fahima, T.; Abbo, S.; Saranga, Y.; Korol, A.B. Transcriptome profiling of wheat glumes in wild emmer, hulled landraces and modern cultivars. BMC Genom. 2015, 16, 777. [Google Scholar] [CrossRef]
- Liu, Y.; Hou, J.; Wang, X.; Li, T.; Majeed, U.; Hao, C.; Zhang, X. The NAC transcription factor NAC019-A1 is a negative regulator of starch synthesis in wheat developing endosperm. J. Exp. Bot. 2020, 71, 5794–5807. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Cao, H.; Liu, J.-d.; Zuo, J.-h.; Fang, Y.; Lin, C.-T.; Sun, R.-z.; Li, W.-l.; Liu, Y.-x. Insights into transcriptional characteristics and homoeolog expression bias of embryo and deembryonated kernels in developing grain through RNA-Seq and Iso-Seq. Funct. Integr. Genom. 2019, 19, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Yu, H.; Han, W.; Gao, F.; Liu, T.; Liu, B.; Kang, X.; Gao, J.; Chen, W. Development of a SCAR marker for molecular detection and diagnosis of Tilletia controversa Kühn, the causal fungus of wheat dwarf bunt. World J. Microbiol. Biotechnol. 2014, 30, 3185–3195. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wei, X.; Gao, L.; Chen, W.; Liu, T.; Liu, B. iTRAQ-based proteomic analysis of wheat bunt fungi Tilletia controversa, T. caries, and T. foetida. Curr. Microbiol. 2018, 75, 1103–1107. [Google Scholar] [CrossRef]
- Berger, S. Visualization of dynamics of plant-pathogen interaction by novel combination of chlorophyll fuorescence imaging and statistical analysis: Diferential efects of virulent and avirulent strains of P. syringae and of oxylipins on A. thaliana. J. Exp. Bot. 2007, 58, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Ehness, R.; Ecker, M.; Godt, D.E.; Roitsch, T. Glucose and stress independently regulate source and sink metabolism and defense mechanisms via signal transduction pathways involving protein phosphorylation. Plant Cell. 1997, 9, 1825–1841. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Sinha, A.K.; Roitsch, T. Plant physiology meets phytopathology: Plant primary metabolism and plant-pathogen interactions. J. Exp. Bot. 2007, 58, 4019–4026. [Google Scholar] [CrossRef]
- Siemens, J. Transcriptome analysis of Arabidopsis clubroots indicate a key role for cytokinins in disease development. Mol. Plant-Microbe Interact. 2006, 19, 480–494. [Google Scholar] [CrossRef]
- Ren, Z.; Liu, J.; Din GM, U.; Zhang, H.; Du, Z.; Chen, W.; Liu, T.; Zhang, J.; Zhao, S.; Gao, L. Transcriptome analysis of wheat spikes in response to Tilletia controversa Kühn which cause wheat dwarf bunt. Sci. Rep. 2020, 10, 21567. [Google Scholar] [CrossRef]
- Gebbing, T.; Schnyder, H. 13C Labeling kinetics of sucrose in glumes indicates significant refixation of respiration CO2 in the wheat ear. Aust. J. Plant Physiol. 2001, 28, 1047–1053. [Google Scholar]
- Feng, B.; Li, H.W.; Wang, F.H.; Kong, L.A.; Zhang, B.; Wang, Z.S.; Li, S.D. Photosynthetic characteristic and contribution of non-foliar photosynthetic organs to grain yield in wheat. Plant Physiol. J. 2019, 55, 32–40. [Google Scholar]
- Chen, T.C.; Yang, G.H.; Jiang, D.X.; Wu, S.L.; Chen, J.Q.; Xie, B.G.; Jiang, Y.J.; Chen, B.Z. Effects of MADS-box transcription factors regulated Phosphatidylinositol signalling system on Volvariella volvacea during storage. Acta Edulis Fungi 2021, 28, 11–17. [Google Scholar]
- Zhou, H.H.; Huang, H.; Xu, B.L.; Su, X.; Deng, R.; Hong, Y.B.; Jiang, M.; Song, F.M.; Zhang, H.J. Biological function of NAC transcription factors in plant abiotic and biotic stress responses. Plant Physiol. J. 2017, 53, 1372–1382. [Google Scholar]
- Sicilia, A.; Russo, R.; Caruso, M.; Arlotta, C.; Di Silvestro, S.; Gmitter, F.G., Jr.; Gentile, A.; Nicolosi, E.; Lo Piero, A.R. Transcriptome analysis of Plenodomus tracheiphilus infecting rough lemon (Citrus jambhiri Lush.) indicates a multifaceted strategy during host pathogenesis. Biology 2022, 11, 761. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Pamphilis, C.W.; Ma, H. Phylogenetic analysis of the plant-specific zinc finger-homeobox and mini zinc finger gene families. J. Integr. Plant Biol. 2008, 50, 1031–1045. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Chao, M.N.; Du, H.Y.; Yu, D.Y.; Huang, F. Cloning and expression analysis of ZF-HD transcription factor GmZHD1 in Glccine max. Acta Agric. Boreali—Sin. 2017, 32, 1–7. [Google Scholar]
- Liu, Y.; Wen, X.; Gu, D.; Guo, Q.; Zeng, A.; Li, C.; Liao, Y. Effect of Polyamine on grain filling of winter wheat and its physiological mechanism. Acta Agron. Sin. 2013, 39, 712–719. [Google Scholar] [CrossRef]
- Liu, K. Regulation of Abscisic Acid and Ethylene to Grain Filling in rice and Wheat and Its Physiological Mechanism. Ph.D. Thesis, Yangzhou University, Yangzhou, China, 2008. (In Chinese). [Google Scholar]
- Yang, J.C.; Wang, Z.Q.; Zhu, Q.S.; Su, B.L. Regulation of ABA and GA to the grain filling of rice. Acta Agron. Sin. 1999, 25, 343–348. [Google Scholar]
- Wang, H.B. Effects of Wxogenous IAA and 6-BA Treatment on Grain Weight and Filling Rate of Different Allelic Variations of TaGW2-6A in Wheat; Northwest A&F University: Xianyang, China, 2020; p. 239. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Wang, G.D.; Zhang, S.; Ma, X.C.; Wang, Y.; Kong, F.Y.; Meng, Q.W. A stress-associated NAC transcription factor (Sl NAC35) from tomato plays a positive role in biotic and abiotic stresses. Physiol. Plant. 2016, 158, 45–64. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Michael, A.; Catherine, A.B.; Judith, A.B.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Gene Ontol. Consort. Nat Genet 2000, 25, 25–29. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. The Clusters of Orthologous Groups (COGs) Database: Phylogenetic classification of proteins from complete genomes. In The NCBI Handbook; Antenna House: Newport, DE, USA, 2002. [Google Scholar]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Aggarwal, R.; Gurjar, M.S.; Sharma, S.; Jain, S.; Saharan, M.S. Identification and expression analysis of pathogenicity-related genes in Tilletia indica inciting Karnal bunt of wheat. Australas. Plant Pathol. 2020, 49, 393–402. [Google Scholar] [CrossRef]
- Pfaf, M.W. A new mathematical model for relative quantifcation in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Numbers | Samples | Clean Reads | Clean Bases | GC Content | Q30 (%) |
|---|---|---|---|---|---|
| 1 | I-YLC1 | 33,846,470 | 10,118,821,246 | 55.69% | 95.46% |
| 2 | I-YLC2 | 38,588,627 | 11,517,825,572 | 54.84% | 95.00% |
| 3 | I-YLC3 | 37,958,897 | 11,343,101,536 | 54.37% | 95.01% |
| 4 | I-YLI1 | 37,731,986 | 11,276,627,552 | 57.82% | 93.91% |
| 5 | I-YLI2 | 35,408,404 | 10,561,674,364 | 58.70% | 93.97% |
| 6 | I-YLI3 | 43,665,455 | 13,042,812,834 | 58.55% | 93.90% |
| 7 | II-YLC1 | 37,424,109 | 11,172,643,120 | 51.80% | 91.54% |
| 8 | II-YLC2 | 39,668,019 | 11,817,094,732 | 52.53% | 91.17% |
| 9 | II-YLC3 | 43,770,044 | 13,052,803,514 | 51.85% | 91.92% |
| 10 | II-YLI1 | 41,044,118 | 12,272,430,466 | 56.42% | 93.91% |
| 11 | II-YLI2 | 41,626,954 | 12,447,001,058 | 56.32% | 93.99% |
| 12 | II-YLI3 | 43,680,494 | 13,053,542,618 | 56.40% | 93.96% |
| 13 | III-YLC1 | 41,024,047 | 12,249,838,096 | 57.04% | 93.24% |
| 14 | III-YLC2 | 42,236,376 | 12,634,731,488 | 56.04% | 94.14% |
| 15 | III-YLC3 | 36,526,906 | 10,892,077,864 | 56.40% | 93.28% |
| 16 | III-YLI1 | 43,832,299 | 13,101,853,390 | 58.51% | 93.54% |
| 17 | III-YLI2 | 43,363,058 | 12,976,495,304 | 58.83% | 93.85% |
| 18 | III-YLI3 | 45,101,790 | 13,488,891,936 | 58.73% | 93.80% |
| Gene ID | I-YLC vs. I-YLI | II-YLC vs. II-YLI | III-YLC vs. III-YLI | KEGG_Pathway_Annotation |
|---|---|---|---|---|
| TraesCS4D02G258000 | up | up | up | - |
| TraesCS5B02G162100 | up | up | up | - |
| TraesCS7D02G419400 | up | up | up | Plant hormone signal transduction (ko04075) |
| TraesCS1B02G229400 | down | down | down | MAPK signaling pathway—plant (ko04016); Plant hormone signal transduction (ko04075) |
| TraesCS1B02G237400 | down | down | down | - |
| TraesCS1D02G146900 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS1D02G218200 | down | down | down | MAPK signaling pathway—plant (ko04016); Plant hormone signal transduction (ko04075) |
| TraesCS2A02G309300 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS2D02G234100 | up | down | down | Protein processing in the endoplasmic reticulum (ko04141) |
| TraesCS3A02G006600 | down | down | down | - |
| TraesCS3A02G145300 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS3A02G233000 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS3A02G307100 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS3A02G371800 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS3A02G372000 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS3A02G372100 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS3B02G007400 | down | down | down | - |
| TraesCS3B02G404300 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS3B02G404400 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS3D02G292100 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS3D02G364900 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS4B02G161800 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS4D02G210900 | down | up | up | MAPK signaling pathway—plant (ko04016); Plant hormone signal transduction (ko04075) |
| TraesCS4D02G231900 | down | down | down | - |
| TraesCS5A02G243900 | down | down | down | - |
| TraesCS5A02G265600 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS5B02G265300 | down | down | down | Plant hormone signal transduction (ko04075) |
| TraesCS5D02G206700 | down | down | down | MAPK signaling pathway—plant (ko04016); Plant hormone signal transduction (ko04075) |
| TraesCS5D02G432700 | down | up | up | Glutathione metabolism (ko00480) |
| TraesCS7B02G019600 | down | up | up | - |
| Triticum_aestivum_newGene_3982 | down | down | down | Plant hormone signal transduction (ko04075) |
| Triticum_aestivum_newGene_8549 | down | down | down | - |
| Gene ID | KEGG_Pathway_Annotation |
|---|---|
| TraesCS1A02G403300 | Photosynthesis–antenna proteins (ko00196) |
| TraesCS1B02G432700 | Photosynthesis–antenna proteins (ko00196) |
| TraesCS1D02G411300 | Photosynthesis–antenna proteins (ko00196) |
| TraesCS2A02G206200 | Photosynthesis–antenna proteins (ko00196) |
| TraesCS2D02G209900 | Photosynthesis–antenna proteins (ko00196) |
| TraesCS3B02G133400 | Galactose metabolism (ko00052) |
| TraesCS4B02G122300 | - |
| TraesCS4B02G200100 | Glutathione metabolism (ko00480) |
| TraesCS4D02G258000 | - |
| TraesCS4D02G319100 | - |
| TraesCS5A02G322500 | Photosynthesis–antenna proteins (ko00196) |
| TraesCS5A02G350600 | Photosynthesis–antenna proteins (ko00196) |
| TraesCS5B02G162100 | - |
| TraesCS5B02G572400 | Glutathione metabolism (ko00480) |
| TraesCS5D02G413300 | Alpha-linolenic acid metabolism (ko00592) |
| TraesCS6A02G372100 | - |
| TraesCS6D02G152700 | Photosynthesis–antenna proteins (ko00196) |
| TraesCS6D02G356100 | - |
| TraesCS7A02G177700 | Protein processing in the endoplasmic reticulum (ko04141) |
| Gene ID | qRT-PCR | FPKM | Validated | Gene ID | qRT-PCR | FPKM | Validated |
|---|---|---|---|---|---|---|---|
| TraesCS1A02G403300 | 0.2 ± 0.10 gh | 44.83 | + | TraesCS5B02G572400 | 0.64 ± 0.03 ef | 30.59 | + |
| TraesCS1B02G432700 | 0.28 ± 0.01 gh | 55.71 | + | TraesCS5D02G413300 | 2.9 ± 1.67 a | 8.4 | + |
| TraesCS2A02G206200 | 0.32 ± 0.13 gh | 32.49 | + | TraesCS6D02G152700 | 1.65 ± 0.15 c | 24.14 | + |
| TraesCS2D02G209900 | 0.37 ± 0.05 fg | 37.23 | + | TraesCS7A02G177700 | 0.95 ± 0.63 d | 24.43 | + |
| TraesCS3B02G133400 | 0.03 ± 0.11 h | 76.43 | + | TraesCS7D02G419400 | 0.22 ± 0.03 gh | 12.13 | + |
| TraesCS4B02G122300 | 0.19 ± 0.29 gh | 4.81 | + | TraesCS1B02G229400 | 0.26 ± 0.2 gh | 5.12 | + |
| TraesCS4B02G200100 | 0.30 ± 0.11 gh | 24.14 | + | TraesCS2A02G309300 | 0.12 ± 0.06 gh | 4.34 | + |
| TraesCS4D02G258000 | 0.13 ± 0.01 gh | 56.0 | + | TraesCS2D02G234100 | 0.16 ± 0.09 gh | 5.29 | + |
| TraesCS4D02G319100 | 1.97 ± 0.72 b | 4.02 | + | TraesCS3D02G364900 | 0.17 ± 0.1 gh | 19.24 | + |
| TraesCS5A02G322500 | 0.43 ± 0.41 fg | 15.25 | + | TraesCS4D02G210900 | 0.3 ± 0.27 gh | 15.05 | + |
| TraesCS5A02G350600 | 1.48 ± 0.02 c | 63.58 | + | TraesCS5D02G206700 | 0.32 ± 0.27 gh | 3.49 | + |
| TraesCS5B02G162100 | 0.76 ± 0.13 de | 38.2 | + | TraesCS5D02G432700 | 0.4 ± 0.96 gh | 214.13 | + |
| Gene ID | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| TraesCS1A02G403300 | ATGTTCGGCTTCTTCGTG | CCAGGCGTTGTTGTTGAC |
| TraesCS1B02G432700 | TTCTCCATGTTCGGCTTCT | CCAGGCGTTGTTGTTGAC |
| TraesCS2A02G206200 | CTGGTGATCGGGTACATC | CAGAGTCTCCTTCTTCTCC |
| TraesCS2D02G209900 | ATCGGGTACATCGAGTTC | CAGAGTCTCCTTCTTCTCC |
| TraesCS3B02G133400 | GCTACAACACCGAGAATG | CTAGACCCGAATCTCCAA |
| TraesCS4B02G122300 | CCTGAAGCTCTCCTACAC | GCTGGTCGTAGTAGAGTG |
| TraesCS4B02G200100 | TGCTGCCTGATGATTCTG | TCGCTGAACTTTCCCAAG |
| TraesCS4D02G258000 | GTGGTGCTCTACGACCTC | TAGGCGTCCAGGTTGTTG |
| TraesCS4D02G319100 | TGGCTCTTCAGTTCCTCT | GACCTTCTTCTTCTCCTTGG |
| TraesCS5A02G322500 | TTCTCCATGTTCGGCTTCT | CCAGGCGTTGTTGTTGAC |
| TraesCS5A02G350600 | ATGTTCGGCTTCTTCGTG | CCAGGCGTTGTTGTTGAC |
| TraesCS5B02G162100 | AGCGGCATATTTACTTCG | TCAGCATCAAGGTAGGTT |
| TraesCS5B02G572400 | CCGAGAAGTTGCTGTCAC | CTGAGGTCTGCGATGGAT |
| TraesCS5D02G413300 | GATCTTGCTGCCAATGCT | AAGTTGATGCGGTCCTTG |
| TraesCS6D02G152700 | AAGAGCGAGAAGGAGATG | TGAGGATGTTGTTGTTGAC |
| TraesCS7A02G177700 | AGCTTCTTCTCGCAGGAC | CCTCCATCAGAGACAGCAG |
| TraesCS7D02G419400 | CCAGTCCAAGAACCAGTA | CGACGACAGGTAGAAAGT |
| TraesCS1B02G229400 | CATACGATTCAAGGAGGTT | GAAAGTAGTTGCTGGAAGA |
| TraesCS2A02G309300 | CTAACTGTCTCAGGTGATG | CTTCTAACTTGGCGACTT |
| TraesCS2D02G234100 | GGCAATAGACTCTGTTCAGG | GGCTCCATCAGGTTCTTC |
| TraesCS3D02G364900 | CGGCAGTCTTCCATCTTC | ACTCCTCGGCATTCCATA |
| TraesCS4D02G210900 | ATACGGCGGCATAGACTG | TACTTGGCGGGCTCAATC |
| TraesCS5D02G206700 | CGTTCATTGGCTGCTTCTA | GGTCAAGATTGCCGACTC |
| TraesCS5D02G432700 | CTGGACATCCTCAAGACC | CTCTCGTAGCTGTGGAAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, S.; Zhang, Z.; Shen, T.; Chen, J.; Liu, Q. Kernel Transcriptome Profiles of Susceptible Wheat Genotypes in Response to Wheat Dwarf Bunt. Int. J. Mol. Sci. 2023, 24, 17281. https://doi.org/10.3390/ijms242417281
Su S, Zhang Z, Shen T, Chen J, Liu Q. Kernel Transcriptome Profiles of Susceptible Wheat Genotypes in Response to Wheat Dwarf Bunt. International Journal of Molecular Sciences. 2023; 24(24):17281. https://doi.org/10.3390/ijms242417281
Chicago/Turabian StyleSu, Shenqiang, Zihao Zhang, Tong Shen, Jing Chen, and Qi Liu. 2023. "Kernel Transcriptome Profiles of Susceptible Wheat Genotypes in Response to Wheat Dwarf Bunt" International Journal of Molecular Sciences 24, no. 24: 17281. https://doi.org/10.3390/ijms242417281
APA StyleSu, S., Zhang, Z., Shen, T., Chen, J., & Liu, Q. (2023). Kernel Transcriptome Profiles of Susceptible Wheat Genotypes in Response to Wheat Dwarf Bunt. International Journal of Molecular Sciences, 24(24), 17281. https://doi.org/10.3390/ijms242417281