3.1. Synthesis
3.1.1. General Information
Methyl 4,6-
O-
p-methoxybenzylidene-α-D-glucopyranoside (
3) [
28], methyl 2,3-di-
O-octyl- and methyl 2,3-di-
O-butyl-4,6-
O-
p-methoxybenzylidene-α-D-glucopyranoside (
4b [
29] and
4c [
30]), methyl 2,3-di-
O-octyl- and methyl 2,3-di-
O-butyl-4-
O-
p-methoxybenzylidene-α-D-glucopyranoside (
5b [
29] and
5c [
30]), the mono-bromo-mono-
p-toluenesulfonic acid derivative of ethylene glycol (
6) [
33],
N-acetyl-2-
S-propargyl-neuraminic acid (
9) [
34], methyl
N-acetyl-2-
S-propargyl-neuraminic acid (
10) [
34], 3-(4,6-di-
O-acetyl-2,3-dideoxy-α-D-
erythro-hex-2-enopyranosyl)-1-propene (
14) [
36], monoallyl-tetraethylene glycol (
19), and methyl
N-acetyl-4,7,8,9-tetra-
O-acetyl-2-dezoxy-2-thio-neuraminic acid (
16) [
37] were synthesized according to the literature. Tri-
O-acetyl-D-glucal (
13) was purchased from Merck KGaA (Darmstadt, Germany).
TLC was performed on Kieselgel 60 F254 (Merck) with detection by immersion into ammonium molybdate-sulfuric acid solution followed by heating. Flash column chromatography was performed using Silica gel 60 (Merck 0.040–0.063 mm). The photoinitiated reactions were carried out in a borosilicate vessel by irradiation with a low-pressure Hg-lamp (Osram Supratec UV, HTC 150–211, 150 W, 230 V, R7s), giving maximum emission at 365 nm, without any caution to exclude air or moisture [
38].
Conventional 1D and 2D
1H and
13C NMR spectra (
1H-COSY,
1H-
13C-HSQC,
1H-
13C-HMBC) were recorded using a Bruker DRX-400 spectrometer (at 298 K or 300 K) and a 500 MHz (Bruker, Billerica, MA, USA) Avance II. spectrometer (at 310 K) equipped with a TXI probe head. Chemical shifts are referenced to Me
4Si (0.00 ppm for
1H) and to the solvent residual signals (CDCl
3, DMSO-d
6 or pyridine-d
5). NMR spectra of the synthesized compounds can be found in
Supplementary Information.
MALDI-TOF MS (BIFLEX) analyses of the compounds were carried out in the positive reflectron mode using a BIFLEX III mass spectrometer (Bruker, Bremen, Germany) equipped with delayed-ion extraction. In all cases, 19 kV acceleration voltage was used with pulsed ion extraction (PIETM). The positive ions were detected in the reflectron mode (20 kV). A nitrogen laser (337 nm, 3 ns pulse width, 106–107 W/cm2), operating at 4 Hz, was applied to produce laser desorption. Bruker Autoflex Speed mass spectrometer equipped with a time-of-flight (TOF) mass analyzer (Bruker, Bremen, Germany) was also used for MALDI-TOF MS (Autoflex) measurements. In all cases, 19 kV (ion source voltage 1) and 16.65 kV (ion source voltage 2) were used. For reflectron mode, 21 kV and 9.55 kV were applied as reflector voltage 1 and reflector voltage 2, respectively. A solid phase laser (355 nm, ≥100 μJ/pulse), operating at 500 Hz, was applied to produce laser desorption. A 2,5-Dihydroxybenzoic acid (DHB) was used as a matrix, and F3CCOONa as a cationising agent in DMF. ESI-QTOF MS measurements were carried out on a maXis II UHR ESI-QTOF MS instrument (Bruker, Bremen, Germany). The following parameters were applied for the electrospray ion source: capillary voltage: 3.5 kV; end plate offset: 500 V; nebulizer pressure: 0.4 bar; dry gas temperature: 200 °C; and dry gas flow rate: 4 L/min. Constant background correction was applied to each spectrum; the background was recorded before each sample by injecting the blank sample matrix (solvent). Na-formate calibrant was injected after each sample, which enabled external calibration during data evaluation. Mass spectra were recorded by micrOTOFcontrol version 2.2 (Bruker, Bremen, Germany) and processed by Compass DataAnalysis version 3.4 (Bruker Daltonik GmbH).
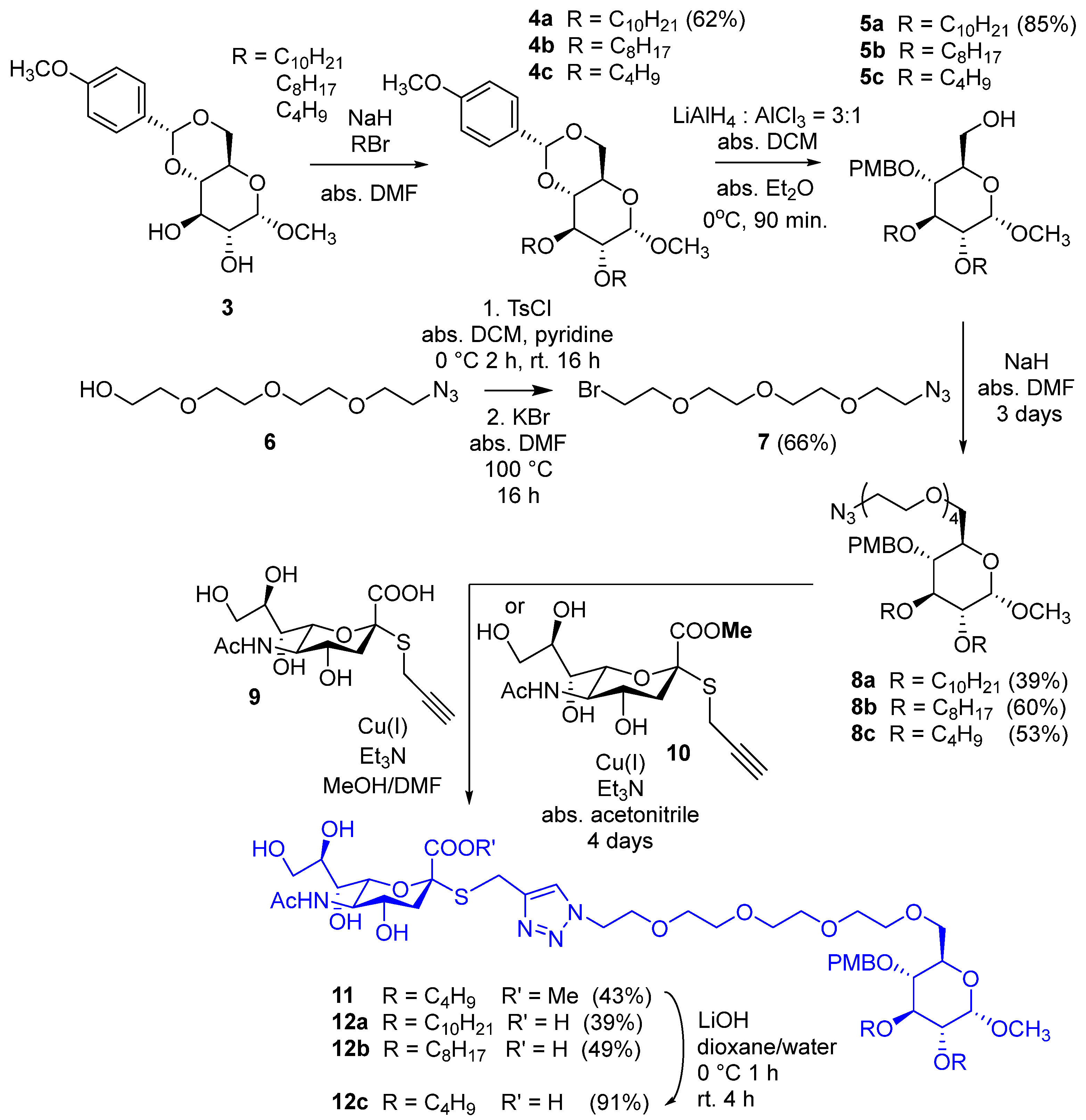
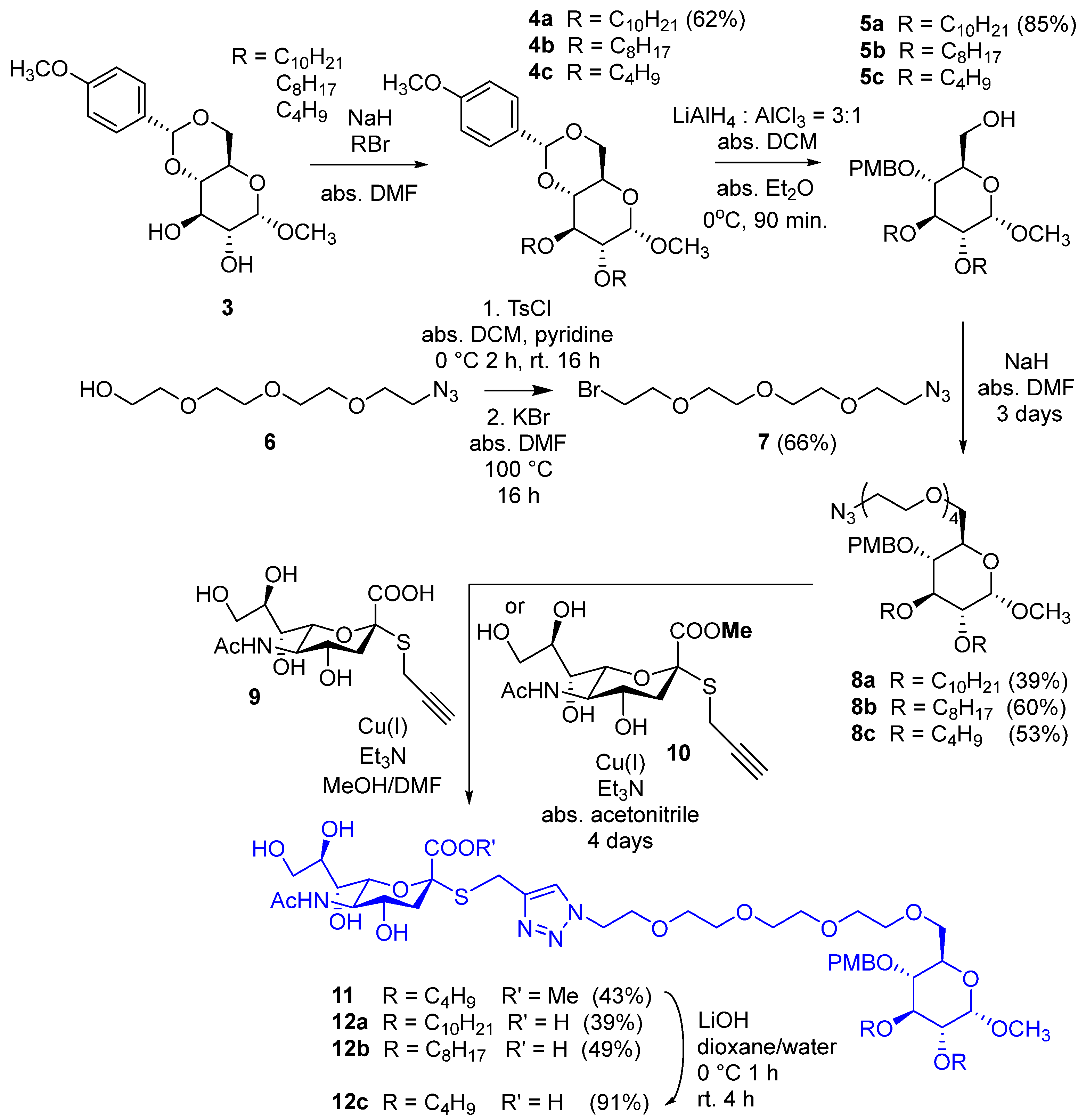
3.1.2. Methyl 2,3-di-O-decyl-4,6-O-p-methoxybenzylidene-α-D-glucopyranoside (4a)
Sodium hydride (180 mg, 4.48 mmol, 2 equiv./OH, 60% in mineral oil) was washed with hexane twice and then dried under argon. N,N-Dimethylformamide (10 mL) and methyl 4,6-O-p-methoxybenzylidene-α-D-glucopyranoside (3, 350 mg, 1.12 mmol) were added, and the mixture was stirred under argon atmosphere for 30 min. Then, n-decyl bromide (0.645 mL, 2.69 mmol, 1.2 equiv./OH) was added, and the mixture was stirred at room temperature overnight. Then, another portion of sodium hydride (50 mg) and n-decyl bromide (100 µL) were added, and the mixture was further stirred at room temperature for 2 days. Methanol (2 mL) and water (1 mL) were added successively to the reaction mixture, and it was stirred for 2 × 15 min. Then, the solvent was evaporated in a vacuum; the residue was dissolved in dichloromethane (100 mL) and was washed with water (2 × 10 mL). The organic phase was dried over Na2SO4 and filtered; then, the solvent was evaporated in a vacuum. The crude product was purified by flash column chromatography (hexane/ethyl acetate 95:5) to yield 4a (414 mg, 62%) as a white powder. Rf = 0.73 (hexane/acetone 7:3); Mp: 80–83 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.41 (d, J = 8.7 Hz, 2H, arom.), 6.88 (d, J = 8.7 Hz, 2H, arom.), 5.49 (s, 1H, Hac), 4.79 (d, J = 3.6 Hz, 1H, H-1), 4.25 (dd, J = 9.8 Hz, J = 4.5 Hz, 1H, H-6a), 3.80 (s, 3H, PMP-OCH3), 3.78–3.61 (m, 7H, H-3, H-5, H-6b, 2 × decyl OCH2), 3.47 (t, J = 9.3 Hz, 1H, H-4), 3.43 (s, 3H, C-1-OCH3), 3.34 (dd, J = 9.3 Hz, J = 3.7 Hz, 1H, H-2), 1.62–1.52 (m, 4H, 2 × decyl CH2), 1.32–1.23 (m, 28H, 14 × decyl CH2), 0.89–0.86 (m, 6H, 2 × decyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 160.1, 130.2 (2C, 2 × Cq arom.), 127.5, 113.6 (4C, arom.), 101.3 (1C, Cac), 99.3 (1C, C-1), 82.1, 80.6, 78.4 (3C, skeleton carbons), 73.6, 72.4 (2C, 2 × decyl OCH2), 69.2 (1C, C-6), 62.4 (1C, C-5), 55.4 (2C, 2 × OCH3), 32.1, 30.5, 30.2, 29.8, 29.7, 29.6, 29.5, 26.3, 26.1, 22.8 (16C, 16 × decyl CH2), 14.3 (2C, 2 × decyl CH3); MALDI-TOF MS (BIFLEX): m/z calcd for C35H60O7Na+: 615.42 [M+Na]+; found: 615.35.
3.1.3. Methyl 2,3-di-O-decyl-4-O-p-methoxybenzyl-α-D-glucopyranoside (5a)
The solution of 4a (400 mg, 0.6747 mmol) in the mixture of anhydrous dichloromethane (10 mL) and anhydrous diethyl ether (3.5 mL) was cooled to 0 °C under an argon atmosphere. Lithium aluminum hydride (115 mg, 3.036 mmol, 4.5 equiv.) was added in three portions. In another flask, anhydrous diethyl ether (3.5 mL) was cooled to 0 °C under an argon atmosphere, and aluminum chloride (135 mg, 1.01 mmol, 1.5 equiv.) was added. It was stirred for 10 min and then added to the solution of 4a. The reaction mixture was stirred at 0 °C under an argon atmosphere for 90 min. Ethyl acetate (15 mL) was added, and the mixture was stirred for 10 min; then, water (4 mL) was added, and it was stirred for another 10 min. The reaction mixture was filtered through Celite® (Sigma-Aldrich, Burlington, MA, USA) and washed with ethyl acetate (2 × 7 mL). The filtrate was diluted with ethyl acetate (50 mL) and washed with water (2 × 10 mL). The organic layer was dried over Na2SO4, filtered, and the solvent was evaporated. The residue was purified by flash column chromatography (hexane/acetone 9:1) to yield 5a (340 mg, 85%) as a colorless syrup. Rf = 0.26 (hexane/acetone 8:2); 1H NMR (400 MHz, CDCl3) δ (ppm) 7.28–7.26 (m, 2H, arom.), 6.89–6.87 (m, 2H, arom.), 4.82 (d, J = 10.7 Hz, 1H, PMB-CH2a), 4.76 (d, J = 3.5 Hz, 1H, H-1), 4.57 (d, J = 10.7 Hz, 1H, PMB-CH2b), 3.88–3.84 (m, 1H, decyl CH2a), 3.80 (s, 3H, PMB-OCH3), 3.76–3.57 (m, 7H, decyl CH2b, decyl CH2, H-3, H-5, H-6a,b), 3.41 (t, J = 9.3 Hz, 1H, H-4), 3.38 (s, 3H, C-1-OCH3), 3.27 (dd, J = 9.6 Hz, J = 3.5 Hz, 1H, H-2), 1.70 (t, J = 6.3 Hz, 1H, C-6-OH), 1.64–1.59 (m, 4H, 2 × decyl CH2,), 1.37–1.25 (m, 28H, 14 × decyl CH2), 0.90–0.86 (m, 6H, 2 × decyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 159.5, 130.6 (2C, 2 × Cq arom.), 129.9, 114.0 (4C, arom.), 98.2 (1C, C-1), 81.8, 81.0, 77.3 (3C, skeleton carbons), 74.7, 73.9, 71.9 (3C, 2 × decyl OCH2, PMB-CH2), 70.7 (1C, C-5), 62.2 (1C, C-6), 55.4, 55.2 (2C, 2 × OCH3), 32.1, 30.8, 30.2, 29.8, 29.6, 29.5, 26.4, 26.2, 22.8 (16C, 16 × decyl CH2), 14.2 (2C, 2 × decyl CH3); MALDI-TOF MS (BIFLEX): m/z calcd for C35H62O7Na+: 617.44 [M+Na]+; found: 617.49.
3.1.4. Compound 7
Tetratehylene glycol monoazide (6, 5 g, 22.8 mmol) was dissolved in anhydrous dichloromethane (50 mL); then, anhydrous pyridine (12 mL) was added, and the mixture was cooled to 0 °C. Tosyl chloride (13.04 g, 68.4 mmol. 3 equiv.) was dissolved in anhydrous dichloromethane (100 mL) and was added dropwise to the solution of 6 in 2 h. The mixture was stirred overnight. After 16 h, dichloromethane was evaporated in a vacuum; distilled water (20 mL) was added to the residue, and the mixture was stirred for 2 h. The solvent was evaporated in a vacuum, and the residue was dissolved in dichloromethane (500 mL); then, it was washed with 10% aqueous NaHSO4 solution (2 × 50 mL) and saturated aqueous NaHCO3 solution (2 × 50 mL). The organic phase was dried over Na2SO4, then filtered, and the solvent was evaporated (Rf = 0.51; hexane/acetone 6:4). The residue was dissolved in anhydrous dimethylformamide (60 mL); powdered KBr (8.14 g, 68.4 mmol, 3 equiv.) was added, and the mixture was stirred overnight. The solvent was evaporated in a vacuum, and toluene was added and evaporated. The residue was dissolved in ethyl acetate (600 mL); it was washed with brine (2 × 50 mL); the organic phase was dried over Na2SO4, then filtered, and the solvent was evaporated in a vacuum. The residue was purified by flash column chromatography to yield 7 (4.25 g, 66% for two steps) as a yellowish syrup. Rf = 0.63 (hexane/acetone 6:4); MALDI-TOF MS (Autoflex): m/z calcd for C8H16BrN3O3Na+: 304.027 [M+Na]+; found: 303.976.
3.1.5. Compound 8a
Sodium hydride (40 mg, 1.0 mmol, 2 equiv., 60% in mineral oil) was washed with hexane and dried under argon. Anhydrous N,N-dimethylformamide (10 mL) and 5a (297.5 mg, 0.5 mmol, 1 equiv.) were added, and the mixture was stirred for 30 min under an argon atmosphere. Then, compound 7 (170 mg, 0.6 mmol, 1.2 equiv.) was added, and the reaction mixture was stirred at 40 °C under an argon atmosphere. After one day, other portions of sodium hydride (20 mg, 0.5 mmol, 1 equiv., 60% in mineral oil) and 7 (85 mg, 0.3 mmol, 0.6 equiv.) were added. After two more days of stirring, methanol (1 mL) and water (1 mL) were added successively, and the reaction mixture was stirred for 30 min. Then, the solvent was evaporated in a vacuum, and the residue was purified by flash column chromatography (hexane/acetone 9:1) to yield 8a (156 mg, 39%) as a yellowish syrup. Rf = 0.38 (hexane/acetone 8:2); 1H NMR (400 MHz, CDCl3) δ (ppm) 7.27 (d, J = 8.6 Hz, 2H, arom.), 6.87 (d, J = 8.6 Hz, 2H, arom.), 4.80 (d, J = 10.5 Hz, 1H, PMB-CH2), 4.78 (d, J = 3.6 Hz, 1H, H-1), 4.56 (d, J = 10.4 Hz, 1H, PMB-CH2), 3.86–3.81 (m, 1H, decyl CH2a), 3.80 (s, 3H, PMB-OCH3), 3.74–3.56 (m, 21H, 7 × OCH2 TEG, decyl CH2b, decyl CH2, H-3, H-5, H-6a,b), 3.49 (t, J = 9.2 Hz, 1H, H-4), 3.37 (s, 3H, C-1-OCH3), 3.36–3.35 (m, 2H, CH2-N3), 3.30 (dd, J = 9.7 Hz, J = 3.5 Hz, 1H, H-2), 1.63–1.58 (m, 4H, 2 × decyl CH2), 1.34–1.25 (m, 28H, 14 × decyl CH2), 0.89–0.86 (m, 6H, 2 × decyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 159.3, 130.9 (2C, 2 × Cq arom.), 129.7, 113.9 (4C, arom.), 98.2 (1C, C-1), 81.8, 80.9, 77.5, 70.2 (4C, skeleton carbons), 74.8, 73.8, 71.9, 71.0, 70.8, 70.7, 70.6, 70.1, 70.0 (11C, 7 × OCH2 TEG, 2 × OCH2 decyl, PMB-OCH2, C-6), 55.4, 55.1 (2C, 2 × OCH3), 50.8 (1C, CH2-N3) 32.0, 30.7, 30.2, 29.9, 29.8, 29.7, 29.6, 29.5, 26.4, 26.1, 22.8 (14C, 7 × decyl CH2), 14.2 (2C, 2 × decyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C43H77N3O10Na+: 818.5501 [M+Na]+; found: 818.5509.
3.1.6. Compound 8b
Compound 5b (269 mg, 0.5 mmol, 1 equiv.) was alkylated with the bromide derivative 7 (170 mg, 0.6 mmol, 1.2 equiv.) according to the synthesis of 8a. The crude product was purified by flash column chromatography (hexane/acetone 8:2) to yield 8b (314 mg, 60%) as a yellowish syrup. Rf = 0.46 (hexane/acetone 7:3); 1H NMR (400 MHz, CDCl3) δ (ppm) 7.26 (d, J = 8.1 Hz, 2H, arom.), 6.87 (d, J = 8.4 Hz, 2H, arom.), 4.80 (d, J = 10.8 Hz, 1H, PMB-CH2), 4.77 (d, J = 3.4 Hz, 1H, H-1), 4.57 (d, J = 10.4 Hz, 1H, PMB-CH2), 3.88–3.80 (m, 1H, otyl OCH2a), 3.80 (s, 3H, PMB-OCH3), 3.74–3.56 (m, 21H, 7 × OCH2 TEG, octyl CH2b, octyl CH2, H-3, H-5, H-6a,b), 3.49 (t, J = 9.2 Hz, 1H, H-4), 3.37 (s, 3H, C-1-OCH3), 3.36–3.35 (m, 2H, CH2-N3), 3.30 (dd, J = 9.6 Hz, J = 3.2 Hz, 1H, H-2), 1.61–1.58 (m, 4H, 2 × octyl CH2), 1.33–1.26 (m, 20H, 10 × octyl CH2), 0.88–0.84 (m, 6H, 2 × octyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 159.4, 130.9 (2C, 2 × Cq arom.), 129.7, 113.9 (4C, arom.), 98.2 (1C, C-1), 81.9, 80.9, 77.5, 70.2 (4C, skeleton carbons), 74.8, 73.9, 71.9, 71.1, 70.8, 70.7, 70.6, 70.1, 70.0 (11C, 7 × OCH2 TEG, 2 × OCH2 octyl, PMB-OCH2, C-6), 55.4, 55.2 (2C, 2 × OCH3), 50.8 (1C, CH2-N3), 32.0, 30.8, 30.2, 29.9, 29.8, 29.7, 29.6, 29.5, 29.4, 26.4, 26.1, 22.8 (10C, 5 × octyl CH2), 14.2 (2C, 2 × octyl CH3); MALDI-TOF MS (BRUKER): m/z calcd for C39H69N3O10Na+: 762.49 [M+Na]+; found: 762.32.
3.1.7. Compound 8c
Compound 5c (213 mg, 0.5 mmol, 1 equiv.) was alkylated with the bromide derivative 7 (170 mg, 0.6 mmol, 1.2 equiv.) according to the synthesis of 8a. The crude product was purified by flash column chromatography (hexane/acetone 7:3) to yield 8c (167 mg, 53%) as a yellowish syrup. Rf = 0.28 (hexane/acetone 8:2); 1H NMR (400 MHz, CDCl3) δ (ppm) 7.27 (d, J = 8.5 Hz, 2H, arom.), 6.88 (d, J = 8.5 Hz, 2H, arom.), 4.80 (d, J = 10.4 Hz, 1H, PMB-CH2), 4.78 (d, J = 3.4 Hz, 1H, H-1), 4.58 (d, J = 10.4 Hz, 1H, PMB-CH2), 3.88–3.84 (m, 1H, butyl OCH2a), 3.79 (s, 3H, PMB-OCH3), 3.78–3.58 (m, 21H, 7 × OCH2 TEG, butyl OCH2b, butyl OCH2, H-3, H-5, H-6a,b), 3.51 (t, J = 9.2 Hz, 1H, H-4), 3.38 (s, 3H, C-1-OCH3), 3.36–3.34 (m, 2H, -CH2-N3), 3.31 (dd, J = 9.7 Hz, J = 3.5 Hz, 1H, H-2), 1.64–1.55 (m, 4H, 2 × butyl CH2), 1.43–1.36 (m, 4H, 2 × butyl CH2), 0.92 (t, J = 7.4 Hz, 6H, 2 × butyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 159.2, 130.7 (2C, 2 × Cq arom.), 129.6, 113.7 (4C, arom.), 98.0 (1C, C-1), 81.6 (1C, C-3), 80.7 (1C, C-2), 77.3 (1C, C-4), 74.6 (1C, PMB-CH2), 73.3, 71.2, 70.8, 70.6, 70.4, 69.9, 69.8 (10C, 7 × OCH2 TEG, 2 × OCH2 butyl, C-6), 70.0 (1C, C-5), 55.2 (1C, PMB-OCH3), 54.9 (1C, C-1-OCH3), 50.5 (1C, CH2-N3), 32.6, 32.0, 19.3, 19.1 (4C, 4 × butyl CH2), 14.0, 13.8 (2C, 2 × butyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C31H53N3O10Na+: 650.3623 [M+Na]+; found: 650.3634.
3.1.8. Compound 12a
Compound 9 (50 mg, 0.138 mmol, 1 equiv.) was dissolved in anhydrous methanol (3 mL), and 8a (131 mg, 0.165 mmol, 1.2 equiv.) in anhydrous N,N-dimethylformamide (3 mL) was added. Then, triethylamine (40 µL, 0.275 mmol, 2 equiv.) and copper(I) iodide (3 mg, 0.0138 mmol, 0.1 equiv.) were added. The reaction mixture was stirred at room temperature overnight, then other portions of triethylamine (40 µL, 0.275 mmol, 2 equiv.) and copper(I) iodide (3 mg, 0.0138 mmol, 0.1 equiv.) were added, and the mixture was stirred at 40 °C for 2 days and at 50 °C for 1 day. Thereafter, the solvent was evaporated in a vacuum, and the residue was purified by flash column chromatography (toluene/methanol 8:2 containing 0.1% acetic acid) to yield 12a (62 mg, 39%) as a yellowish syrup. Rf = 0.18 (toluene/methanol 7:3 containing 0.1% acetic acid); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.82 (d, J = 7.3 Hz, 1H, NH), 7.96 (s, 1H, triazole C=CH), 7.22 (d, J = 8.6 Hz, 2H, arom.), 6.89 (d, J = 8.6 Hz, 2H, arom.), 4.74 (d, J = 3.3 Hz, 1H, H-1), 4.64 (d, J = 10.7 Hz, 1H, PMB-CH2a), 4.49 (d, J = 10.8 Hz, 1H, PMB-CH2b), 4.44 (t, J = 5.3 Hz, 2H, NCH2), 4.00–3.92 (m, 2H, SCH2,), 3.89–3.20 (m, 33H, 7 × OCH2 TEG, 2 × decyl OCH2, H-3, H-4, H-5, H-6a,b, H-4′, H-5′, H-6′, H-7′, H-8′, H-9′a,b, PMB-OCH3), 3.17 (s, 3H, C-1-OCH3), 3.16–3.13 (m, 1H, H-2), 2.72 (dd, J = 11.2 Hz, J = 3.8 Hz, 1H, H-3′a), 1.89 (s, 3H, NAc CH3), 1.50–1.43 (m, 4H, 2 × decyl CH2), 1.38 (t, J = 11.4 Hz, 1H, H-3′b), 1.24–1.21 (m, 32H, 4 × OH, 14 × decyl CH2), 0.87–0.83 (m, 6H, 2 × decyl CH3); 13C NMR (100 MHz, DMSO-d6) δ (ppm) 177.0 (1C, Ac C=O), 173.0 (1C, C-1′), 159.2 (1C, Cq arom.), 144.6 (1C, triazole C=CH), 131.1 (1C, Cq arom.), 129.7 (2C, arom.), 124.0 (1C, triazole C=CH), 114.0 (2C, arom.), 98.4 (1C, C-1), 85.7 (1C, C-2′), 81.7 (1C, C-3), 80.4 (1C, C-2), 77.5 (1C, C-4), 75.6 (1C, C-6′), 74.0 (1C, PMB-CH2), 73.0 (1C, OCH2 decyl), 71.7 (1C, C-8′), 70.6 (1C, OCH2 decyl), 70.3, 70.2, 70.1, 70.0, 69.9, 69.1 (8C, C-6, 7 × OCH2 TEG), 69.7 (2C, C-5, C-7′), 67.6 (1C, C-4′), 63.8 (1C, C-9′), 55.5 (1C, PMB-OCH3), 54.9 (1C, C-1-OCH3), 53.7 (1C, C-5′), 49.7 (1C, NCH2), 42.5 (1C, C-3′), 31.8, 30.5, 30.1, 29.6, 29.5, 29.4, 29.2, 26.2, 26.1 (14C, 14 × decyl CH2), 23.9 (1C, SCH2), 22.3 (1C, NAc CH3), 22.6 (2C, 2 × decyl CH2), 14.4 (2C, 2 × decyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C57H97N4O18SNa2+: 1203.631 [M-H+2Na]+; found: 1203.638.
3.1.9. Compound 12b
Compound 9 (50 mg, 0.138 mmol) was reacted with compound 8b (122 mg, 0.165 mmol, 1.2 equiv.) in the presence of triethylamine (40 µL, 0.2752 mmol, 2 equiv.) and copper(I) iodide (3 mg, 0.0138 mmol, 0.1 equiv.) according to the procedure applied for the synthesis of 12a. The crude product was purified by column chromatography (1st Silicagel 60, 40–63 μm, dichloromethane/methanol 92:8, 2nd Sephadex LH20, MeOH) to yield 12b (50 mg, 33%) as a yellowish syrup. Rf = 0.22 (toluene/methanol 7:3 containing 0.1% acetic acid); 1H NMR (400 MHz, Pyridine-d5) δ (ppm) 9.21 (s, 1H, NH), 8.27 (s, 1H, triazole C=CH), 7.63 (d, J = 8.4 Hz, 2H, arom.), 7.18 (d, J = 8.4 Hz, 2H, arom.), 5.18–5.14 (m, 2H, H-1, PMB-CH2a), 4.96 (d, J = 10.8 Hz, 1H, PMB-CH2b), 4.69–4.64 (m, 5H, H-5′, NCH2, SCH2,), 4.51–4.48 (m, 1H, H-9′a), 4.39–4.37 (m, 1H, H-9′b), 4.14–3.65 (m, 31H, 7 × OCH2 TEG, 2 × octyl OCH2, H-2, H-3, H-4, H-5, H-6a,b, H-4′, H-6′, H-7′, H-8′, PMB-OCH3), 3.56 (s, 3H, C-1-OCH3), 2.45–2.42 (m, 1H, H-3′a), 2.14 (s, 3H, NAc CH3), 1.83–1.28 (m, 29H, H-3′b, 4 × OH, 12 × octyl CH2), 0.91–0.90 (m, 6H, 2 × octyl CH3); 13C NMR (100 MHz, Pyridine-d5) δ (ppm) 175.1 (1C, Ac C=O), 172.7 (1C, C-1′), 160.7 (1C, Cq arom.), 144.8 (1C, triazole C=CH), 132.6 (1C, Cq arom.), 131.0 (2C, arom.), 115.2 (2C, arom.), 99.3 (1C, C-1), 86.7 (1C, C-2′), 83.3 (1C, C-3), 82.0 (1C, C-2), 79.0 (1C, C-4), 77.8 (1C, C-6′), 75.7 (1C, PMB-CH2), 74.7 (1C, OCH2 octyl), 74.0 (1C, C-8′), 72.1 (1C, OCH2 octyl), 71.9 (2C, C-5, C-7′), 71.7, 71.6, 71.5, 70.5 (8C, C-6, 7 × OCH2 TEG), 70.2 (1C, C-4′), 65.4 (1C, C-9′), 56.4 (1C, PMB-OCH3), 56.2 (1C, C-1-OCH3), 55.1 (1C, C-5′), 51.2 (1C, NCH2), 44.2 (1C, C-3′), 33.1, 33.0, 32.0, 30.8, 30.7, 30.5, 27.6, 27.4 (10C, 10 × octyl CH2), 25.6 (1C, SCH2), 23.9 (1C, NAc CH3), 23.8 (2C, 2 × octyl CH2), 15.2 (2C, 2 × octyl CH3); ESI-QTOF MS: m/z calcd for C53H89N4O18S−: 1101.589 [M-H]−; found: 1101.599.
3.1.10. Compound 12c
Compounds 10 (70 mg, 0.185 mmol, 1 equiv.) and 8c (138 mg, 0.278 mmol, 1.5 equiv.) were dissolved in anhydrous acetonitrile (10 mL). Triethylamine (52 µL, 0.37 mmol, 2 equiv.) and copper(I) iodide (3.5 mg, 0.0185 mmol, 0.1 equiv.) were added. The reaction mixture was stirred at room temperature overnight; then, additional triethylamine (26 µL, 0.185 mmol, 1 equiv.) and copper(I) iodide (3.5 mg, 0.0185 mmol, 0.1 equiv.) were added. After 3 more days, the solvent was evaporated in a vacuum, and the residue was purified by flash column chromatography (dichloromethane/methanol 95:5) to yield 11 (73.2 mg, 43%) as a yellowish syrup (Rf = 0.62 in dichloromethane/methanol 7:3 containing 0.1% acetic acid).
Compound 11 (60 mg, 0.06 mmol) was dissolved in anhydrous dioxane (9 mL) and water (1 mL), and the solution was cooled to 0 °C. Then, a 0.06 M solution of lithium hydroxide in water (300 µL, 0.179 mmol, 3 equiv) was added. The reaction mixture was stirred at 0 °C for 1 h and at room temperature for 4 h. Thereafter, the solvent was evaporated, and the residue was purified by flash column chromatography (toluene/methanol 7:3) to yield 12c (54 mg, 91%) as a yellowish syrup. Rf = 0.64 (toluene/methanol 6:4); 1H NMR (400 MHz, Pyridine-D5) δ (ppm) 8.06 (s, 1H, triazole C=CH), 7.51 (d, J = 8.0 Hz, 2H, arom.), 7.07 (d, J = 8.1 Hz, 2H, arom.), 6.16 (s, 1H, NH), 5.06–4.82 (m, 3H, H-1, PMB-CH2a,b), 4.73–4.44 (m, 4H, SCH2, NCH2), 4.01–3.53 (m, 33H, 7 × OCH2 TEG, 2 × butyl OCH2, H-3, H-4, H-5, H-6a,b, H-4′, H-5′, H-6′, H-7′, H-8′, H-9′a,b, PMB-OCH3), 3.45–3.42 (m, 1H, H-2), 3.39 (s, 3H, C-1-OCH3), 2.34–2.30 (m, 1H, H-3′a), 2.03 (s, 3H, NAc), 1.66–1.54 (m, 5H, H-3′b, 2 × butyl CH2), 1.44–1.37 (m, 4H, 2 × butyl CH2), 0.89–0.83 (m, 6H, 2 × butyl CH3); 13C NMR (100 MHz, Pyridine-D5) δ (ppm) 177.4 (1C, Ac C=O), 174.0 (1C, C-1′), 160.1 (1C, Cq arom.), 144.5 (1C, triazole C=CH), 132.2 (1C, Cq arom.), 130.3 (2C, arom.), 124.7 (1C, triazole C=CH), 114.6 (2C, arom.), 98.7 (1C, C-1), 86.7 (1C, C-2′), 82.7 (1C, C-3), 81.6 (1C, C-2), 78.5 (1C, C-4), 77.3 (1C, C-6′), 75.0 (1C, PMB-CH2), 73.5 (1C, OCH2 butyl), 73.0 (1C, C-8′), 71.6 (1C, OCH2 butyl), 71.4 (1C- C-5), 71.2, 71.1, 71.0, 70.9 (9C, C-7′, 7 × OCH2 TEG, C-6), 68.1 (1C, C-4′), 63.1 (1C, C-9′), 55.6 (1C, PMB-OCH3), 55.4 (1C, C-1-OCH3), 54.5 (1C, C-5′), 50.6 (1C, NCH2), 41.7 (1C, C-3′), 33.5, 32.9 (2C, 2 × butyl CH2), 25.1 (1C, SCH2), 23.4 (1C, NAc), 20.1, 19.9 (2C, 2 × butyl CH2), 14.6, 14.4 (2C, 2 × butyl CH3); ESI-QTOF MS: m/z calcd for C45H74N4O18SH+: 991.479 [M+H]+; found: 991.490.
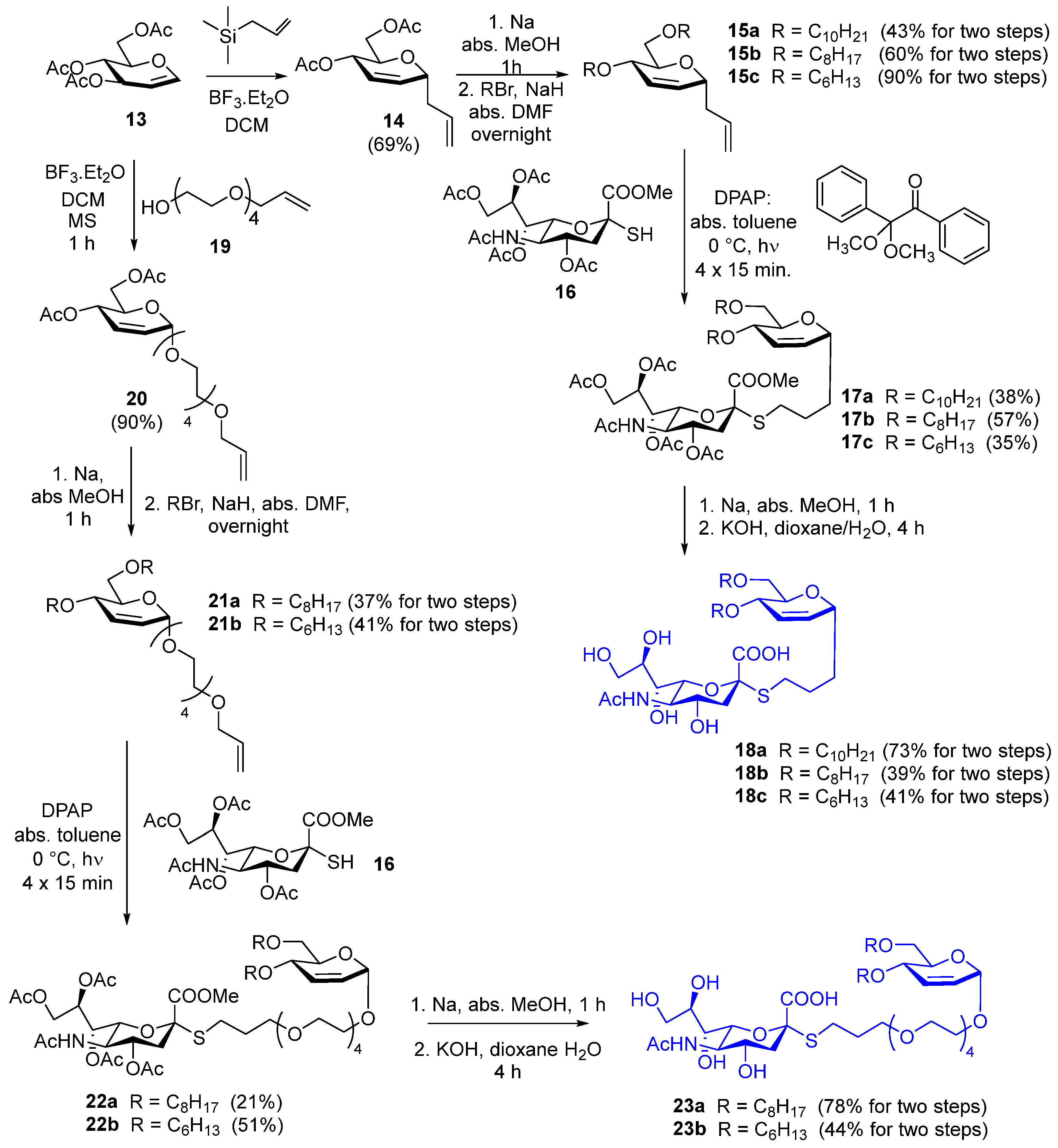
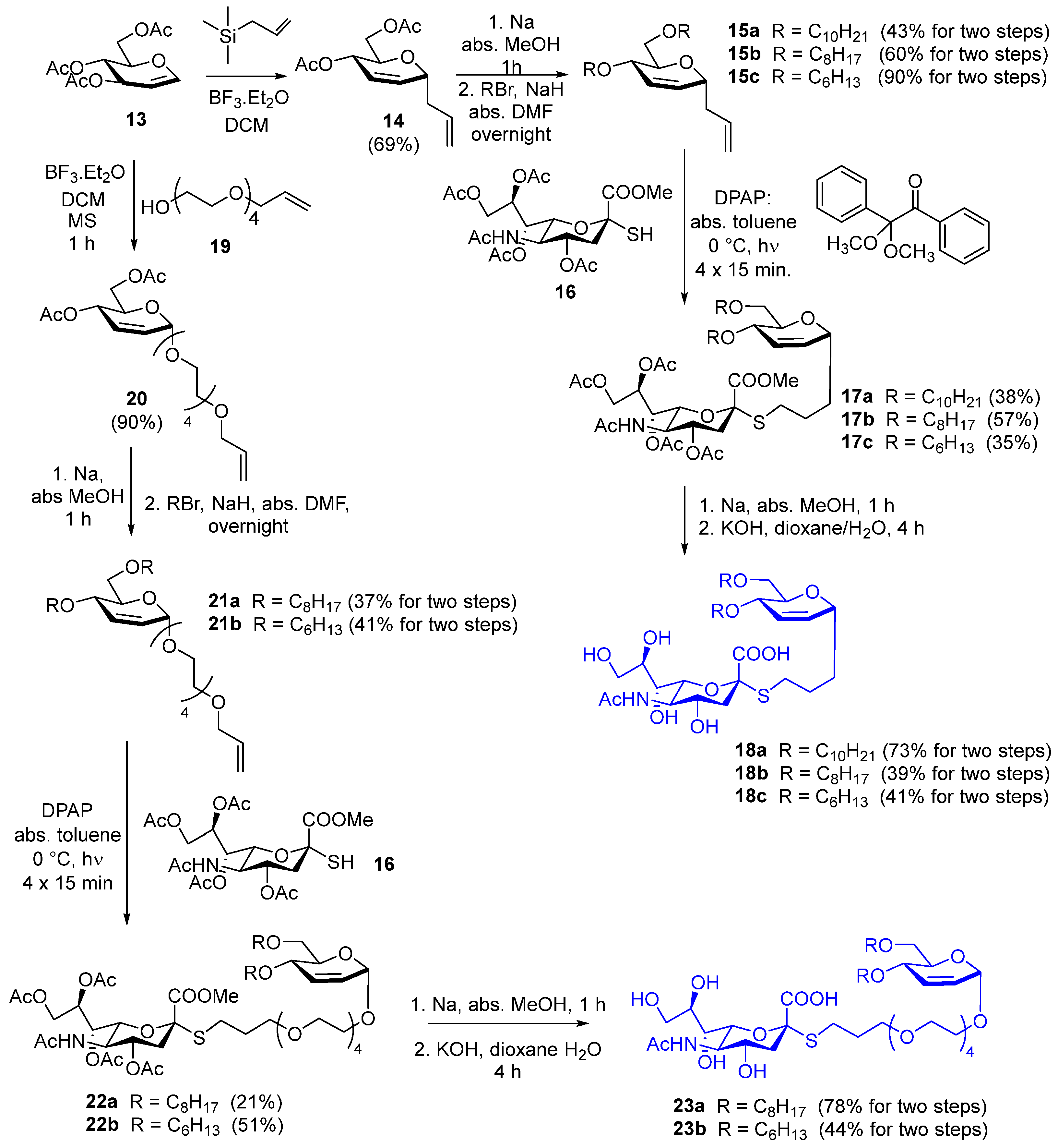
3.1.11. Compound 15a
Compound 14 (500 mg, 1.97 mmol) was dissolved in anhydrous methanol (10 mL); small pieces of sodium were added to make the solution alkaline (pH ~ 11). The reaction mixture was stirred for 1 h; then, Serdolit Red H+ ion exchange resin was added to make the pH neutral. Thereafter, the reaction mixture was filtered, and the solvent was evaporated. A dichloromethane/methanol 9:1 mixture was used for TLC (Rf = 0.60).
Sodium hydride (315 mg, 7.88 mmol, 2 equiv./OH, 60% in mineral oil) was washed with hexane and dried under an argon atmosphere; then, anhydrous N,N-dimethylformamide (DMF) (3 mL) was added. The crude product of the previous step was dissolved in anhydrous DMF (2 mL) and added to the sodium hydride. The reaction mixture was stirred under an argon atmosphere for 20 min; then, decyl bromide (980 μL, 4.72 mmol, 1.2 equiv/OH) was added in two portions, and the reaction was further stirred at room temperature overnight. Then, methanol (1 mL) and water (1 mL) were added successively, and the reaction mixture was stirred for 30 min. The solvent was evaporated, and the residue was purified by flash column chromatography (hexane/ethylacetate 97:3) to yield 15a (378 mg, 43%) as a colorless syrup. Rf = 0.89 (hexane/ethylacetate 8:2); 1H NMR (400 MHz, CDCl3) δ (ppm) 5.92–5.89 (m, 1H, H-3), 5.88–5.82 (m, 1H, allyl CH=CH2), 5.82–5.79 (m, 1H, H-2), 5.12–5.05 (m, 2H, allyl CH=CH2), 4.23 (ddd, J = 8.1 Hz, J = 6.2 Hz, J = 2.2 Hz, 1H, H-1), 3.82 (dq, J = 7.3 Hz, J = 1.9 Hz, 1H, H-4), 3.69–3.65 (m, 1H, H-5), 3.64–3.61 (m, 3H, decyl OCH2a, H-6a,b), 3.54–3.39 (m, 3H, decyl CH2b, decyl CH2), 2.51–2.44 (m, 1H, allyl CH2a), 2.32–2.25 (m, 1H, allyl CH2b), 1.63–1.53 (m, 4H, 2 × decyl CH2), 1.30–1.26 (m, 28H, 14 × decyl CH2), 0.88 (t, J = 6.7 Hz, 6H, 2 × decyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 134.8 (1C, allyl CH=CH2), 130.7 (1C, C-2), 126.3 (1C, C-3), 117.3 (1C, allyl CH=CH2), 72.4 (1C, C-1), 71.8 (1C, decyl OCH2), 71.2 (1C, C-5), 70.6 (1C, C-4), 70.2 (1C, C-6), 69.5 (1C, decyl OCH2), 38.1 (1C, allyl CH2), 32.0, 30.2, 29.8, 29.7, 29.6, 29.5, 26.3, 22.8 (16C, 16 × decyl CH2), 14.2 (2C, 2 × decyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C29H54O3Na+: 473.3965 [M+Na]+; found: 473.3960.
3.1.12. Compound 15b
Deacetylation of compound 14 (500 mg, 1.97 mmol) and subsequent alkylation with octyl bromide (820 μL, 4.72 mmol, 1.2 equiv./OH) in the presence of sodium hydride (315 mg, 7.88 mmol, 2 equiv./OH, 60% in mineral oil) were performed according to the synthesis of 15a. The crude product was purified by flash column chromatography (hexane/ethylacetate 97:3→9:1) to yield 15b (466 mg, 60%) as a colorless syrup. Rf = 0.24 (hexane/ethylacetate 95:5); 1H NMR (400 MHz, CDCl3) δ (ppm) 5.92–5.88 (m, 1H, H-3), 5.87–5.83 (m, 1H, allyl CH=CH2), 5.82–5.78 (m, 1H, H-2), 5.12–5.05 (m, 2H, allyl CH=CH2), 4.25–4.21 (m, 1H, H-1), 3.83–3.80 (m, 1H, H-4), 3.69–3.66 (m, 1H, H-5), 3.64–3.58 (m, 3H, octyl OCH2a, H-6a,b), 3.54–3.40 (m, 3H, octyl CH2b, octyl CH2), 2.47 (dt, J = 14.4 Hz, J = 7.3 Hz, 1H, allyl CH2a), 2.29 (dt, J = 13.9 Hz, J = 6.8 Hz, 1H, allyl CH2b), 1.62–1.53 (m, 4H, 2 × octyl CH2), 1.30–1.26 (m, 20H, 10 × octyl CH2), 0.89–0.86 (m, 6H, 2 × octyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 134.8 (1C, allyl CH=CH2), 130.7 (1C, C-2), 126.3 (1C, C-3), 117.2 (1C, allyl CH=CH2), 72.4 (1C, C-1), 71.8 (1C, octyl OCH2), 71.2 (1C, C-5), 70.6 (1C, C-4), 70.2 (1C, C-6), 69.4 (1C, octyl OCH2), 38.1 (1C, allyl CH2), 31.9, 30.2, 29.7, 29.6, 29.5, 29.4, 26.3, 22.8 (12C, 12 × octyl CH2), 14.2 (2C, 2 × octyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C25H46O3Na+: 417.3339 [M+Na]+; found: 417.3343.
3.1.13. Compound 15c
Deacetylation of compound 14 (500 mg, 1.97 mmol) and subsequent alkylation with hexyl bromide (664 μL, 4.72 mmol, 1.2 equiv./OH) in the presence of sodium hydride (315 mg, 7.88 mmol, 2 equiv./OH, 60% in mineral oil) were performed according to the synthesis of 15a. The crude product was purified by flash column chromatography (hexane/ethylacetate 97:3→9:1) to yield 15c (600 mg, 90%) as a colorless syrup. Rf = 0.28 (hexane/ethyl acetate 9:1); 1H NMR (400 MHz, CDCl3) δ (ppm) 5.92–5.89 (m, 1H, H-3), 5.88–5.82 (m, 1H, allyl CH=CH2), 5.82–5.79 (m, 1H, H-2), 5.12–5.05 (m, 2H, allyl CH=CH2), 4.23 (ddd, J = 8.1 Hz, J = 6.1 Hz, J = 2.2 Hz, 1H, H-1), 3.82 (dq, J = 7.4 Hz, J = 2.0 Hz, 1H, H-4), 3.68 (q, J = 3.7 Hz, J = 3.1 Hz, 1H, H-5), 3.64–3.59 (m, 3H, hexyl OCH2a, H-6a,b), 3.54–3.40 (m, 3H, hexyl CH2b, hexyl CH2), 2.51–2.44 (m, 1H, allyl CH2a), 2.32–2.25 (m, 1H, allyl CH2b), 1.63–1.53 (m, 4H, 2 × hexyl CH2), 1.39–1.21 (m, 12H, 6 × hexyl CH2), 0.91–0.87 (m, 6H, 2 × hexyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 134.8 (1C, allyl CH=CH2), 130.7 (1C, C-2), 126.3 (1C, C-3), 117.2 (1C, allyl CH=CH2), 72.4 (1C, C-1), 71.8 (1C, hexyl OCH2), 71.2 (1C, C-5), 70.6 (1C, C-4), 70.2 (1C, C-6), 69.4 (1C, hexyl OCH2), 38.1 (1C, allyl CH2), 31.8, 31.7, 30.1, 29.7, 26.0, 22.8 (8C, 8 × hexyl CH2), 14.2 (2C, 2 × hexyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C21H38O3Na+: 361.2713 [M+Na]+; found: 361.2691.
3.1.14. Compound 17a
Compound 15a (100 mg, 0.22 mmol) and compound 16 (135 mg, 0.266 mmol, 1.2 equiv.) were dissolved in anhydrous toluene (5 mL), and 2,2-dimethoxy-2-phenylacetophenone (DPAP) (5.6 mg, 0.022 mmol, 0.1 equiv) was added. The reaction mixture was cooled to 0 °C and irradiated with UV light 4 times for 15 min, and DPAP (5.6 mg, 0.022 mmol, 0.1 equiv) was added before each irradiation cycle. Then, the solvent was evaporated and the crude product was purified by flash column chromatography (dichloromethane/acetone 94:6→9:1) to yield 17a (81 mg, 38%) as a yellowish syrup. Rf = 0.28 (dichloromethane/acetone 9:1); 1H NMR (400 MHz, CDCl3) δ (ppm) 5.87 (d, J = 10.4 Hz, 1H, H-3), 5.77 (d, J = 10.9 Hz, 1H, H-2), 5.39 (d, J = 10.1 Hz, 1H, NHAc), 5.33–5.31 (m, 2H, H-7′, H-8′), 4.86 (td, J = 11.5 Hz, J = 4.6 Hz, 1H, H-4′), 4.31 (dd, J = 12.4 Hz, J = 2.0 Hz, 1H, H-9′a), 4.17–4.15 (m, 1H, H-1), 4.11 (dd, J = 12.6 Hz, J = 4.6 Hz, 1H, H-9′b), 4.07–4.02 (m, 1H, H-5′), 3.85–3.81 (m, 2H, H-4, H-6′), 3.79 (s, 3H, COOCH3), 3.63–3.58 (m, 4H, H-5, H-6a,b, decyl OCH2a), 3.55–3.37 (m, 3H, decyl CH2b, decyl CH2), 2.76 (dd, J = 13.0 Hz, J = 7.1 Hz, 1H, SCH2a), 2.71 (dd, J = 12.8 Hz, J = 4.6 Hz, 1H, H-3′a), 2.58 (dt, J = 12.8 Hz, J = 7.3 Hz, 1H, SCH2b), 2.15, 2.14, 2.04, 2.03 (4 × s, 12H, 4 × Ac CH3), 1.97 (t, J = 12.3 Hz, 1H, H-3′b), 1.88 (s, 3H, NHAc CH3), 1.77–1.52 (m, 8H, 2 × CH2, 2 × decyl CH2), 1.28–1.24 (m, 28H, 14 × decyl CH2), 0.88 (t, J = 6.8 Hz, 6H, 2 × decyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 171.0, 170.7, 170.3, 170.2, 170.1 (5C, 5 × Ac C=O), 168.5 (1C, C-1′), 131.1 (1C, C-2), 126.1 (1C, C-3), 83.2 (1C, C-2′), 74.2 (1C, C-6′), 72.3 (1C, C-1), 71.8 (1C, decyl OCH2), 70.8 (1C, C-5), 70.5 (1C, C-4), 70.1 (1C, C-6), 69.8 (1C, C-4′), 69.4 (1C, decyl OCH2), 68.8 (1C, C-8′), 67.4 (1C, C-7′), 62.3 (1C, C-9′), 53.0 (1C, COOCH3), 49.4 (1C, C-5′), 38.1 (1C, C-3′), 32.0, 30.1, 29.7, 29.6, 29.4, 28.8, 26.3, 25.8, 22.8 (19C, 2 × CH2, SCH2, 16 × decyl CH2), 23.2 (1C, NHAc), 21.3, 20.9, 20.8 (4C, 4 × Ac CH3), 14.2 (2C, 2 × decyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C49H83NO15SNa+: 980.5376 [M+Na]+; found: 980.5368.
3.1.15. Compound 17b
Compound 15b (95 mg, 0.24 mmol) was reacted with compound 16 (146 mg, 0.288 mmol, 1.2 equiv.) in the presence of DPAP (6.2 mg, 0.024 mmol, 0.1 equiv) according to the synthesis of 15a. The crude product was purified by flash column chromatography (dichloromethane/methanol 99:1) to yield 17b (125 mg, 57%) as a yellowish syrup. Rf = 0.46 (dichloromethane/acetone 8:2); 1H NMR (500 MHz, CDCl3) δ (ppm) 5.87 (dt, J = 10.4 Hz, J = 1.8 Hz, 1H, H-3), 5.77 (dt, J = 10.5 Hz, J = 2.0 Hz, 1H, H-2), 5.36 (dd, J = 5.8 Hz, J = 2.5 Hz, 1H, H-8′), 5.33 (td, J = 8.4 Hz, J = 7.8 Hz, J = 2.2 Hz, 1H, H-7′), 5.19 (d, J = 10.1 Hz, 1H, NHAc), 4.86 (td, J = 11.5 Hz, J = 4.6 Hz, 1H, H-4′), 4.30 (dd, J = 12.5 Hz, J = 2.4 Hz, 1H, H-9′a), 4.16–4.15 (m, 1H, H-1), 4.11 (dd, J = 12.5 Hz, J = 5.0 Hz, 1H, H-9′b), 4.04 (q, J = 10.4 Hz, 1H, H-5′), 3.85–3.81 (m, 2H, H-4, H-6′), 3.80 (s, 3H, COOCH3), 3.63–3.58 (m, 4H, H-5, H-6a,b, octyl OCH2a), 3.54–3.38 (m, 3H, octyl CH2b, octyl CH2), 2.76 (dd, J = 13.1 Hz, J = 5.7 Hz, 1H, SCH2a), 2.71 (dd, J = 12.8 Hz, J = 4.7 Hz, 1H, H-3′a), 2.61–2.56 (m, 1H, SCH2b), 2.15, 2.14, 2.04, 2.03 (4 × s, 12H, 4 × Ac CH3), 1.97 (t, J = 12.3 Hz, 1H, H-3′b), 1.87 (s, 3H, NHAc CH3), 1.79–1.53 (m, 8H, 2 × CH2, 2 × octyl CH2), 1.34–1.26 (m, 20H, 10 × octyl CH2), 0.89–0.87 (m, 6H, 2 × octyl CH3); 13C NMR (125 MHz, CDCl3) δ (ppm) 170.9, 170.6, 170.1, 169.9 (5C, 5 × Ac C=O), 168.5 (1C, C-1′), 131.0 (1C, C-2), 126.0 (1C, C-3), 83.1 (1C, C-2′), 74.1 (1C, C-6′), 72.2 (1C, C-1), 71.7 (1C, octyl OCH2), 70.8 (1C, C-5), 70.5 (1C, C-4), 70.1 (1C, C-6), 69.7 (1C, C-4′), 69.3 (1C, octyl OCH2), 68.6 (1C, C-8′), 67.3 (1C, C-7′), 62.2 (1C, C-9′), 52.9 (1C, COOCH3), 49.4 (1C, C-5′), 38.1 (1C, C-3′), 32.0, 31.8, 30.1, 29.6, 29.5, 29.3, 28.7, 26.2, 25.8, 22.7 (15C, 2 × CH2, SCH2, 12 × decyl CH2), 23.2 (1C, NHAc), 21.2, 20.8, 20.7 (4C, 4 × Ac CH3), 14.1 (2C, 2 × decyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C45H75NO15SNa+: 924.4750 [M+Na]+; found: 924.4753.
3.1.16. Compound 17c
Compound 15c (97 mg 0.25 mmol) was reacted with compound 16 (152 mg, 0.30 mmol, 1.2 equiv.) in the presence of DPAP (6.5 mg, 0.025 mmol, 0.1 equiv) according to the synthesis of 15a. The crude product was purified by flash column chromatography (dichloromethane/methanol 99:1) to yield 17c (85 mg, 35%) as a yellowish syrup. Rf = 0.35 (dichloromethane/acetone 8:2); 1H NMR (400 MHz, CDCl3) δ (ppm) 5.87 (d, J = 10.4 Hz, 1H, H-3), 5.77 (d, J = 10.5 Hz, 1H, H-2), 5.38–5.31 (m, 2H, H-7′, H-8′), 5.28 (d, J = 10.1 Hz, 1H, NHAc), 4.86 (td, J = 11.3 Hz, J = 4.6 Hz, 1H, H-4′), 4.30 (dd, J = 12.4 Hz, J = 1.9 Hz, 1H, H-9′a), 4.16–4.15 (m, 1H, H-1), 4.11 (dd, J = 12.5 Hz, J = 4.6 Hz, 1H, H-9′b), 4.09–4.01 (m, 1H, H-5′), 3.83–3.81 (m, 2H, H-4, H-6′), 3.80 (s, 3H, COOCH3), 3.63–3.58 (m, 4H, H-5, H-6a,b, hexyl OCH2a), 3.55–3.38 (m, 3H, hexyl CH2b, hexyl CH2), 2.76 (dd, J = 12.7 Hz, J = 6.7 Hz, 1H, SCH2a), 2.71 (dd, J = 12.8 Hz, J = 4.5 Hz, 1H, H-3′a), 2.58 (dt, J = 12.9 Hz, J = 7.2 Hz, 1H, SCH2b), 2.15, 2.14, 2.04, 2.03 (4 × s, 12H, 4 × Ac CH3), 1.97 (t, J = 12.3 Hz, 1H, H-3′b), 1.88 (s, 3H, NHAc CH3), 1.77–1.52 (m, 8H, 2 × CH2, 2 × hexyl CH2), 1.34–1.24 (m, 12H, 6 × hexyl CH2), 0.90–0.87 (m, 6H, 2 × hexyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 171.1, 170.7, 170.3, 170.2, 170.1 (5C, 5 × Ac C=O), 168.6 (1C, C-1′), 131.1 (1C, C-2), 126.1 (1C, C-3), 83.2 (1C, C-2′), 74.2 (1C, C-6′), 72.3 (1C, C-1), 71.8 (1C, hexyl OCH2), 70.8 (1C, C-5), 70.6 (1C, C-4), 70.2 (1C, C-6), 69.8 (1C, C-4′), 69.4 (1C, hexyl OCH2), 68.7 (1C, C-8′), 67.4 (1C, C-7′), 62.3 (1C, C-9′), 53.1 (1C, COOCH3), 49.5 (1C, C-5′), 38.2 (1C, C-3′), 32.1, 31.8, 30.1, 29.7, 28.8, 26.0, 25.9, 22.7 (11C, 2 × CH2, SCH2, 8 × decyl CH2), 23.3 (1C, NHAc), 21.3, 21.0, 20.9 (4C, 4 × Ac CH3), 14.2 (2C, 2 × hexyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C41H67NO15SNa+: 868.4124 [M+Na]+; found: 868.4126.
3.1.17. Compound 18a
Compound 17a (75 mg, 0.078 mmol) was dissolved in anhydrous methanol (10 mL), and little pieces of sodium were added to make the pH basic (pH ~ 11). After 1 h of stirring, Amberlite IR-50 H+ ion exchange resin was added, and the pH was adjusted to neutral. Then, the reaction mixture was filtered, and the solvent was evaporated.
The crude product of the previous step was dissolved in 1,4-dioxane (9 mL) and water (1 mL), and the reaction mixture was cooled to 0 °C. Then, an aqueous solution of potassium hydroxide (0.2 M, 1.95 mL, 0.39 mmol, 5 equiv.) was added, and the reaction mixture was stirred at room temperature for 4 h; then, Amberlite IR-50 H+ ion exchange resin was added, and the pH was adjusted to neutral. The reaction mixture was filtered, and the solvent was evaporated in a vacuum. The residue was purified by flash column chromatography (acetonitrile/water 95:5→9:1) to yield 18a (44.3 mg, 73%) as a yellowish syrup. Rf = 0.45 (acetonitrile/water 8:2); 1H NMR (400 MHz, MeOD) δ (ppm) 5.89–5.82 (m, 2H, H-2, H-3), 4.14–4.10 (m, 1H, H-1), 3.85–3.67 (m, 6H, H-4, H-4′, H-5′, H-8′, H-9′a,b), 3.64–3.59 (m, 4H, H-5, H-6a,b, H-7′), 3.55–3.40 (m, 5H, H-6′, 2 × decyl OCH2), 2.89–2.82 (m, 2H, H-3′a, SCH2a), 2.74–2.67 (m, 1H, SCH2b), 2.01 (s, 3H, NHAc CH3), 1.82–1.52 (m, 9H, H-3′b, 2 × CH2, 2 × decyl CH2), 1.32–1.28 (m, 28H, 14 × decyl CH2), 0.90 (t, J = 6.6 Hz, 6H, 2 × decyl CH3); 13C NMR (100 MHz, MeOD) δ (ppm) 175.5, 175.1 (2C, C-1′, NAc C=O), 132.5 (1C, C-2), 126.4 (1C, C-3), 87.2 (1C, C-2′), 76.6 (1C, C-6′), 73.5 (1C, C-1), 73.0 (1C, C-8′), 72.6 (1C, decyl OCH2), 72.3 (1C, C-5), 71.8 (1C, C-4), 71.1 (1C, C-6), 70.2 (1C, C-7′), 70.0 (1C, decyl OCH2), 69.6 (1C, C-4′), 64.3 (1C, C-9′), 54.0 (1C, C-5′), 43.2 (1C, C-3′), 33.4, 33.1, 31.1, 30.8, 30.7, 30.6, 30.5, 30.4, 27.7, 27.3, 23.7 (19C, 2 × CH2, SCH2, 16 × decyl CH2), 22.6 (1C, NHAc), 14.5 (2C, 2 × decyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C40H73NO11SNa+: 798.4797 [M+Na]+; found: 798.4800.
3.1.18. Compound 18b
Compound 17b (124 mg, 0.135 mmol) was deacetylated; then, methyl ester hydrolysis was performed with potassium hydroxide solution (0.2 M, 3.38 mL, 0.675 mmol, 5 equiv.) according to the procedure for 18a. The crude product was purified by flash column chromatography (acetonitrile/water 95:5→9:1) to yield 18b (37 mg, 39%) as a yellowish syrup. Rf = 0.47 (acetonitrile/water 9:1); 1H NMR (500 MHz, MeOD) δ (ppm) 5.89–5.82 (m, 2H, H-2, H-3), 4.12–4.11 (m, 1H, H-1), 3.84–3.61 (m, 7H, H-4, H-4′, H-5′, H-5, H-8′, H-9′a,b), 3.60–3.58 (m, 2H, H-6a,b), 3.54–3.41 (m, 6H, H-6′, H-7′, 2 × octyl OCH2), 2.88–2.83 (m, 2H, H-3′a, SCH2a), 2.74–2.68 (m, 1H, SCH2b), 2.01 (s, 3H, NHAc CH3), 1.81–1.52 (m, 9H, H-3′b, 2 × CH2, 2 × octyl CH2), 1.37–1.25 (m, 20H, 10 × octyl CH2), 0.90 (t, J = 6.8 Hz, 6H, 2 × octyl CH3); 13C NMR (125 MHz, MeOD) δ (ppm) 175.5 (1C, NAc C=O), 175.1 (1C, C-1′), 132.6 (1C, C-2), 126.4 (1C, C-3), 87.2 (1C, C-2′), 76.6 (1C, C-6′), 73.4 (1C, C-1), 73.0 (1C, C-8′), 72.6 (1C, octyl OCH2), 72.3 (1C, C-5), 71.8 (1C, C-4), 71.1 (1C, C-6), 70.2 (1C, C-7′), 70.0 (1C, octyl OCH2), 69.6 (1C, C-4′), 64.3 (1C, C-9′), 54.0 (1C, C-5′), 43.2 (1C, C-3′), 33.4 (1C, CH2), 33.0, 31.1, 30.7, 30.6, 30.5 (9C, SCH2, 8 × octyl CH2), 27.5 (1C, CH2), 27.3, 23.7 (4C, 4 × octyl CH2), 22.6 (1C, NHAc), 14.5 (2C, 2 × octyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C36H65NO11SNa+: 742.417 [M+Na]+; found: 742.406.
3.1.19. Compound 18c
Compound 17c (78 mg, 0.092 mmol) was deacetylated; then, methyl ester hydrolysis was performed with potassium hydroxide solution (0.2 M, 2.3 mL, 0.46 mmol, 5 equiv.) according to the procedure for 18a. The crude product was purified by flash column chromatography (acetonitrile/water 95:5→9:1) to yield 18c (25.3 mg, 41%) as a yellowish syrup. Rf = 0.38 (acetonitrile/water 8:2); 1H NMR (400 MHz, MeOD) δ (ppm) 5.89–5.82 (m, 2H, H-2, H-3), 4.14–4.10 (m, 1H, H-1), 3.82–3.67 (m, 6H, H-4, H-4′, H-5′, H-8′, H-9′a,b), 3.65–3.59 (m, 4H, H-5, H-6a,b, H-7′), 3.54–3.39 (m, 5H, H-6′, 2 × hexyl OCH2), 2.88–2.85 (m, 2H, H-3′a, SCH2a), 2.72–2.70 (m, 1H, SCH2b), 2.00 (s, 3H, NHAc CH3), 1.83–1.51 (m, 9H, H-3′b, 2 × CH2, 2 × hexyl CH2), 1.40–1.29 (m, 12H, 6 × hexyl CH2), 0.91 (t, J = 6.7 Hz, 6H, 2 × hexyl CH3); 13C NMR (100 MHz, MeOD) δ (ppm) 175.5 (2C, C-1′, NAc C=O), 132.6 (1C, C-2), 126.4 (1C, C-3), 86.2 (1C, C-2′), 76.6 (1C, C-6′), 73.4 (1C, C-1), 73.0 (1C, C-8′), 72.6 (1C, hexyl OCH2), 72.3 (1C, C-5), 71.8 (1C, C-4), 71.2 (1C, C-6), 70.3 (1C, C-7′), 70.0 (1C, decyl OCH2), 69.7 (1C, C-4′), 64.4 (1C, C-9′), 54.0 (1C, C-5′), 43.2 (1C, C-3′), 33.4 (1C, CH2), 32.9, 32.8, 31.8, 30.7 (4C, 4 × hexyl CH2), 30.5 (1C, SCH2), 27.5 (1C, CH2), 27.0, 23.7 (4C, 4 × hexyl CH2), 22.6 (1C, NHAc), 14.4 (2C, 2 × hexyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C32H56NO11SNa2+: 708.3364 [M-H+2Na]+; found: 708.3367.
3.1.20. Compound 20
Tri-O-acetyl-D-glucal (13, 1.8 g, 6.6 mmol) and monoallyl tetraethylene glycol (19, 2.319 g, 9.9 mmol, 1.5 equiv.) were dissolved in anhydrous dichloromethane (15 mL). A 4Å molecular sieve (1.8 g) was added, and the flask was plugged with a drying tube filled with anhydrous CaCl2. The mixture was stirred at room temperature for 15 min; then, boron trifluoride diethyl etherate (405 µL, 3.3 mmol, 0.5 equiv.) was added. After 60 min, a saturated aqueous solution of NaHCO3 (5 mL) was added, and the reaction mixture was stirred for 15 min. Then, the solvent was evaporated in a vacuum, and the residue was dissolved in dichloromethane (500 mL). This solution was washed three times with aqueous saturated NaHCO3 solution (3 × 50 mL) and twice with water (2 × 50 mL). The organic phase was dried over Na2SO4, then filtered, and the solvent was evaporated in a vacuum. The residue was purified by flash column chromatography (hexane/acetone 8:2) to yield 20 (1.855 g, 90%) as a colorless syrup. Rf = 0.24 (hexane/acetone 7:3); 1H NMR (400 MHz, CDCl3) δ (ppm) 5.96–5.89 (m, 1H, CH=CH2), 5.87 (s, 2H, H-2, H-3), 5.32 (d, J = 9.6 Hz, 1H, H-4), 5.27 (dd, J = 17.2 Hz, J = 1.6 Hz, 1H, allyl CH=CH2a), 5.19–5.16 (m, 1H, allyl CH=CH2b), 5.08 (s, 1H, H-1), 4.26 (dd, J = 12.1 Hz, J = 5.1 Hz, 1H, H-6a), 4.17 (dd, J = 12.1 Hz, J = 2.3 Hz, 1H, H-6b), 4.12 (ddd, J = 9.3 Hz, J = 5.1 Hz, J = 2.2 Hz, 1H, H-5), 4.03–4.02 (m, 2H, allyl CH2), 3.91–3.59 (m, 16H, 8 × TEG OCH2), 2.10, 2.09 (2 × s, 6H, 2 × Ac CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 170.8, 170.3 (2C, 2 × Ac C=O), 134.8 (1C, allyl CH=CH2), 129.2, 127.8 (2C, C-2, C-3), 117.1 (1C, allyl CH=CH2), 94.7 (1C, C-1), 72.8, 70.6, 70.5, 69.5, 67.9 (9C, allyl CH2, 8 × TEG OCH2), 66.9 (1C, C-5), 65.3 (1C, C-4), 63.0 (1C, C-6), 21.0, 20.9 (2C, 2 × Ac CH3); MALDI-TOF MS (Autoflex): m/z calcd for C21H34O10Na+: 469.2044 [M+Na]+; found: 469.2035.
3.1.21. Compound 21a
Compound 20 (500 mg, 1.12 mmol) was dissolved in anhydrous methanol (20 mL), and small pieces of sodium were added to make the solution alkaline (pH~11). The reaction mixture was stirred for 1 h; then, Serdolit Red H+ ion exchange resin was added to make the pH neutral. Thereafter, the reaction mixture was filtered, and the solvent was evaporated.
In the second step, sodium hydride (0.179 g, 4.48 mmol, 2 equiv./OH, 60% in mineral oil) was washed with hexane and dried under argon for 30 min. The crude product of the previous step was dissolved in anhydrous N,N-dimethylformamide (5 mL), and this solution was added to the washed sodium hydride. The mixture was stirred under an argon atmosphere for 30 min; then, octyl bromide (0.585 mL, 3.36 mmol, 3 equiv.) was added. The reaction mixture was stirred at room temperature overnight; then, methanol (1 mL) and water (1 mL) were added successively, and the reaction mixture was stirred for 2 × 15 min. The solvent was evaporated, and the residue was purified by flash column chromatography (hexane/acetone 9:1) to yield 21a (240 mg, 37%) as a colorless syrup. Rf = 0.6 (hexane/acetone 7:3); 1H NMR (400 MHz, CDCl3) δ (ppm) 6.03 (d, J = 10.3 Hz, 1H, H-3), 5.91 (ddt, J = 16.1 Hz, J = 10.7 Hz, J = 5.7 Hz, 1H, CH=CH2), 5.75 (dt, J = 10.3 Hz, J = 2.0 Hz, 1H, H-2), 5.29–5.25 (m, 1H, allyl CH=CH2a), 5.19–5.15 (m, 1H, allyl CH=CH2b), 5.03 (s, 1H, H-1), 4.03–4.02 (m, 2H, allyl CH2), 3.98–3.96 (m, 1H, H-4), 3.91–3.87 (m, 1H, H-6a), 3.85–3.82 (m, 1H, H-5), 3.71–3.60 (m, 17H, H-6b, 8 × TEG OCH2), 3.59–3.37 (m, 4H, 2 × octyl OCH2), 1.63–1.51 (m, 4H, 2 × octyl CH2), 1.41–1.27 (m, 20H, 10 × octyl CH2), 0.89–0.86 (m, 6H, 2 × octyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 134.8 (1C, allyl CH=CH2), 131.2 (1C, C-3), 126.2 (1C, C-2), 117.1 (1C, allyl CH=CH2), 94.9 (1C, C-1), 72.3, 71.8, 70.7, 70.6, 70.5, 69.7, 69.5 (12C, C-4, allyl CH2, 2 × octyl OCH2, 8 × TEG OCH2), 69.3 (1C, C-5), 67.6 (1C, C-6), 31.9, 30.1, 29.7, 29.5, 29.4, 26.2, 22.7 (12C, 12 × octyl CH2), 14.2 (2C, 2 × octyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C33H62O8Na+: 609.4337 [M+Na]+; found: 609.4340.
3.1.22. Compound 21b
Compound 20 (500 mg, 1.12 mmol) was deacetylated, then alkylated with hexyl bromide (0.473 mL, 3.36 mmol, 3 equiv.) in the presence of sodium hydride (0.179 g, 4.48 mmol, 2 equiv./OH, 60% in mineral oil) according to the procedure for 21a. The crude product was purified by flash column chromatography (hexane/acetone 9:1) to yield 21b (303 mg, 41%) as a colorless syrup. Rf = 0.59 (hexane/acetone 6:4); 1H NMR (400 MHz, CDCl3) δ (ppm) 6.03 (d, J = 10.2 Hz, 1H, H-3), 5.96–5.82 (m, 1H, CH=CH2), 5.77–5.73 (m, 1H, H-2), 5.27 (dq, J = 17.2 Hz, J = 1.6 Hz, 1H, allyl CH=CH2a), 5.19–5.14 (m, 1H, allyl CH=CH2b), 5.04 (s, 1H, H-1), 4.02 (dt, J = 5.7 Hz, J = 1.4 Hz, 2H, allyl CH2), 3.98–3.96 (m, 1H, H-4), 3.91–3.88 (m, 1H, H-6a), 3.86–3.81 (m, 1H, H-5), 3.72–3.60 (m, 17H, H-6b, 8 × TEG OCH2), 3.59–3.37 (m, 4H, 2 × hexyl CH2), 1.65–1.51 (m, 4H, 2 × hexyl CH2), 1.37–1.26 (m, 12H, 6 × hexyl CH2), 0.91–0.87 (m, 6H, 2 × hexyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 134.8 (1C, allyl CH=CH2), 131.2 (1C, C-3), 126.2 (1C, C-2), 117.1 (1C, allyl CH=CH2), 94.9 (1C, C-1), 72.3, 71.8, 70.7, 70.6, 70.5, 69.7, 69.5 (12C, C-4, allyl CH2, 2 × hexyl OCH2, 8 × TEG OCH2), 69.2 (1C, C-5), 67.6 (1C, C-6), 31.8, 31.7, 30.1, 29.6, 25.9, 22.7 (8C, 8 × hexyl CH2), 14.1 (2C, 2 × hexyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C29H54O8Na+: 553.3711 [M+Na]+; found: 553.3692.
3.1.23. Compound 22a
Compound 21a (183 mg, 0.312 mmol) and compound 16 (174 mg, 0.343 mmol, 1.1 equiv.) were dissolved in anhydrous toluene (3 mL), and 2,2-dimethoxy-2-phenylacetophenone (DPAP) (8 mg, 0.0312 mmol, 0.1 equiv.) was added. The reaction mixture was cooled to 0 °C and irradiated with UV light for 4 × 15 min; DPAP (8 mg, 0.0312 mmol, 0.1 equiv.) was added before each irradiation cycle. Then, the solvent was evaporated in a vacuum, and the residue was purified by flash column chromatography (hexane/acetone 9:1) to yield 22a (72 mg, 21%) as a colorless syrup. It was used for further conversion without NMR characterization. Rf = 0.38 (hexane/acetone 6:4); MALDI-TOF MS (Autoflex): m/z calcd for C53H91N1O20SNa+: 1116.5747 [M+Na]+; found: 1116.5711.
3.1.24. Compound 22b
Compound 21b (150 mg, 0.283 mmol) and compound 16 (172 mg, 0.339 mmol, 1.2 equiv.) were reacted in the presence of DPAP (7 mg, 0.0283 mmol, 0.1 equiv.) according to the procedure applied for the synthesis of 22a. The crude product was purified by flash column chromatography (hexane/acetone 8:2→6:4) to yield 22b (170 mg, 51%) as a colorless syrup. It was used for further conversion without NMR characterization. Rf = 0.33 (dichloromethane/acetone 8:2); MALDI-TOF MS (Autoflex): m/z calcd for C49H83N1O20SNa+: 1060.5121 [M+Na]+; found: 1060.5128.
3.1.25. Compound 23a
Compound 22a (71 mg, 0.065 mmol) was dissolved in anhydrous methanol (10 mL), and little pieces of sodium were added to make the pH basic (pH~11). After 1 h of stirring, Amberlite IR-50 H+ ion exchange resin was added, and the pH was adjusted to neutral. Then, the reaction mixture was filtered, and the solvent was evaporated.
The residue was dissolved in 1,4-dioxane (9 mL) and water (1 mL), and the reaction mixture was cooled to 0 °C. Then, an aqueous solution of potassium hydroxide (0.2 M, 1.625 mL, 0.325 mmol, 5 equiv.) was added, and the reaction mixture was stirred at room temperature for 4 h; then, Amberlite IR-50 H+ ion exchange resin was added, and the pH was adjusted to neutral. The reaction mixture was filtered; the solvent was evaporated in a vacuum, and the residue was purified by flash column chromatography (acetonitrile/water 9:1) to yield 23a (46.4 mg, 78%) as a colorless syrup. Rf = 0.43 (acetonitrile/water 8:2); 1H NMR (400 MHz, CDCl3) δ (ppm) 5.99 (d, J = 10.3 Hz, 1H, H-3), 5.69 (d, J = 10.3 Hz, 1H, H-2), 4.93 (s, 1H, H-1), 3.81–3.68 (m, 4H, H-4, H-5, H-6a, H-9′a), 3.66–3.30 (m, 29H, H-6b, H-4′, H-5′, H-6′, H-7′, H-8′, H-9′b, OCH2, 2 × octyl OCH2, 8 × TEG OCH2), 2.79–2.72 (m, 2H, H-3′a, SCH2a), 2.66–2.59 (m, 1H, SCH2b), 1.92 (s, 3H, NHAc), 1.87–1.75 (m, 2H, CH2), 1.57–1.54 (m, 1H, H-3′b), 1.51–1.43 (m, 4H, 2 × octyl CH2), 1.21–1.12 (m, 20H, 10 × octyl CH2), 0.82–0.79 (m, 6H, 2 × octyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 175.5 (2C, Ac C=O, C-1′), 131.9 (1C, C-3), 127.3 (1C, C-2), 96.0 (1C, C-1), 87.4 (1C, C-2′), 76.6 (1C, C-6′), 73.0 (1C, C-8′), 72.6 (2C, 2 × octyl OCH2), 72.1 (1C, C-4), 71.4, 71.3, 71.2, 71.1, 70.9, 70.3 (9C, OCH2, 8 × TEG OCH2), 70.7 (2C, C-5, C-7′), 69.6 (1C, C-4′), 68.5 (1C, C-6), 64.4 (1C, C-9′), 54.0 (1C, C-5′), 43.2 (1C, C-3′), 33.0, 31.1, 31.0, 30.7, 30.6, 30.5, 27.4, 27.3, 23.7 (14C, CH2, SCH2, 12 × octyl CH2), 22.6 (1C, NHAc), 14.5 (2C, 2 × octyl CH3); MALDI-TOF MS (Autoflex): m/z calcd for C44H80N1O16SNa2+: 956.4988 [M-H+2Na]+; found: 956.4993.
3.1.26. Compound 23b
Compound 22b (170 mg, 0.164 mmol) was deacetylated, and then, the methyl ester was hydrolyzed with an aqueous solution of potassium hydroxide (0.2 M, 4.1 mL, 0.82 mmol, 5 equiv.) according to the procedure applied for the synthesis of 23a. The crude product was purified by flash column chromatography (acetonitrile/water 9:1) to yield 23b (61 mg, 44%) as a colorless syrup. Rf = 0.56 (acetonitrile/water 8:2); 1H NMR (400 MHz, CDCl3) δ (ppm) 6.08 (d, J = 10.3 Hz, 1H, H-3), 5.77 (dt, J = 10.3 Hz, J = 2.1 Hz, 1H, H-2), 5.02 (s, 1H, H-1), 3.90–3.83 (m, 4H, H-4, H-5, H-6a, H-9′a), 3.78–3.39 (m, 29H, H-6b, H-4′, H-5′, H-6′, H-7′, H-8′, H-9′b, OCH2, 2 × hexyl OCH2, 8 × TEG OCH2), 2.89–2.81 (m, 2H, H-3′a, SCH2a), 2.76–2.71 (m, 1H, SCH2b), 2.02 (s, 3H, NHAc), 1.94–1.86 (m, 2H, CH2), 1.66–1.63 (m, 1H, H-3′b), 1.60–1.52 (m, 4H, 2 × hexyl CH2), 1.39–1.28 (m, 12H, 6 × hexyl CH2), 0.93–0.89 (m, 6H, 2 × hexyl CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 175.5, 175.0 (2C, Ac C=O, C-1′), 131.9 (1C, C-3), 127.3 (1C, C-2), 96.0 (1C, C-1), 87.3 (1C, C-2′), 76.6 (1C, C-6′), 73.0 (1C, C-8′), 72.6 (2C, 2 × hexyl OCH2), 72.1 (1C, C-4), 71.4, 71.3, 71.2, 71.1, 71.0, 70.9, 70.7, 70.3 (9C, OCH2, 8 × TEG OCH2), 70.7 (2C, C-5, C-7′), 69.5 (1C, C-4′), 68.5 (1C, C-6), 64.4 (1C, C-9′), 54.0 (1C, C-5′), 43.2 (1C, C-3′), 32.8, 32.7, 31.1, 31.0, 30.7, 27.4, 26.9, 23.7 (10C, CH2, SCH2, 8 × hexyl CH2), 22.7 (1C, NHAc), 14.4 (2C, 2 × hexyl CH3); ESI-QTOF MS: m/z calcd for C40H72NO16S−: 854.457 [M-H]−; found: 854.455.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}