
Investigating the Human Intestinal DNA Virome and Predicting Disease-Associated Virus–Host Interactions in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)

 ,
,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
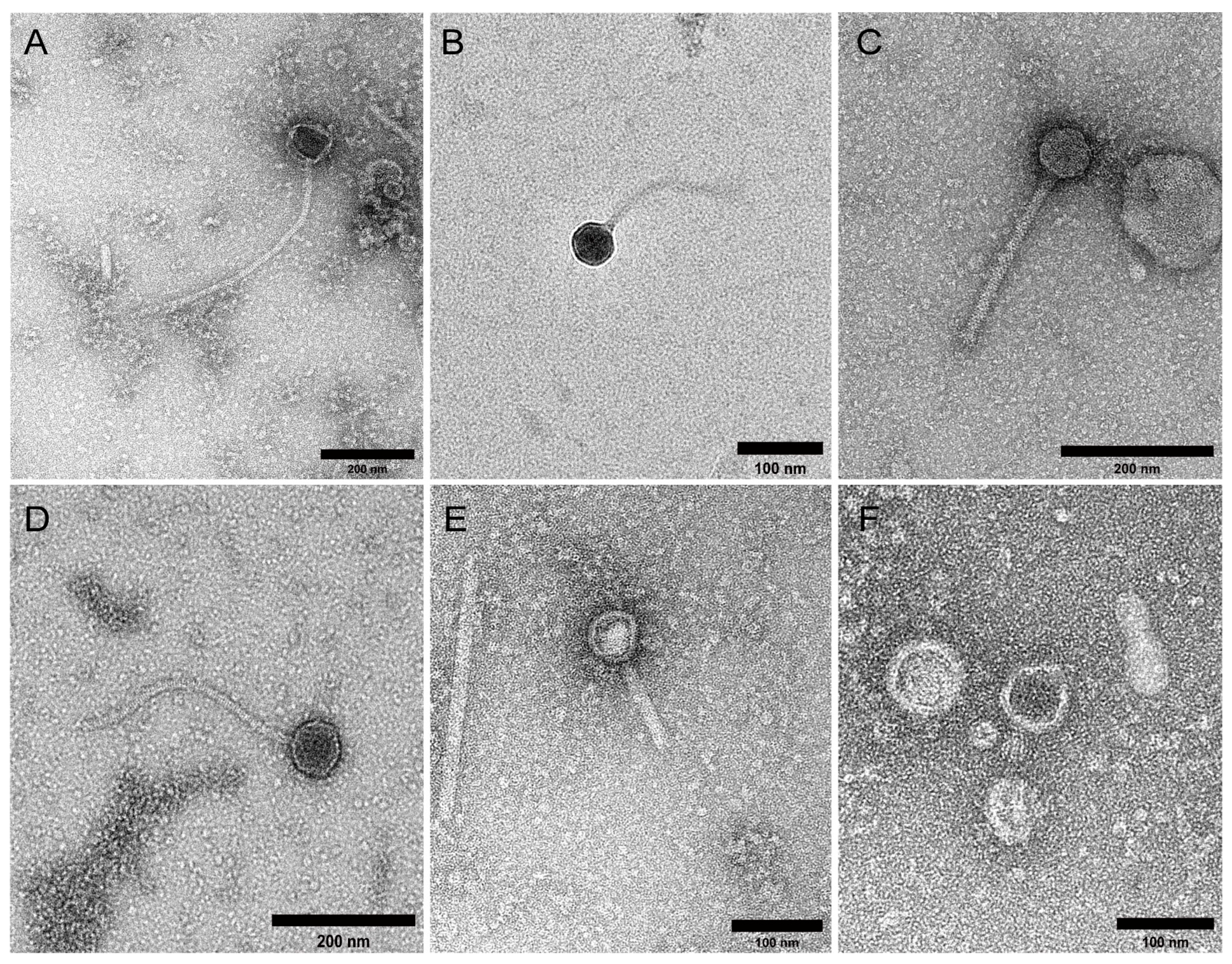
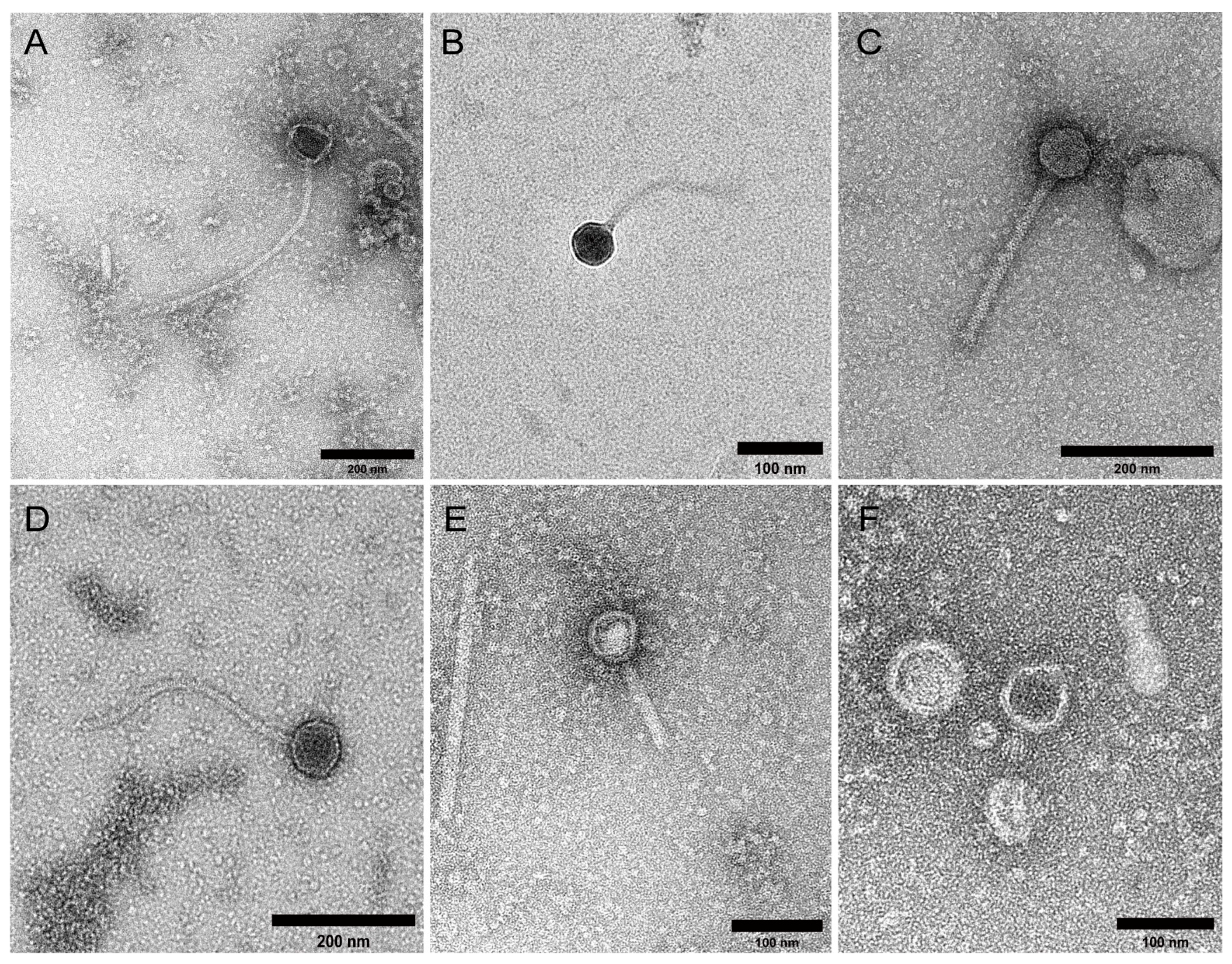
2.1. Study Cohort and VLP Characterisation
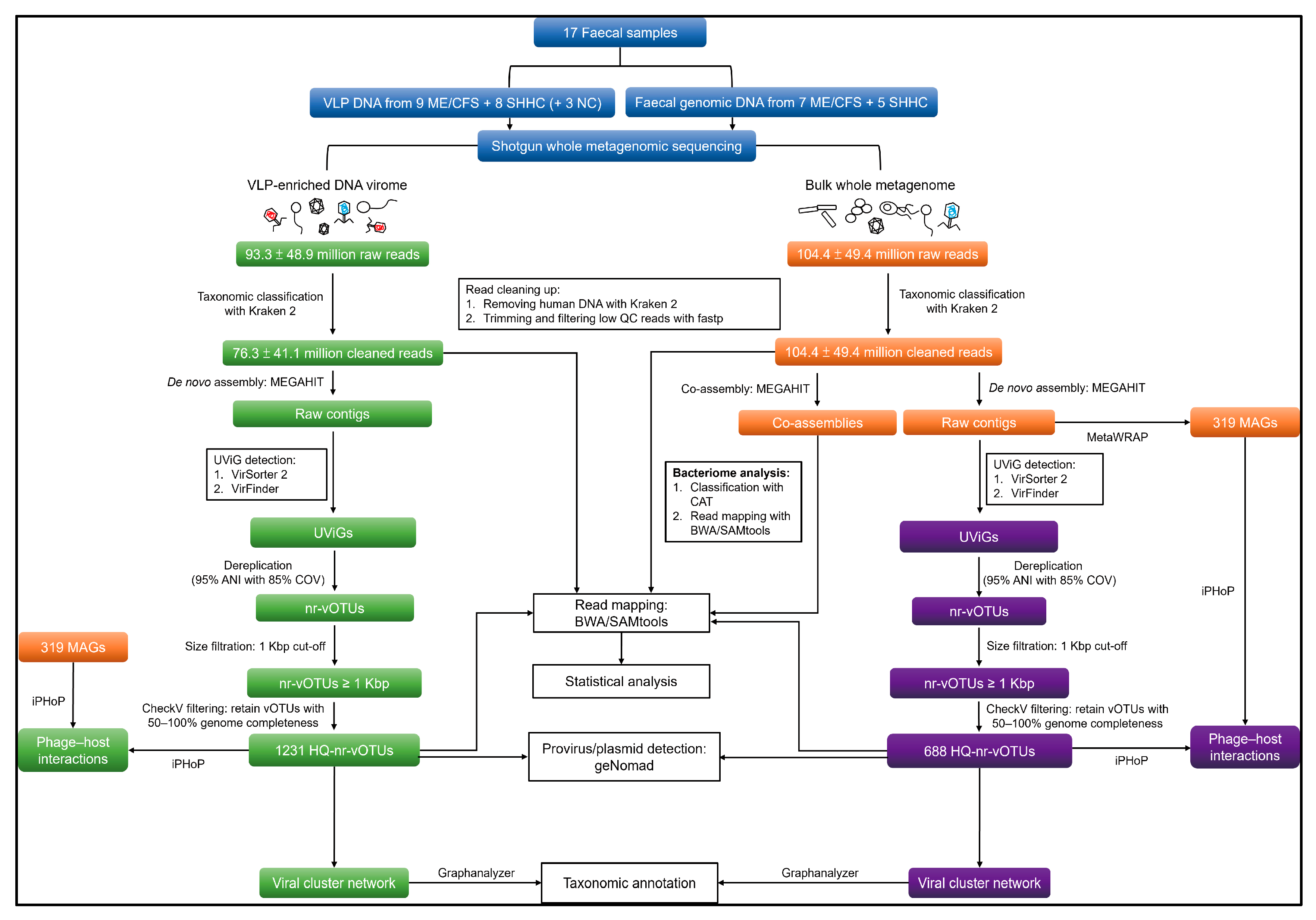
2.2. Quantitative and Qualitative Assessment of Enriched VLP- and WMS-Derived Viral Genomes
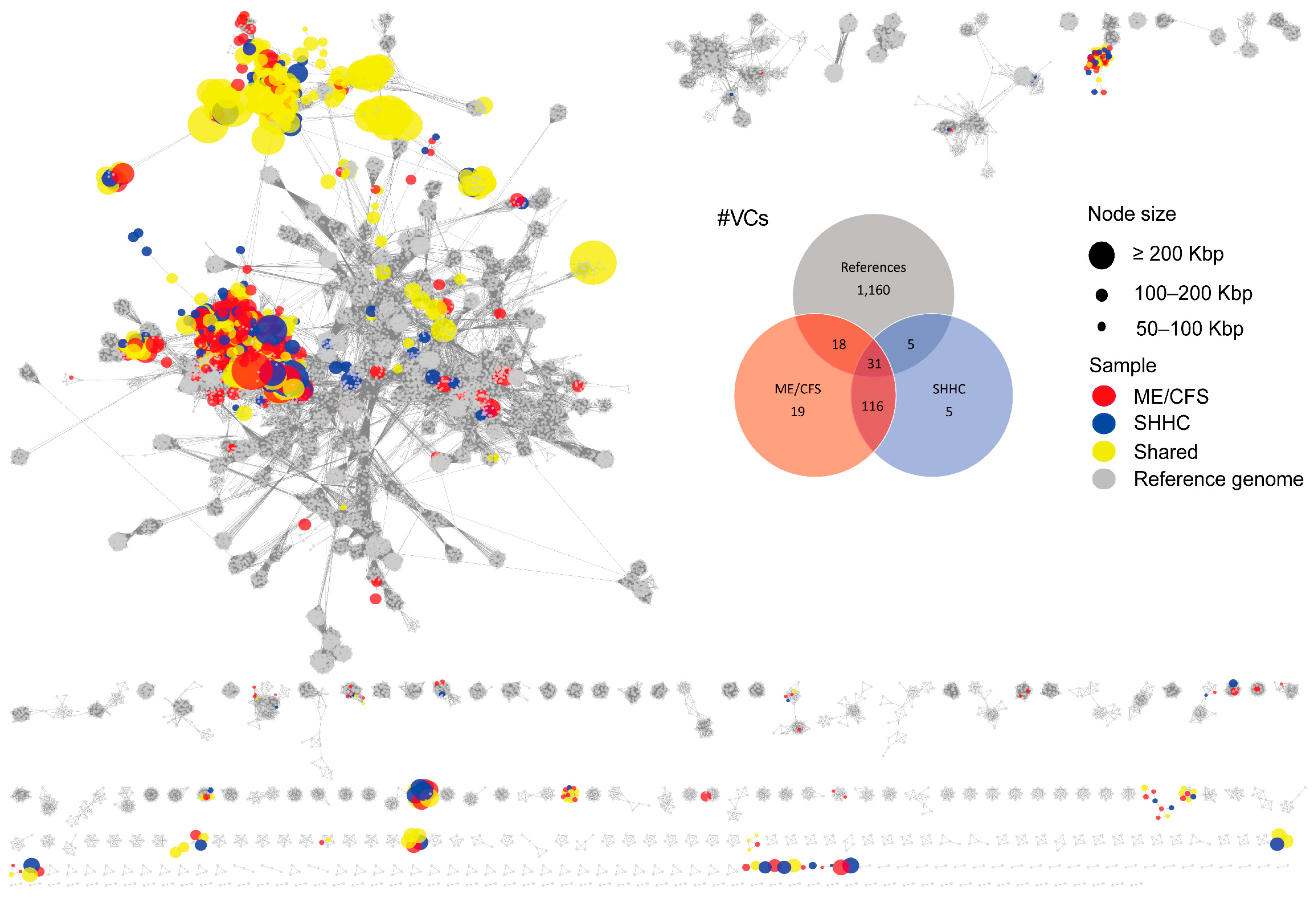
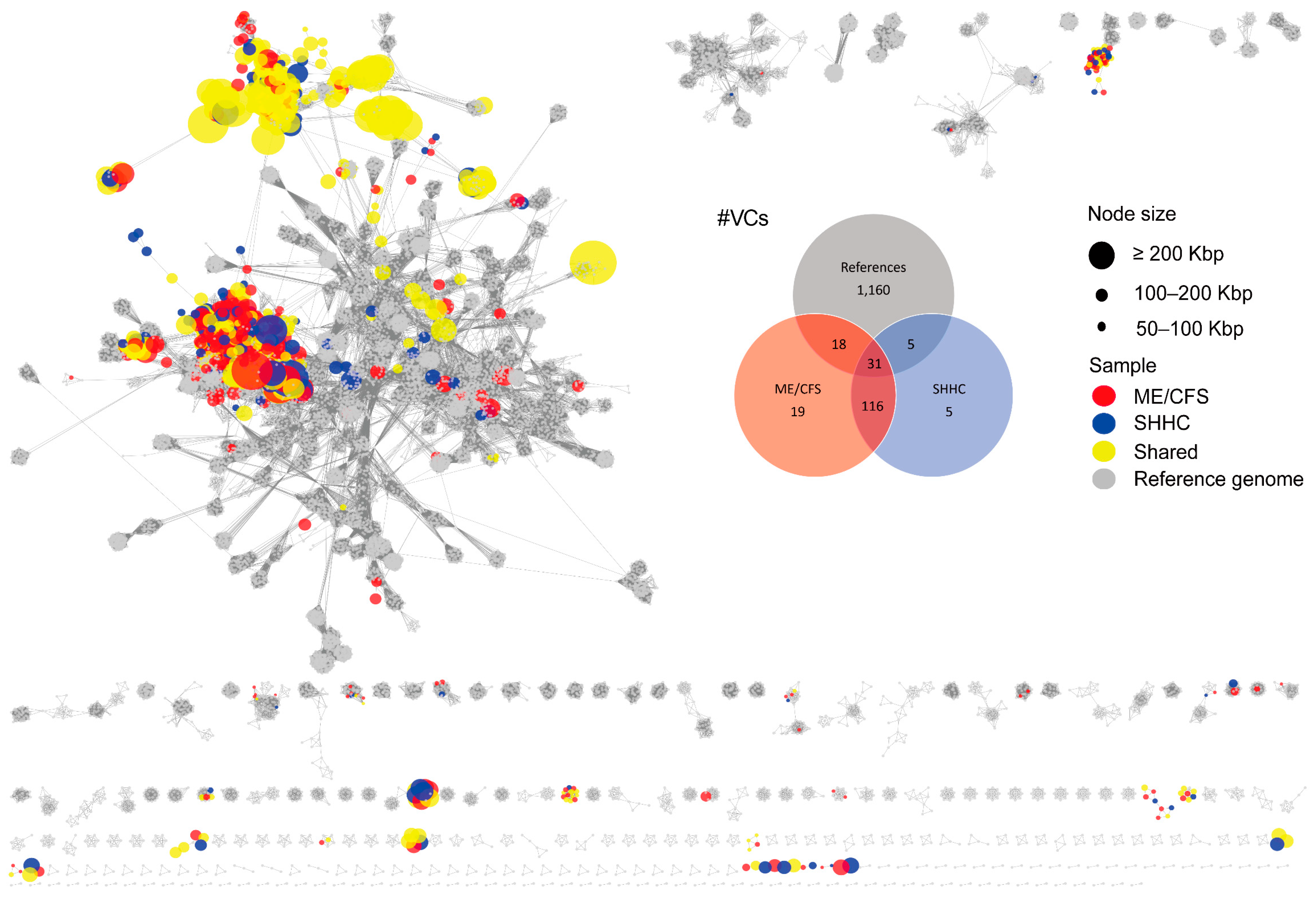
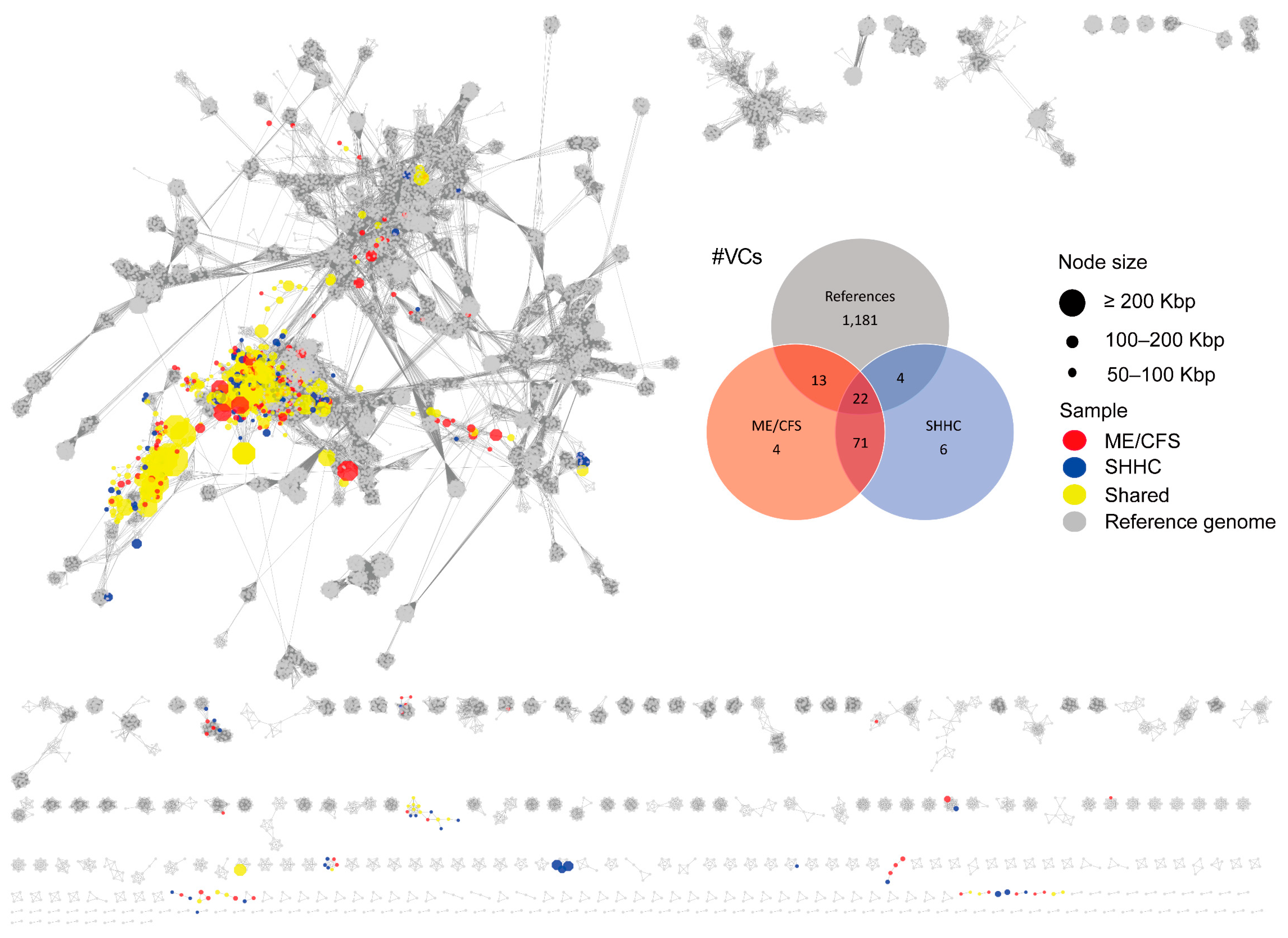
2.3. Cluster Analysis and Taxonomic Assignment of the HQ-vOTUs
2.4. Detection of GI Eukaryotic DNA Viruses
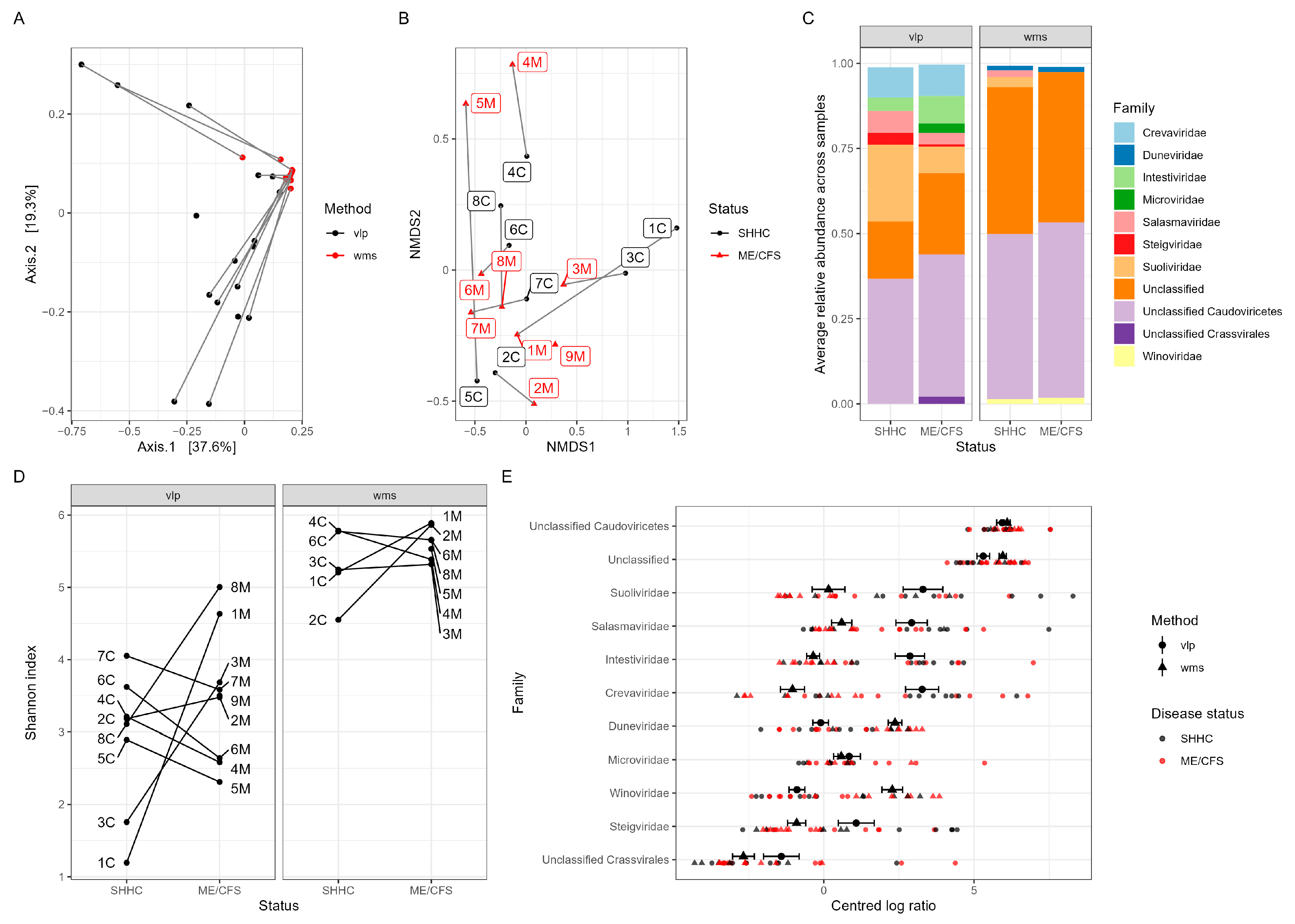
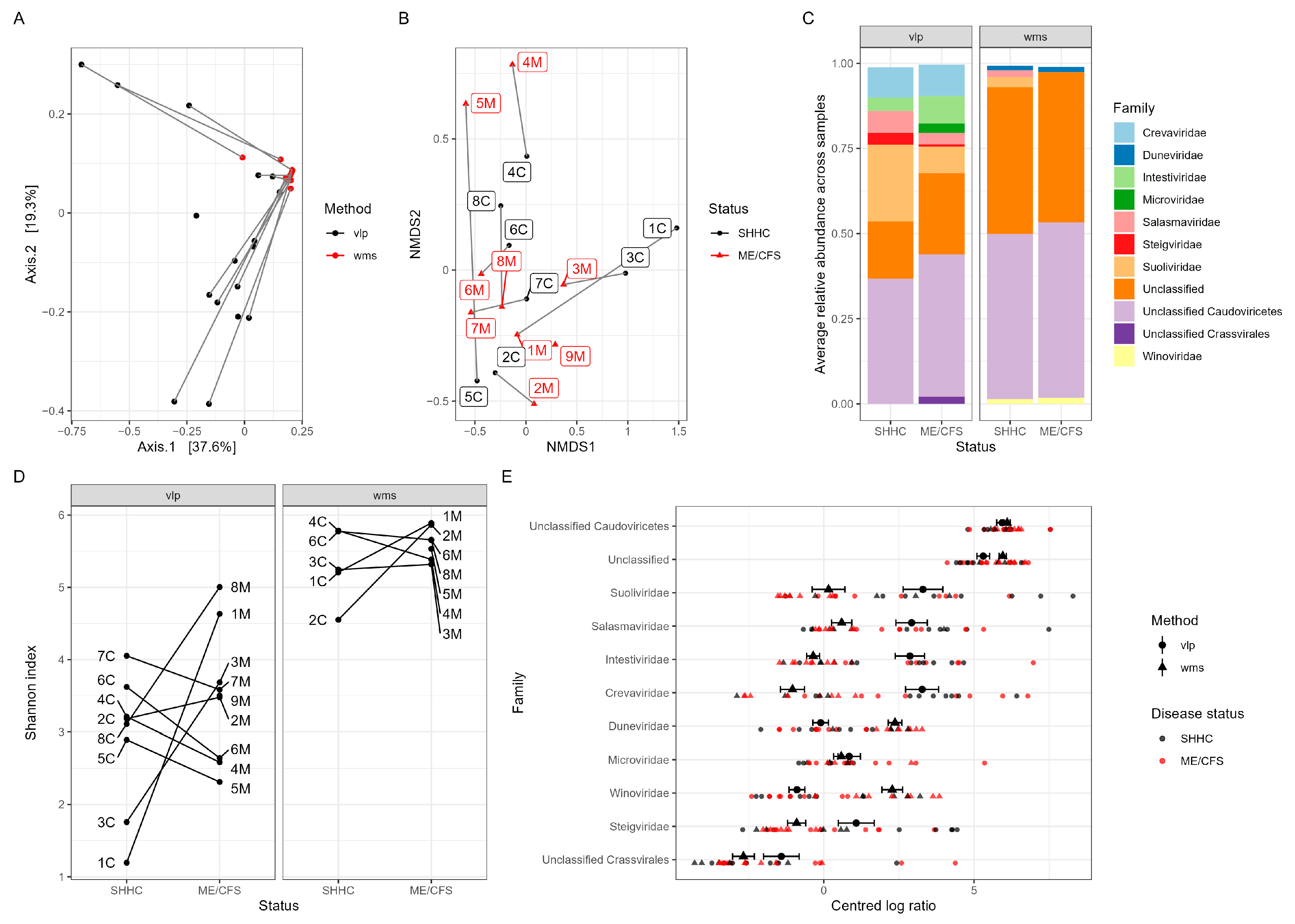
2.5. Macrodiversity of Intestinal Viromes
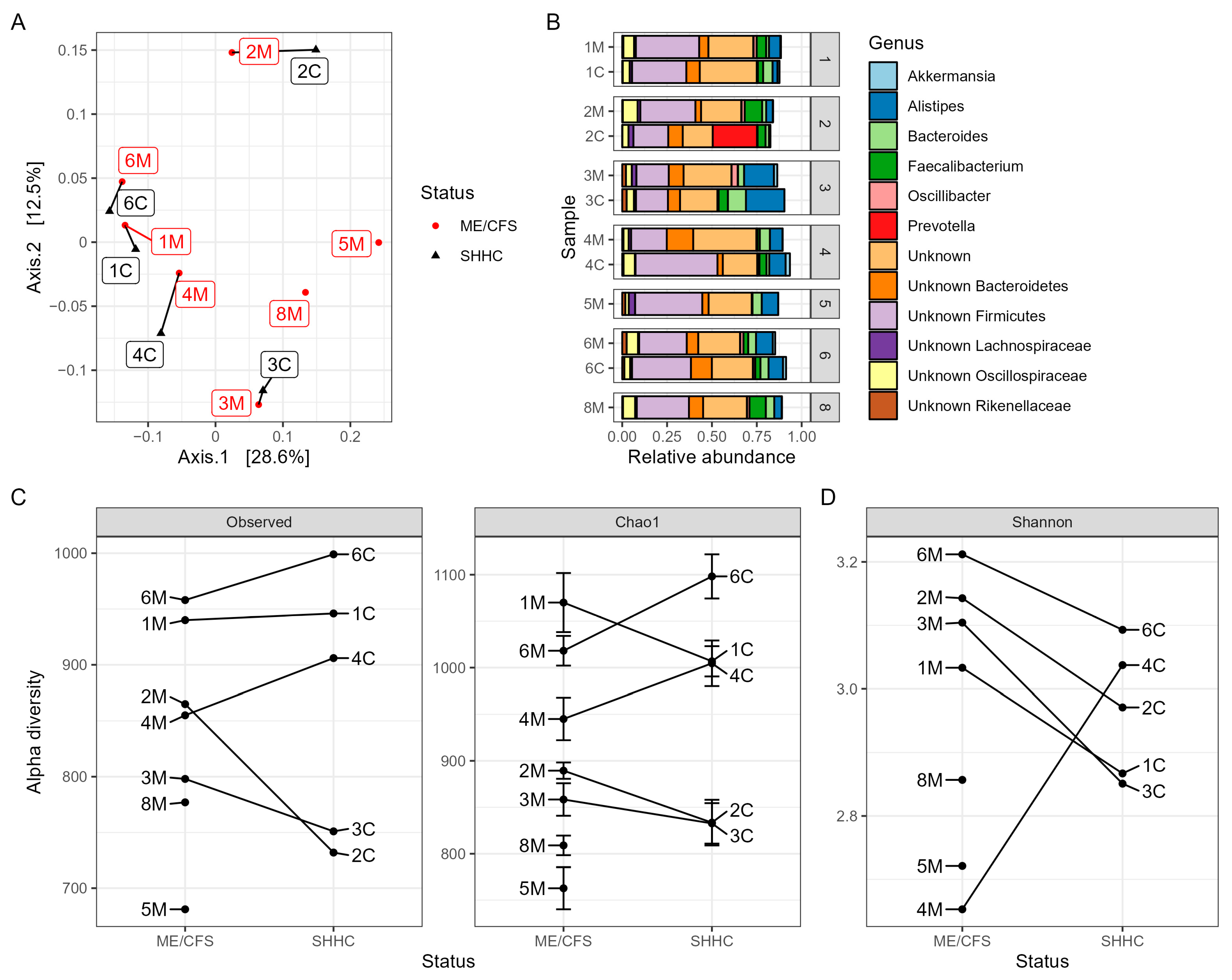
2.6. Macrodiversity of Intestinal Bacteriomes
2.7. Predicting Disease-Associated Bacterial Hosts for the HQ-vOTUs
3. Discussion
4. Materials and Methods
4.1. Study Participants and Sample Collection
4.2. Faecal VLP and VLP DNA Isolation
4.3. Epifluorescence Microscopy (EFM)-Based Enumeration of Enriched Faecal VLPs
4.4. Transmission Electron Microscopy (TEM)
4.5. Library Preparation and Whole Metagenomic Sequencing of Enriched VLP DNA
4.6. Faecal Genomic DNA Isolation, Library Preparation and Whole Metagenomic Sequencing
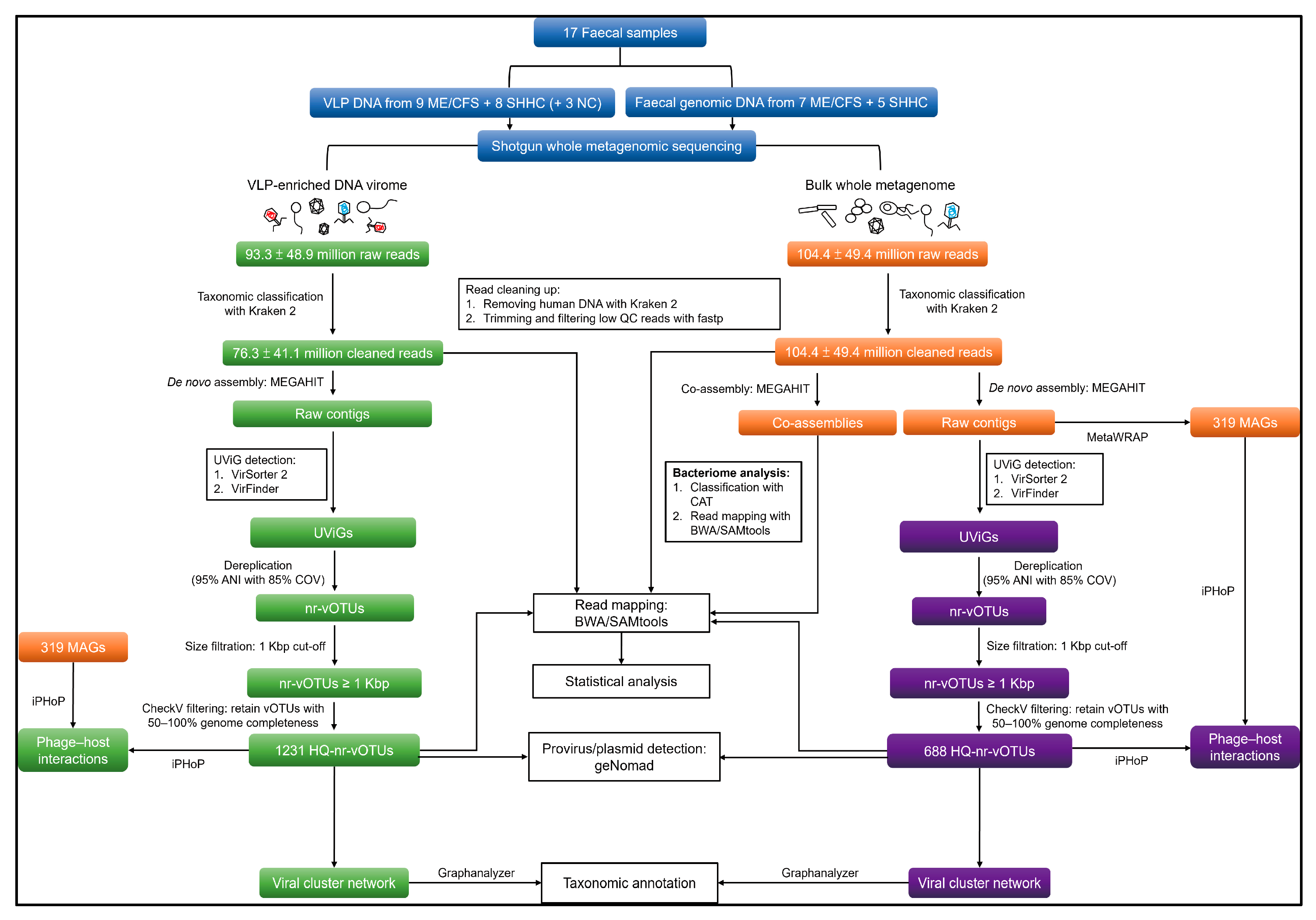
4.7. Analysis of VLP and WMS HQ-vOTUs
4.7.1. Genome Assembly and Viral Mining
4.7.2. Dereplicating and Generating Non-Redundant Viral Operational Taxonomic Units (vOTUs)
4.7.3. Similarity Identification between VLP and WMS HQ-vOTU Datasets
4.7.4. Detecting Provirus and Plasmid from the VLP and WMS vOTU Datasets
4.7.5. Read Mapping, Average Coverage Calculation and Count Table Construction
4.7.6. vOTU Clustering and Taxonomic Annotation
4.7.7. Detection of Human Eukaryotic DNA Viruses
4.8. Analysis of Bacteriome
4.9. Host Predictions for VLP and WMS HQ-vOTUs
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W. The virome in mammalian physiology and disease. Cell 2014, 157, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M. Phage Diversity in the Human Gut Microbiome: A Taxonomist’s Perspective. mSystems 2021, 6, e0079921. [Google Scholar] [CrossRef]
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment. Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Camarillo-Guerrero, L.F.; Almeida, A.; Rangel-Pineros, G.; Finn, R.D.; Lawley, T.D. Massive expansion of human gut bacteriophage diversity. Cell 2021, 184, 1098–1109.e1099. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 2020, 28, 724–740. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef]
- Carruthers, B.M.; van de Sande, M.I.; De Meirleir, K.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; Powles, A.C.; Speight, N.; Vallings, R.; et al. Myalgic encephalomyelitis: International Consensus Criteria. J. Intern. Med. 2011, 270, 327–338. [Google Scholar] [CrossRef]
- Fukuda, K.; Straus, S.E.; Hickie, I.; Sharpe, M.C.; Dobbins, J.G.; Komaroff, A.; Schluederberg, A.; Jones, J.F.; Lloyd, A.R.; Wessely, S.; et al. The Chronic Fatigue Syndrome—A Comprehensive Approach to Its Definition and Study. Ann. Intern. Med. 1994, 121, 953–959. [Google Scholar] [CrossRef]
- National Institutes of Health (N.I.H.). State of the Knowledge Workshop. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) Research: Workshop Report; Office of Research on Women’s Health; NIH; U.S. Department of Health and Human Services: Bethesda, MD, USA, 2011; p. 28.
- Rasa, S.; Nora-Krukle, Z.; Henning, N.; Eliassen, E.; Shikova, E.; Harrer, T.; Scheibenbogen, C.; Murovska, M.; Prusty, B.K.; European Network on, M.C. Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J. Transl. Med. 2018, 16, 268. [Google Scholar] [CrossRef]
- Lidbury, B.A. Ross River Virus Immune Evasion Strategies and the Relevance to Post-Viral Fatigue, and Myalgic Encephalomyelitis Onset. Front. Med. 2021, 8, 662513. [Google Scholar] [CrossRef] [PubMed]
- Lupo, G.F.D.; Rocchetti, G.; Lucini, L.; Lorusso, L.; Manara, E.; Bertelli, M.; Puglisi, E.; Capelli, E. Potential role of microbiome in Chronic Fatigue Syndrome/Myalgic Encephalomyelits (CFS/ME). Sci. Rep. 2021, 11, 7043. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Che, X.; Briese, T.; Ranjan, A.; Allicock, O.; Yates, R.A.; Cheng, A.; March, D.; Hornig, M.; Komaroff, A.L.; et al. Deficient butyrate-producing capacity in the gut microbiome is associated with bacterial network disturbances and fatigue symptoms in ME/CFS. Cell Host Microbe 2023, 31, 288–304.e8. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.W.; McGregor, N.R.; Lewis, D.P.; Butt, H.L.; Gooley, P.R. The association of fecal microbiota and fecal, blood serum and urine metabolites in myalgic encephalomyelitis/chronic fatigue syndrome. Metabolomics 2017, 13, 8. [Google Scholar] [CrossRef]
- Giloteaux, L.; Goodrich, J.K.; Walters, W.A.; Levine, S.M.; Ley, R.E.; Hanson, M.R. Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 2016, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Germain, A.; Ruppert, D.; Levine, S.M.; Hanson, M.R. Metabolic profiling of a myalgic encephalomyelitis/chronic fatigue syndrome discovery cohort reveals disturbances in fatty acid and lipid metabolism. Mol. Biosyst. 2017, 13, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, J.R.; Wettenhall, R.E.; Scanlon, D.; Gooley, P.R.; Lewis, D.P.; McGregor, N.; Stapleton, D.I.; Butt, H.L.; De Meirleir, K.L. Increased d-lactic Acid intestinal bacteria in patients with chronic fatigue syndrome. In Vivo 2009, 23, 621–628. [Google Scholar]
- Myhill, S.; Booth, N.E.; McLaren-Howard, J. Chronic fatigue syndrome and mitochondrial dysfunction. Int. J. Clin. Exp. Med. 2009, 2, 1–16. [Google Scholar]
- Behan, W.M.; More, I.A.; Behan, P.O. Mitochondrial abnormalities in the postviral fatigue syndrome. Acta Neuropathol. 1991, 83, 61–65. [Google Scholar] [CrossRef]
- Tanaka, S.; Kuratsune, H.; Hidaka, Y.; Hakariya, Y.; Tatsumi, K.I.; Takano, T.; Kanakura, Y.; Amino, N. Autoantibodies against muscarinic cholinergic receptor in chronic fatigue syndrome. Int. J. Mol. Med. 2003, 12, 225–230. [Google Scholar] [CrossRef]
- Plioplys, A.V. Antimuscle and anti-CNS circulating antibodies in chronic fatigue syndrome. Neurology 1997, 48, 1717–1719. [Google Scholar] [CrossRef] [PubMed]
- Klimas, N.G.; Salvato, F.R.; Morgan, R.; Fletcher, M.A. Immunologic abnormalities in chronic fatigue syndrome. J. Clin. Microbiol. 1990, 28, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Underhill, R.A. Myalgic encephalomyelitis, chronic fatigue syndrome: An infectious disease. Med. Hypotheses 2015, 85, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Bested, A.C.; Marshall, L.M. Review of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: An evidence-based approach to diagnosis and management by clinicians. Rev. Environ. Health 2015, 30, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Chapenko, S.; Krumina, A.; Kozireva, S.; Nora, Z.; Sultanova, A.; Viksna, L.; Murovska, M. Activation of human herpesviruses 6 and 7 in patients with chronic fatigue syndrome. J. Clin. Virol. 2006, 37 Suppl. 1, S47–S51. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.M.; Beqaj, S.H.; Deeter, R.G.; Fitzgerald, J.T. IgM serum antibodies to Epstein-Barr virus are uniquely present in a subset of patients with the chronic fatigue syndrome. In Vivo 2004, 18, 101–106. [Google Scholar] [PubMed]
- Lerner, A.M.; Zervos, M.; Dworkin, H.J.; Chang, C.-H.; Fitzgerald, J.T.; Goldstein, J.; Lawrie-Hoppen, C.; Franklin, B.; Krotkin, S.M.; Brodsky, M.; et al. New Cardiomyopathy: Pilot Study of Intravenous Ganciclovir in a Subset of the Chronic Fatigue Syndrome. Infect. Dis. Clin. Pract. 1997, 6, 110–117. [Google Scholar] [CrossRef]
- Fremont, M.; Metzger, K.; Rady, H.; Hulstaert, J.; De Meirleir, K. Detection of herpesviruses and parvovirus B19 in gastric and intestinal mucosa of chronic fatigue syndrome patients. In Vivo 2009, 23, 209–213. [Google Scholar]
- Kerr, J.R.; Cunniffe, V.S.; Kelleher, P.; Bernstein, R.M.; Bruce, I.N. Successful intravenous immunoglobulin therapy in 3 cases of parvovirus B19-associated chronic fatigue syndrome. Clin. Infect. Dis. 2003, 36, e100–e106. [Google Scholar] [CrossRef]
- Kerr, J.R.; Bracewell, J.; Laing, I.; Mattey, D.L.; Bernstein, R.M.; Bruce, I.N.; Tyrrell, D.A. Chronic fatigue syndrome and arthralgia following parvovirus B19 infection. J. Rheumatol. 2002, 29, 595–602. [Google Scholar]
- Chia, J.K. The role of enterovirus in chronic fatigue syndrome. J. Clin. Pathol. 2005, 58, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.K.; Chia, A.Y. Chronic fatigue syndrome is associated with chronic enterovirus infection of the stomach. J. Clin. Pathol. 2008, 61, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Giloteaux, L.; Hanson, M.R.; Keller, B.A. A Pair of Identical Twins Discordant for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Differ in Physiological Parameters and Gut Microbiome Composition. Am. J. Case Rep. 2016, 17, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Aaron, L.A.; Burke, M.M.; Buchwald, D. Overlapping conditions among patients with chronic fatigue syndrome, fibromyalgia, and temporomandibular disorder. Arch. Intern. Med. 2000, 160, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, S.; Das, A.; O’Herlihy, E.; Shanahan, F.; O’Toole, P.W.; Jeffery, I.B. The gut virome in Irritable Bowel Syndrome differs from that of controls. Gut Microbes 2021, 13, 1887719. [Google Scholar] [CrossRef] [PubMed]
- Lax, S.; Smith, D.P.; Hampton-Marcell, J.; Owens, S.M.; Handley, K.M.; Scott, N.M.; Gibbons, S.M.; Larsen, P.; Shogan, B.D.; Weiss, S.; et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 2014, 345, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Leite, G.; Pimentel, M.; Barlow, G.M.; Chang, C.; Hosseini, A.; Wang, J.; Parodi, G.; Sedighi, R.; Rezaie, A.; Mathur, R. Age and the aging process significantly alter the small bowel microbiome. Cell Rep. 2021, 36, 109765. [Google Scholar] [CrossRef]
- de la Cuesta-Zuluaga, J.; Kelley, S.T.; Chen, Y.; Escobar, J.S.; Mueller, N.T.; Ley, R.E.; McDonald, D.; Huang, S.; Swafford, A.D.; Knight, R.; et al. Age- and Sex-Dependent Patterns of Gut Microbial Diversity in Human Adults. mSystems 2019, 4, 1–12. [Google Scholar] [CrossRef]
- Hoyles, L.; McCartney, A.L.; Neve, H.; Gibson, G.R.; Sanderson, J.D.; Heller, K.J.; van Sinderen, D. Characterization of virus-like particles associated with the human faecal and caecal microbiota. Res. Microbiol. 2014, 165, 803–812. [Google Scholar] [CrossRef]
- Liang, G.; Zhao, C.; Zhang, H.; Mattei, L.; Sherrill-Mix, S.; Bittinger, K.; Kessler, L.R.; Wu, G.D.; Baldassano, R.N.; DeRusso, P.; et al. The stepwise assembly of the neonatal virome is modulated by breastfeeding. Nature 2020, 581, 470–474. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Ryan, F.J.; Draper, L.A.; Forde, A.; Stockdale, S.R.; Daly, K.M.; McDonnell, S.A.; Nolan, J.A.; Sutton, T.D.S.; Dalmasso, M.; et al. Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome 2018, 6, 68. [Google Scholar] [CrossRef]
- Shah, S.A.; Deng, L.; Thorsen, J.; Pedersen, A.G.; Dion, M.B.; Castro-Mejia, J.L.; Silins, R.; Romme, F.O.; Sausset, R.; Jessen, L.E.; et al. Expanding known viral diversity in the healthy infant gut. Nat. Microbiol. 2023, 8, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Thingstad, T.F. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Oceanogr. 2000, 45, 1320–1328. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe 2019, 26, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, S.G.; Reyes, A.; Maurice, C.F. Bacteriophages playing nice: Lysogenic bacteriophage replication stable in the human gut microbiota. iScience 2023, 26, 106007. [Google Scholar] [CrossRef] [PubMed]
- Czyz, A.; Los, M.; Wrobel, B.; Wegrzyn, G. Inhibition of spontaneous induction of lambdoid prophages in Escherichia coli cultures: Simple procedures with possible biotechnological applications. BMC Biotechnol. 2001, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Diard, M.; Bakkeren, E.; Cornuault, J.K.; Moor, K.; Hausmann, A.; Sellin, M.E.; Loverdo, C.; Aertsen, A.; Ackermann, M.; De Paepe, M.; et al. Inflammation boosts bacteriophage transfer between Salmonella spp. Science 2017, 355, 1211–1215. [Google Scholar] [CrossRef]
- Brown, T.L.; Charity, O.J.; Adriaenssens, E.M. Ecological and functional roles of bacteriophages in contrasting environments: Marine, terrestrial and human gut. Curr. Opin. Microbiol. 2022, 70, 102229. [Google Scholar] [CrossRef]
- Lin, D.M.; Lin, H.C. A theoretical model of temperate phages as mediators of gut microbiome dysbiosis. F1000Research 2019, 8, 997. [Google Scholar] [CrossRef]
- Silveira, C.B.; Rohwer, F.L. Piggyback-the-Winner in host-associated microbial communities. NPJ Biofilms Microbiomes 2016, 2, 16010. [Google Scholar] [CrossRef]
- Kim, K.H.; Bae, J.W. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 2011, 77, 7663–7668. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. PhiCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef] [PubMed]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.S.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and Taxonomy of crAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe 2018, 24, 653–664.e656. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.B.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Breitbart, M.; Lee, W.H.; Run, J.Q.; Wei, C.L.; Soh, S.W.; Hibberd, M.L.; Liu, E.T.; Rohwer, F.; Ruan, Y. RNA viral community in human feces: Prevalence of plant pathogenic viruses. PLoS Biol. 2006, 4, e3. [Google Scholar] [CrossRef] [PubMed]
- Fremont, M.; Coomans, D.; Massart, S.; De Meirleir, K. High-throughput 16S rRNA gene sequencing reveals alterations of intestinal microbiota in myalgic encephalomyelitis/chronic fatigue syndrome patients. Anaerobe 2013, 22, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Virgin, H.W. Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Science 2016, 351, aad5872. [Google Scholar] [CrossRef]
- Xiong, R.; Gunter, C.; Fleming, E.; Vernon, S.D.; Bateman, L.; Unutmaz, D.; Oh, J. Multi-’omics of gut microbiome-host interactions in short- and long-term myalgic encephalomyelitis/chronic fatigue syndrome patients. Cell Host Microbe 2023, 31, 273–287.e5. [Google Scholar] [CrossRef]
- Nagy-Szakal, D.; Williams, B.L.; Mishra, N.; Che, X.; Lee, B.; Bateman, L.; Klimas, N.G.; Komaroff, A.L.; Levine, S.; Montoya, J.G.; et al. Fecal metagenomic profiles in subgroups of patients with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 2017, 5, 44. [Google Scholar] [CrossRef]
- Togo, A.H.; Diop, A.; Dubourg, G.; Khelaifia, S.; Richez, M.; Armstrong, N.; Maraninchi, M.; Fournier, P.E.; Raoult, D.; Million, M. Anaerotruncus massiliensis sp. nov., a succinate-producing bacterium isolated from human stool from an obese patient after bariatric surgery. New Microbes New Infect. 2019, 29, 100508. [Google Scholar] [CrossRef]
- Pham, T.P.; Cadoret, F.; Alou, M.T.; Brah, S.; Diallo, B.A.; Diallo, A.; Sokhna, C.; Delerce, J.; Fournier, P.E.; Million, M.; et al. ‘Urmitella timonensis’ gen. nov., sp. nov., ‘Blautia marasmi’ sp. nov., ‘Lachnoclostridium pacaense’ sp. nov., ‘Bacillus marasmi’ sp. nov. and ‘Anaerotruncus rubiinfantis’ sp. nov., isolated from stool samples of undernourished African children. New Microbes New Infect. 2017, 17, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Woo, P.C.; Woo, G.K.; Fung, A.M.; Ngan, A.H.; Song, Y.; Liu, C.; Summanen, P.; Finegold, S.M.; Yuen, K. Bacteraemia caused by Anaerotruncus colihominis and emended description of the species. J. Clin. Pathol. 2006, 59, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Satokari, R.; Fuentes, S.; Mattila, E.; Jalanka, J.; de Vos, W.M.; Arkkila, P. Fecal transplantation treatment of antibiotic-induced, noninfectious colitis and long-term microbiota follow-up. Case Rep. Med. 2014, 2014, 913867. [Google Scholar] [CrossRef] [PubMed]
- Zupancic, M.L.; Cantarel, B.L.; Liu, Z.; Drabek, E.F.; Ryan, K.A.; Cirimotich, S.; Jones, C.; Knight, R.; Walters, W.A.; Knights, D.; et al. Analysis of the gut microbiota in the old order Amish and its relation to the metabolic syndrome. PLoS ONE 2012, 7, e43052. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Huang, L.K.; Tsai, M.J.; Liao, Y.T.; Lin, Y.S.; Hu, C.J.; Hsu, Y.H. Cognitive Function Associated with Gut Microbial Abundance in Sucrose and S-Adenosyl-L-Methionine (SAMe) Metabolic Pathways. J. Alzheimer’s Dis. 2022, 87, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Bianchimano, P.; Britton, G.J.; Wallach, D.S.; Smith, E.M.; Cox, L.M.; Liu, S.; Iwanowski, K.; Weiner, H.L.; Faith, J.J.; Clemente, J.C.; et al. Mining the microbiota to identify gut commensals modulating neuroinflammation in a mouse model of multiple sclerosis. Microbiome 2022, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Narushima, S.; Sugiura, Y.; Oshima, K.; Atarashi, K.; Hattori, M.; Suematsu, M.; Honda, K. Characterization of the 17 strains of regulatory T cell-inducing human-derived Clostridia. Gut Microbes 2014, 5, 333–339. [Google Scholar] [CrossRef]
- Carruthers, B.M.; Jain, A.K.; De Meirleir, K.L.; Peterson, D.L.; Klimas, N.G.; Lerner, A.M.; Bested, A.C.; Flor-Henry, P.; Joshi, P.; Powles, A.C.P.; et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Chronic Fatigue Syndr. 2003, 11, 7–115. [Google Scholar] [CrossRef]
- National Institute for Health and Care Excellence (N.I.C.E.). NICE Guideline (2007/CG53): Chronic Fatigue Syndrome/Myalgic Encephalomyelitis (or Encephalopathy): Diagnosis and Management; National Institute for Health and Care Excellence: Manchester, UK, 2007; p. 53. [Google Scholar]
- Zigmond, A.S.; Snaith, R.P. The hospital anxiety and depression scale. Acta Psychiatr. Scand. 1983, 67, 361–370. [Google Scholar] [CrossRef]
- Hsieh, S.Y.; Tariq, M.A.; Telatin, A.; Ansorge, R.; Adriaenssens, E.M.; Savva, G.M.; Booth, C.; Wileman, T.; Hoyles, L.; Carding, S.R. Comparison of PCR versus PCR-Free DNA Library Preparation for Characterising the Human Faecal Virome. Viruses 2021, 13, 2093. [Google Scholar] [CrossRef] [PubMed]
- Budinoff, C.R.; Loar, S.N.; LeCleir, G.R.; Wilhelm, S.W.; Buchan, A. A protocol for enumeration of aquatic viruses by epifluorescence microscopy using Anodisc 13 membranes. BMC Microbiol. 2011, 11, 168. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Rio Deiros, D.; Pedersen, B.; Svensson, V. readfq. Available online: https://github.com/lh3/readfq (accessed on 1 November 2021).
- hewm2008. FxTools. Available online: https://github.com/hewm2008/FxTools (accessed on 1 November 2021).
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46:41-12. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Telatin, A.; Fariselli, P.; Birolo, G. SeqFu: A Suite of Utilities for the Robust and Reproducible Manipulation of Sequence Files. Bioengineering 2021, 8, 59. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitua, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.P.; Roux, S.; Schulz, F.; Babinski, M.; Xu, Y.; Hu, B.; Chain, P.S.G.; Nayfach, S.; Kyrpides, N.C. Identification of mobile genetic elements with geNomad. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- apcamargo. geNomad: Identification of Mobile Genetic Elements. 2023. Available online: https://github.com/apcamargo/genomad (accessed on 15 February 2023).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Birolo, G.; Telatin, A. BamToCov, an efficient toolkit for sequence coverage calculations. Bioinformatics 2022, 38, 2617–2618. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef]
- Cook, R.; Brown, N.; Redgwell, T.; Rihtman, B.; Barnes, M.; Clokie, M.; Stekel, D.J.; Hobman, J.; Jones, M.A.; Millard, A. INfrastructure for a PHAge REference Database: Identification of Large-Scale Biases in the Current Collection of Cultured Phage Genomes. Phage 2021, 2, 214–223. [Google Scholar] [CrossRef]
- Bouras, G.; Nepal, R.; Houtak, G.; Psaltis, A.J.; Wormald, P.J.; Vreugde, S. Pharokka: A fast scalable bacteriophage annotation tool. Bioinformatics 2023, 39, btac776. [Google Scholar] [CrossRef]
- RyanCook94. single_GenBank_to_vConTACT_inputs.pl. Available online: https://github.com/RyanCook94/Random-Perl-Scripts/blob/main/single_GenBank_to_vConTACT_inputs.pl (accessed on 15 December 2022).
- RyanCook94. Inphared. Available online: https://github.com/RyanCook94/inphared (accessed on 15 December 2022).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Pandolfo, M.; Telatin, A.; Lazzari, G.; Adriaenssens, E.M.; Vitulo, N. MetaPhage: An Automated Pipeline for Analyzing, Annotating, and Classifying Bacteriophages in Metagenomics Sequencing Data. mSystems 2022, 7, e0074122. [Google Scholar] [CrossRef] [PubMed]
- lazzarigioele. graphanalyzer. Available online: https://github.com/lazzarigioele/graphanalyzer (accessed on 18 December 2022).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Rangel-Pineros, G.; Almeida, A.; Beracochea, M.; Sakharova, E.; Marz, M.; Reyes Munoz, A.; Holzer, M.; Finn, R.D. VIRify: An integrated detection, annotation and taxonomic classification pipeline using virus-specific protein profile hidden Markov models. PLoS Comput. Biol. 2023, 19, e1011422. [Google Scholar] [CrossRef] [PubMed]
- von Meijenfeldt, F.A.B.; Arkhipova, K.; Cambuy, D.D.; Coutinho, F.H.; Dutilh, B.E. Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol. 2019, 20, 217. [Google Scholar] [CrossRef]
- Roux, S.; Camargo, A.P.; Coutinho, F.H.; Dabdoub, S.M.; Dutilh, B.E.; Nayfach, S.; Tritt, A. iPHoP: An integrated machine learning framework to maximize host prediction for metagenome-derived viruses of archaea and bacteria. PLoS Biol. 2023, 21, e3002083. [Google Scholar] [CrossRef] [PubMed]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Wu, Y.W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Li, X.; Stokholm, J.; Brejnrod, A.; Vestergaard, G.A.; Russel, J.; Trivedi, U.; Thorsen, J.; Gupta, S.; Hjelmso, M.H.; Shah, S.A.; et al. The infant gut resistome associates with E. coli, environmental exposures, gut microbiome maturity, and asthma-associated bacterial composition. Cell Host Microbe 2021, 29, 975–987.e4. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ME/CFS | SHHC | |
|---|---|---|
| Age; mean ± SD (median) | 33.8 ± 13.0 (37.0) | 53.6 ± 15.5 (58.5) |
| Gender (male:female) | 0:9 | 3:5 |
| Antibiotic use < 4 weeks | 0 | 0 |
| Probiotic capsule supplement use < 4 weeks | 0 | 0 |
| Illness duration > 6 months a | 9 | N/A b |
| Self-reported with GI disorder | Four self-reported; Five non-reported | 0 |
| Proviruses | Plasmids | |||
|---|---|---|---|---|
| All VLP-nr-vOTUs (n = 192,146) | All WMS-nr-vOTUs (n = 184,317) | All VLP-nr-vOTUs (n = 192,146) | All WMS-nr-vOTUs (n = 184,317) | |
| Pre-CheckV filtration | 254 | 537 | 4119 | 5359 |
| VLP-HQ-vOTUs (n = 1231) | WMS-HQ-vOTUs (n = 688) | VLP-HQ-vOTUs (n = 1231) | WMS-HQ-vOTUs (n = 688) | |
| Post-CheckV filtration | 123 | 254 | 6 | 6 |
| Top 20 Bacterial Genera | Hosts Shared in Both VLP and WMS | Hosts in VLP Only | Hosts in WMS Only | ||
|---|---|---|---|---|---|
| Alistipes | √ | ||||
| Faecalibacterium | √ | ||||
| Bacteroides | √ | √ d | |||
| Oscillibacter a | − | − | − | ||
| Bifidobacterium | √ | ||||
| Ruminococcus b | √ | √ | √ | ||
| Clostridium | √ | √ | |||
| Parabacteroides | √ | ||||
| Akkermansia | √ | ||||
| Collinsella | √ | ||||
| Dialister | √ | ||||
| Roseburia | √ | ||||
| Odoribacter | √ | ||||
| Coprococcus | √ | ||||
| Sutterella | √ | ||||
| Eggerthella | √ | ||||
| Anaerotruncus | √ | ||||
| Adlercreutzia | √ | ||||
| Eubacterium c | √ | √ | √ | √ | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, S.-Y.; Savva, G.M.; Telatin, A.; Tiwari, S.K.; Tariq, M.A.; Newberry, F.; Seton, K.A.; Booth, C.; Bansal, A.S.; Wileman, T.; et al. Investigating the Human Intestinal DNA Virome and Predicting Disease-Associated Virus–Host Interactions in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Int. J. Mol. Sci. 2023, 24, 17267. https://doi.org/10.3390/ijms242417267
Hsieh S-Y, Savva GM, Telatin A, Tiwari SK, Tariq MA, Newberry F, Seton KA, Booth C, Bansal AS, Wileman T, et al. Investigating the Human Intestinal DNA Virome and Predicting Disease-Associated Virus–Host Interactions in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). International Journal of Molecular Sciences. 2023; 24(24):17267. https://doi.org/10.3390/ijms242417267
Chicago/Turabian StyleHsieh, Shen-Yuan, George M. Savva, Andrea Telatin, Sumeet K. Tiwari, Mohammad A. Tariq, Fiona Newberry, Katharine A. Seton, Catherine Booth, Amolak S. Bansal, Thomas Wileman, and et al. 2023. "Investigating the Human Intestinal DNA Virome and Predicting Disease-Associated Virus–Host Interactions in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)" International Journal of Molecular Sciences 24, no. 24: 17267. https://doi.org/10.3390/ijms242417267
APA StyleHsieh, S.-Y., Savva, G. M., Telatin, A., Tiwari, S. K., Tariq, M. A., Newberry, F., Seton, K. A., Booth, C., Bansal, A. S., Wileman, T., Adriaenssens, E. M., & Carding, S. R. (2023). Investigating the Human Intestinal DNA Virome and Predicting Disease-Associated Virus–Host Interactions in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). International Journal of Molecular Sciences, 24(24), 17267. https://doi.org/10.3390/ijms242417267