
Rational Design of Novel Peptidomimetics against Influenza A Virus: Biological and Computational Studies

 , ,
, ,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Design
2.1.1. Design of N-Methyl Peptides
2.1.2. Design of Peptoids
2.2. Chemistry
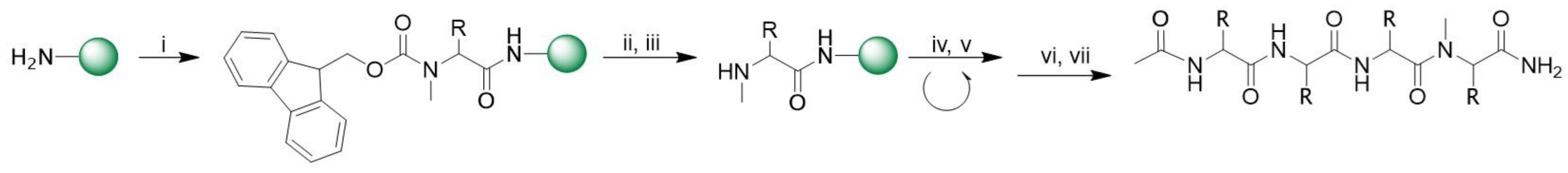
2.2.1. N-Methyl Peptides
2.2.2. Peptoids
2.3. Direct Binding Assays
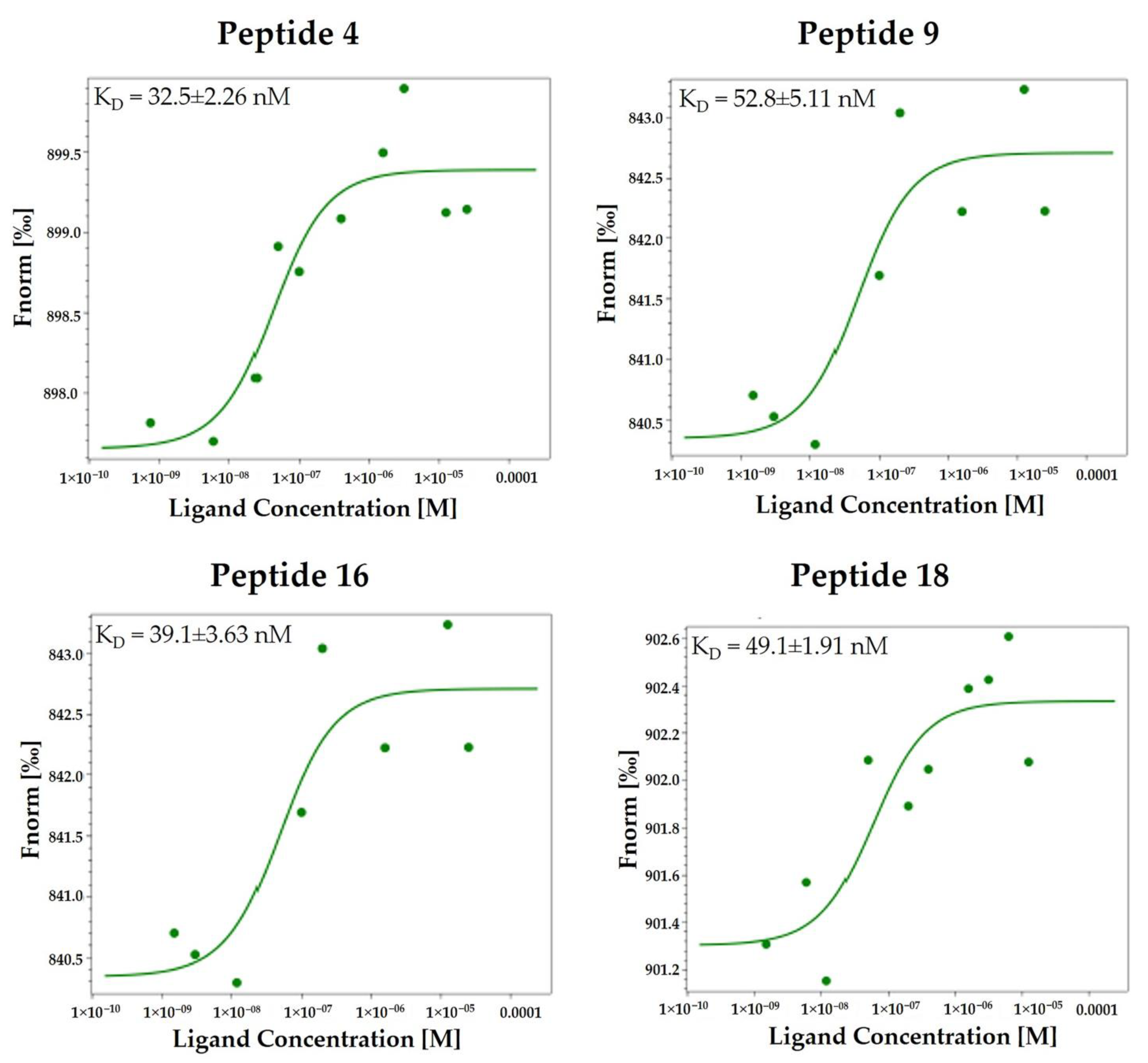
2.3.1. Microscale Thermophoresis (MST)
2.3.2. Surface Plasmon Resonance (SPR)
2.4. Antiviral Activity
2.4.1. Toxicity of Compounds in MDCK cells
2.4.2. Hemagglutination Inhibition Assay (HI)
2.4.3. Neutralization Assay (NT)
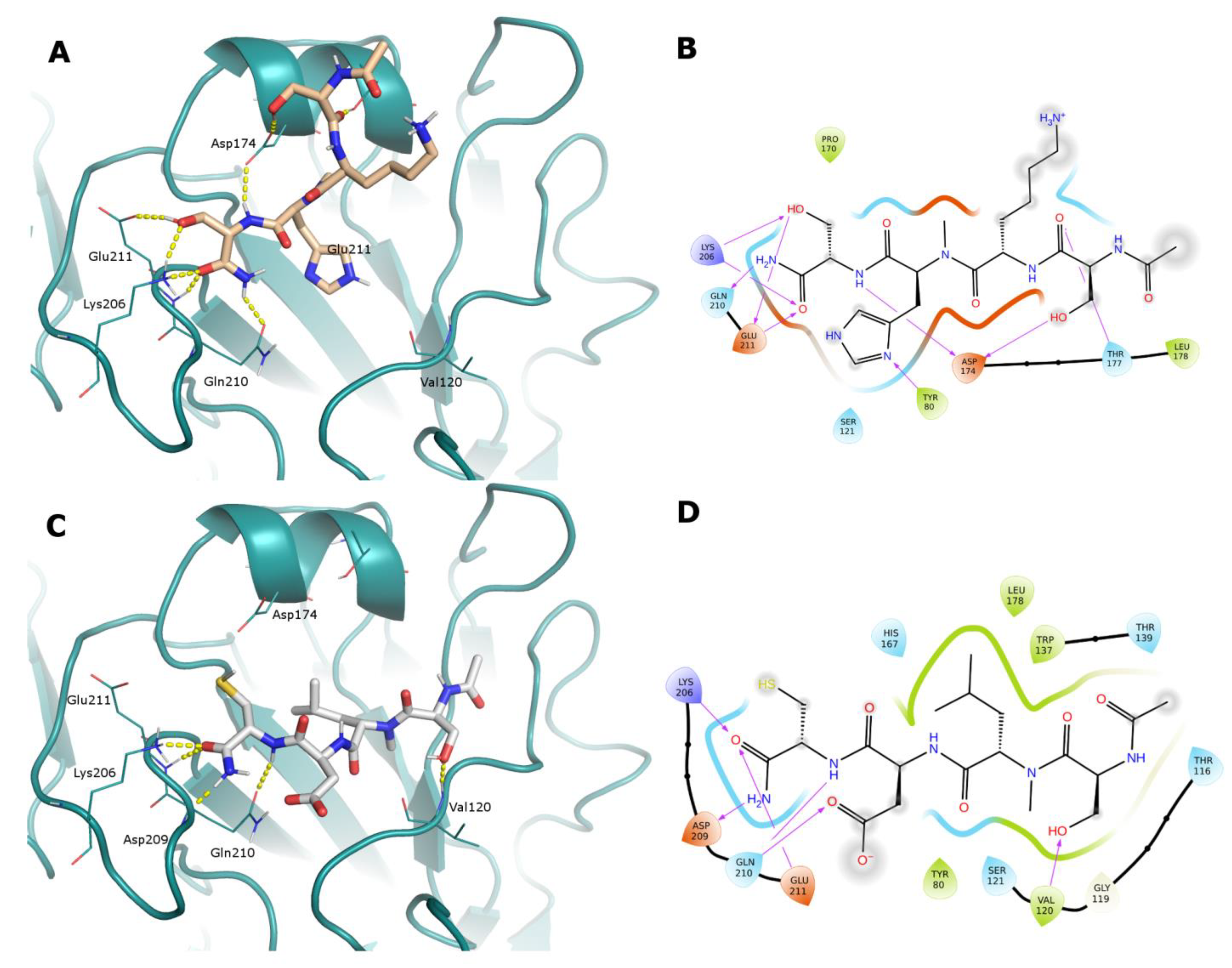
2.5. Computational Studies
3. Materials and Methods
3.1. Peptide Synthesis
3.1.1. Synthesis of N-Methyl Peptides
3.1.2. Synthesis of Peptoids
3.1.3. Purification and Characterization
3.2. Direct Binding Assay
3.2.1. Microscale Thermophoresis (MST)
3.2.2. Surface Plasmon Resonance (SPR)
3.3. Biological Procedures
3.3.1. Cells Line and VIRUS
3.3.2. Cytotoxicity Assay
3.3.3. Hemagglutination Inhibition Assay (HI)
3.3.4. Neutralization Assay (NT)
3.4. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salomon, R.; Webster, R.G. The influenza virus enigma. Cell 2009, 136, 402–410. [Google Scholar] [CrossRef]
- Uyeki, T.M. Influenza. Ann. Intern. Med. 2017, 167, ITC33–ITC48. [Google Scholar] [CrossRef]
- de Graaf, M.; Fouchier, R.A. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J. 2014, 33, 823–841. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.J.; Wang, G.L.; Anderson, B.D.; Bi, Z.Q.; Lu, B.; Wang, X.J.; Wang, C.X.; Chen, S.H.; Qian, Y.H.; Song, S.X.; et al. Evidence for Cross-species Influenza A Virus Transmission Within Swine Farms, China: A One Health, Prospective Cohort Study. Clin. Infect. Dis. 2018, 66, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G. Respiratory viral threats. Curr. Opin. Infect. Dis. 2006, 19, 169–178. [Google Scholar] [CrossRef]
- Hurt, A.C. The epidemiology and spread of drug resistant human influenza viruses. Curr. Opin. Virol. 2014, 8, 22–29. [Google Scholar] [CrossRef]
- Alves Galvão, M.G.; Rocha Crispino Santos, M.A.; Alves da Cunha, A.J. Amantadine and rimantadine for influenza A in children and the elderly. Cochrane Database Syst. Rev. 2014, 11, CD002745. [Google Scholar] [CrossRef] [PubMed]
- Kamali, A.; Holodniy, M. Influenza treatment and prophylaxis with neuraminidase inhibitors: A review. Infect. Drug. Resist. 2013, 6, 187–198. [Google Scholar] [CrossRef]
- Shie, J.J.; Fang, J.M. Development of effective anti-influenza drugs: Congeners and conjugates—A review. J. Biomed. Sci. 2019, 26, 84. [Google Scholar] [CrossRef]
- Agamennone, M.; Fantacuzzi, M.; Vivenzio, G.; Scala, M.C.; Campiglia, P.; Superti, F.; Sala, M. Antiviral Peptides as Anti-Influenza Agents. Int. J. Mol. Sci. 2022, 23, 11433. [Google Scholar] [CrossRef]
- Air, G.M.; Brouillette, W.J. Influenza virus antiviral targets. In Antiviral Research: Strategies in Antiviral Drug Discovery; LaFemina, R.L., Ed.; ASM Press: Washington, DC, USA, 2009; pp. 187–207. [Google Scholar] [CrossRef]
- Berkhout, B.; Sanders, R.W. Molecular strategies to design an escape-proof antiviral therapy. Antivir. Res. 2011, 92, 7–14. [Google Scholar] [CrossRef]
- Krol, E.; Rychowska, M.; Szewczyk, B. Antivirals—Current trends in fighting influenza. Acta Biochim. Pol. 2014, 61, 495–504. [Google Scholar] [CrossRef]
- Wilson, J.C.; von Itzstein, M. Recent strategies in the search for new anti-influenza therapies. Curr. Drug Targets. 2003, 4, 389–408. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef] [PubMed]
- García, J.; Sovero, M.; Torres, A.L.; Gomez, J.; Douce, R.; Barrantes, M.; Sanchez, F.; Jimenez, M.; Comach, G.; de Rivera, I.; et al. Antiviral resistance in influenza viruses circulating in Central and South America based on the detection of established genetic markers. Influenza Other Respir Viruses 2009, 3, 69–74. [Google Scholar] [CrossRef]
- Meijer, A.; Lackenby, A.; Hungnes, O.; Lina, B.; van-der-Werf, S.; Schweiger, B.; Opp, M.; Paget, J.; van-de-Kassteele, J.; Hay, A.; et al. Oseltamivir-resistant influenza virus A (H1N1), Europe, 2007–2008 season. Emerg. Infect. Dis. 2009, 15, 552–560. [Google Scholar] [CrossRef]
- Beigel, J.H.; Farrar, J.; Han, A.M.; Hayden, F.G.; Hyer, R.; de Jong, M.D.; Lochindarat, S.; Nguyen, T.K.; Nguyen, T.H.; Tran, T.H.; et al. Avian influenza A (H5N1) infection in humans. N. Engl. J. Med. 2005, 353, 1374–1385. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, S.; Song, Z.; Wang, W.; Hao, P.; Li, J.; Zhang, X.; Yen, H.-L.; Shi, B.; Li, T.; et al. Association between adverse clinical outcome in human disease caused by novel influenza A H7N9 virus and sustained viral shedding and emergence of antiviral resistance. Lancet 2013, 381, 2273–2279. [Google Scholar] [CrossRef]
- Hurt, A.C.; Selleck, P.; Komadina, N.; Shaw, R.; Brown, L.; Barr, I.G. Susceptibility of highly pathogenic A(H5N1) avian influenza viruses to the neuraminidase inhibitors and adamantanes. Antivir. Res. 2007, 73, 228–231. [Google Scholar] [CrossRef]
- Skog, E.; Nykvist, M.; Naguib, M.M.; Wille, M.; Bröjer, C.; Agarwal, V.; Ellström, P.; Westman, G.; Lundkvist, Å.; Järhult, J.D. An oseltamivir-resistant avian H1N1 influenza A virus can transmit from mallards to chickens similarly to a wild-type strain: Implications for the risk of resistance transmission to humans. J. Gen. Virol. 2023, 104, 001835. [Google Scholar] [CrossRef]
- Ammendolia, M.G.; Agamennone, M.; Pietrantoni, A.; Lannutti, F.; Siciliano, R.A.; De Giulio, B.; Amici, C.; Superti, F. Bovine lactoferrin-derived peptides as novel broad-spectrum inhibitors of influenza virus. Pathog. Glob. Health 2012, 106, 12–19. [Google Scholar] [CrossRef]
- Scala, M.C.; Sala, M.; Pietrantoni, A.; Spensiero, A.; Di Micco, S.; Agamennone, M.; Bertamino, A.; Novellino, E.; Bifulco, G.; Gomez-Monterrey, I.M.; et al. Lactoferrin-derived Peptides Active towards Influenza: Identification of Three Potent Tetrapeptide Inhibitors. Sci. Rep. 2017, 7, 10593. [Google Scholar] [CrossRef]
- Haviv, F.; Fitzpatrick, T.D.; Swenson, R.E.; Nichols, C.J.; Mort, N.A.; Bush, E.N.; Diaz, G.; Bammert, G.; Nguyen, A.; Rhutasel, N.S.; et al. Effect of N-methyl substitution of the peptidebonds in luteinizing hormone-releasing hormone agonists. J. Med. Chem. 1993, 36, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Cody, W.L.; He, J.X.; Reily, M.D.; Haleen, S.J.; Walker, D.M.; Reyner, E.L.; Stewart, B.H.; Doherty, A.M. Design of a potent combined pseudopeptide endothelin-A/endothelin-B receptor antagonist, Ac-DBhg(16)-Leu-Asp-Ile-[NMe]Ile-Trp(21) (PD 156252): Examination of itspharmacokinetic and spectralproperties. J. Med. Chem. 1997, 40, 2228–2240. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.C.; Espina, J.R.; Jackson, S.A.; Stouten, P.F.; Duke, J.L.; Mousa, S.A.; DeGrado, W.F. Type II to type I beta-turn swap changes specificity for integrins. J. Am. Chem. Soc. 1996, 118, 293–294. [Google Scholar] [CrossRef]
- Rajeswaran, W.G.; Hocart, S.J.; Murphy, W.A.; Taylor, J.E.; Coy, D.H. Highly potent and subtype selective ligands derived by N-methyl scan of a somatostatin antagonist. J. Med. Chem. 2001, 44, 1305–1311. [Google Scholar] [CrossRef]
- Laufer, R.; Wormser, U.; Friedman, Z.Y.; Gilon, C.; Chorev, M.; Selinger, Z. Neurokinin-B is a preferred agonist for a neuronal substance- P receptor and its action is antagonized by enkephalin. Proc. Natl. Acad. Sci. USA 1985, 82, 7444–7448. [Google Scholar] [CrossRef] [PubMed]
- Samanen, J.; Ali, F.; Romoff, T.; Calvo, R.; Sorenson, E.; Vasko, J.; Storer, B.; Berry, D.; Bennett, D.; Strohsacker, M.; et al. Development of a small RGD peptide fibrinogen receptor antagonist with potent antiaggregatory activity in vitro. J. Med. Chem. 1991, 34, 3114–3125. [Google Scholar] [CrossRef]
- Dechantsreiter, M.A.; Planker, E.; Mathä, B.; Lohof, E.; Hölzemann, G.; Jonczyk, A.; Goodman, S.L.; Kessler, H. N-methylated cyclic RGD peptides as highly active and selective αvβ3 integrin antagonists. J. Med. Chem. 1999, 42, 3033–3040. [Google Scholar] [CrossRef] [PubMed]
- Barker, P.L.; Bullens, S.; Bunting, S.; Burdick, D.J.; Chan, K.S.; Deisher, T.; Eigenbrot, C.; Gadek, T.R.; Gantzos, R.; Lipari, M.T.; et al. Cyclic RGD peptide analogs as antiplatelet antithrombotics. J. Med. Chem. 1992, 35, 2040–2048. [Google Scholar] [CrossRef]
- Romero, F.; Espliego, F.; Pérez Baz, J.; García de Quesada, T.; Gravalos, D.; De la Calle, F.; Fernández-Puentes, J.L. Thiocoraline, a new depsipeptide with antitumor activity produced by a marine micromonospora. I. Taxonomy, fermentation, isolation, and biological activities. J. Antibiot. 1997, 50, 734–737. [Google Scholar] [CrossRef]
- Soto, P.; Griffin, M.A.; Shea, J.E. New insights into the mechanism of alzheimer amyloid-β fibrillogenesis inhibition by N-methylated peptides. Biophys. J. 2007, 93, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Wipf, P. Synthetic studies of biologically active marine cyclopeptides. Chem. Rev. 1995, 95, 2115–2134. [Google Scholar] [CrossRef]
- Zuckermann, R.N.; Kerr, J.M.; Kent, S.B.H.; Moos, W.H. Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J. Am. Chem. Soc. 1992, 114, 10646–10647. [Google Scholar] [CrossRef]
- Miller, S.M.; Simon, R.J.; Ng, S.; Zuckermann, R.N.; Kerr, J.M.; Moos, W.H. Comparison of the proteolytic susceptibilities of homologous L-amino acid, D-amino acid, and N-substituted glycine peptide and peptoid oligomers. Drug Dev. Res. 1995, 35, 20–32. [Google Scholar] [CrossRef]
- Patch, J.A.; Kirshenbaum, K.; Seurynck, S.L.; Zuckermann, R.N.; Barron, A.E. Versatile Oligo(N-Subsitituted) Glycines: The Many Roles of Peptoids in Drug Discovery. In Pseudopeptides in Drug Discovery; Nielsen, P.E., Ed.; Wiley-VCH Verlag, GmbH & Co. KgaA: Weinheim, Germany, 2004; pp. 1–31. [Google Scholar]
- Zuckermann, R.N.; Kodadek, T. Peptoids as potential therapeutics. Curr. Opin. Mol. Therap. 2009, 11, 299–307. [Google Scholar]
- Miller, S.M.; Simon, R.J.; Ng, S.; Zuckermann, R.N.; Kerr, J.M.; Moos, W.H. Proteolytic studies of homologous peptide and N-substituted glycine peptoid oligomers. Bioorg. Med. Chem. Lett. 1994, 4, 2657–2662. [Google Scholar] [CrossRef]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C.K. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367–9371. [Google Scholar] [CrossRef]
- Wisén, S.; Androsavich, J.; Evans, C.G.; Chang, L.; Gestwicki, J.E. Chemical modulators of heat shock protein 70 (Hsp70) by sequential, microwave-accelerated reactions on solid phase. Bioorg. Med. Chem. Lett. 2008, 18, 60–65. [Google Scholar] [CrossRef]
- Zhang, H.B.; Chi, Y.S.; Huang, W.L.; Ni, S.J. Total synthesis of human urotension by microwave-assisted solid phase method. Chin. Chem. Lett. 2007, 18, 902–904. [Google Scholar] [CrossRef]
- Gobbo, M.; Benincasa, M.; Bertoloni, G.; Biondi, B.; Dosselli, R.; Papini, E.; Reddi, E.; Rocchi, R.; Tavano, R.; Gennaro, R. Substitution of the Arginine/Leucine Residues in Apidaecin Ib with Peptoid Residues: Effect on Antimicrobial Activity, Cellular Uptake, and Proteolytic Degradation. J. Med. Chem. 2009, 52, 5197–5206. [Google Scholar] [CrossRef] [PubMed]
- Clapperton, A.M.; Babi, J.; Tran, H. A Field Guide to Optimizing Peptoid Synthesis. ACS Polym. Au 2022, 2, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Navratilova, I.; Hopkins, A.L. Fragment screening by surface plasmon resonance. ACS Med. Chem. Lett. 2010, 1, 44–48. [Google Scholar] [CrossRef]
- Scala, M.C.; Agamennone, M.; Pietrantoni, A.; Di Sarno, V.; Bertamino, A.; Superti, F.; Campiglia, P.; Sala, M. Discovery of a novel tetrapeptide against influenza a virus: Rational design, synthesis, bioactivity evaluation and computational studies. Pharmaceuticals 2021, 14, 959. [Google Scholar] [CrossRef] [PubMed]
- Monolith NT Protein Labeling Kit RED-NHS. User Manual. Available online: https://manualzz.com/doc/7334293/monolithnt%E2%84%A2-protein-labeling-kit-red-nhs (accessed on 29 October 2020).
- Gaush, C.R.; Smith, T.F. Replication and plaque assay of influenza virus in an established line of canine kidney cells. Appl. Microbiol. 1968, 16, 588–594. [Google Scholar] [CrossRef]
- Rimmelzwaan, G.F.; Baars, M.; Claas, E.C.; Osterhaus, A.D. Comparison of RNA hybridization, hemagglutination assay, titration of infectious virus and immunofluorescence as methods for monitoring influenza virus replication in vitro. J. Virol. Methods 1998, 74, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Superti, F.; Marchetti, M.; Rapetti Mogol, G. Inactivation of influenza virus by a blend of essential plant oil vapour and aerosol. J. Biol. Regul. Homeost. Agents. 2021, 35, 1667–1675. [Google Scholar] [CrossRef]
- Marchetti, M.; De Berardis, B.; Bigioni, I.; Mariano, A.; Superti, F.; Scotto d’Abusco, A. In Vitro Antiviral and Anti-Inflammatory Activities of N-Acetylglucosamine: Development of an Alternative and Safe Approach to Fight Viral Respiratory Infections. Int. J. Mol. Sci. 2023, 24, 5129. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pep | Sequence | MST KD (µM) | SPR KD (µM) |
|---|---|---|---|
| 1 a | SLDC | 10.4 ± 0.23 | 7.12 ± 0.26 |
| 2 a | SKHS | 7.26 ± 0.06 | 4.53 ± 0.08 |
| 3 | (N-Me)SLDC | 24.8 ± 0.71 | 7.60 ± 2.69 |
| 4 | S(N-Me)LDC | 0.0325 ± 0.002 | 0.58 ± 0.34 |
| 5 | SL(N-Me)DC | 8.8 ± 0.23 | NPD b |
| 6 | SLD(N-Me)C | 0.142 ± 0.03 | 3.80 ± 1.06 |
| 7 | (N-Me)SKHS | 0.336 ± 0.02 | 4.19 ± 0.86 |
| 8 | S(N-Me)KHS | 7.93 ± 0.73 | 8.33 ± 1.73 |
| 9 | SK(N-Me)HS | 0.0528 ± 0.005 | 1.26 ± 0.36 |
| 10 | SKH(N-Me)S | 3.04 ± 0.85 | NPD b |
| 11 | NhSLDC c | 0.0672 ± 0.002 | 6.55 ± 1.52 |
| 12 | SNLDC c | 0.701 ± 0.03 | 3.81 ± 2.13 |
| 13 | SLNDC c | 1.34 ± 0.02 | NPD b |
| 14 | SLDNhC c | 0.0459 ± 0.01 | NPD b |
| 15 | NhSKHS c | 7.4 ± 0.35 | 2.09 ± 2.41 |
| 16 | SNKHS c | 0.0391 ± 0.003 | 1.70 ± 0.13 |
| 17 | SKNHS c | 3.07 ± 0.79 | NPD b |
| 18 | SKHNhS c | 0.0491 ± 0.001 | 0.43 ± 0.21 |
| Pep | Sequence | IC50 b (nM) |
|---|---|---|
| A/Parma/24/09 H1N1 | ||
| 1 a | SLDC | 6 ± 3 |
| 2 a | SKHS | 1.5 ± 0.7 |
| 4 | S(N-Me)LDC | 4.6 ± 2.3 |
| 9 | SK(N-Me)HS | 3.2 ± 1.6 |
| 16 | SNKHS | 1.2 ± 0.6 |
| 18 | SKHNhS | 3.3 ±1.7 |
| Pep | Sequence | EC50 a (µM) | SI b |
|---|---|---|---|
| 1 c | SLDC | 4.6 ± 0.05 × 10−6 | >5.4 × 106 |
| 2 c | SKHS | 4.8 ± 0.12 × 10−8 | >5.2 × 108 |
| 4 | S(N-Me)LDC | 8.5 ± 0.04 × 10−5 | >2.9 × 105 |
| 9 | SK(N-Me)HS | 1.425 ± 0.02 × 10−4 | >1.78 × 103 |
| 16 | SNKHS | 4.7 ± 0.02 × 10−4 | >5.3 × 104 |
| 18 | SKHNhS | 5 ± 0.04 × 10−5 | >5 × 105 |
| Pep | Docking Score Kcal/mol |
|---|---|
| 1 | −6.760 a |
| 2 | −6.520 a |
| 4 | −5.881 |
| 9 | −5.972 |
| 16 | −6.328 |
| 18 | −6.788 |
| 6′-sialyl-N-acetyllactosamine | −6597 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scala, M.C.; Marchetti, M.; Superti, F.; Agamennone, M.; Campiglia, P.; Sala, M. Rational Design of Novel Peptidomimetics against Influenza A Virus: Biological and Computational Studies. Int. J. Mol. Sci. 2023, 24, 14268. https://doi.org/10.3390/ijms241814268
Scala MC, Marchetti M, Superti F, Agamennone M, Campiglia P, Sala M. Rational Design of Novel Peptidomimetics against Influenza A Virus: Biological and Computational Studies. International Journal of Molecular Sciences. 2023; 24(18):14268. https://doi.org/10.3390/ijms241814268
Chicago/Turabian StyleScala, Maria Carmina, Magda Marchetti, Fabiana Superti, Mariangela Agamennone, Pietro Campiglia, and Marina Sala. 2023. "Rational Design of Novel Peptidomimetics against Influenza A Virus: Biological and Computational Studies" International Journal of Molecular Sciences 24, no. 18: 14268. https://doi.org/10.3390/ijms241814268
APA StyleScala, M. C., Marchetti, M., Superti, F., Agamennone, M., Campiglia, P., & Sala, M. (2023). Rational Design of Novel Peptidomimetics against Influenza A Virus: Biological and Computational Studies. International Journal of Molecular Sciences, 24(18), 14268. https://doi.org/10.3390/ijms241814268