
Adipose Tissue, Non-Communicable Diseases, and Physical Exercise: An Imperfect Triangle



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Adipose Tissue
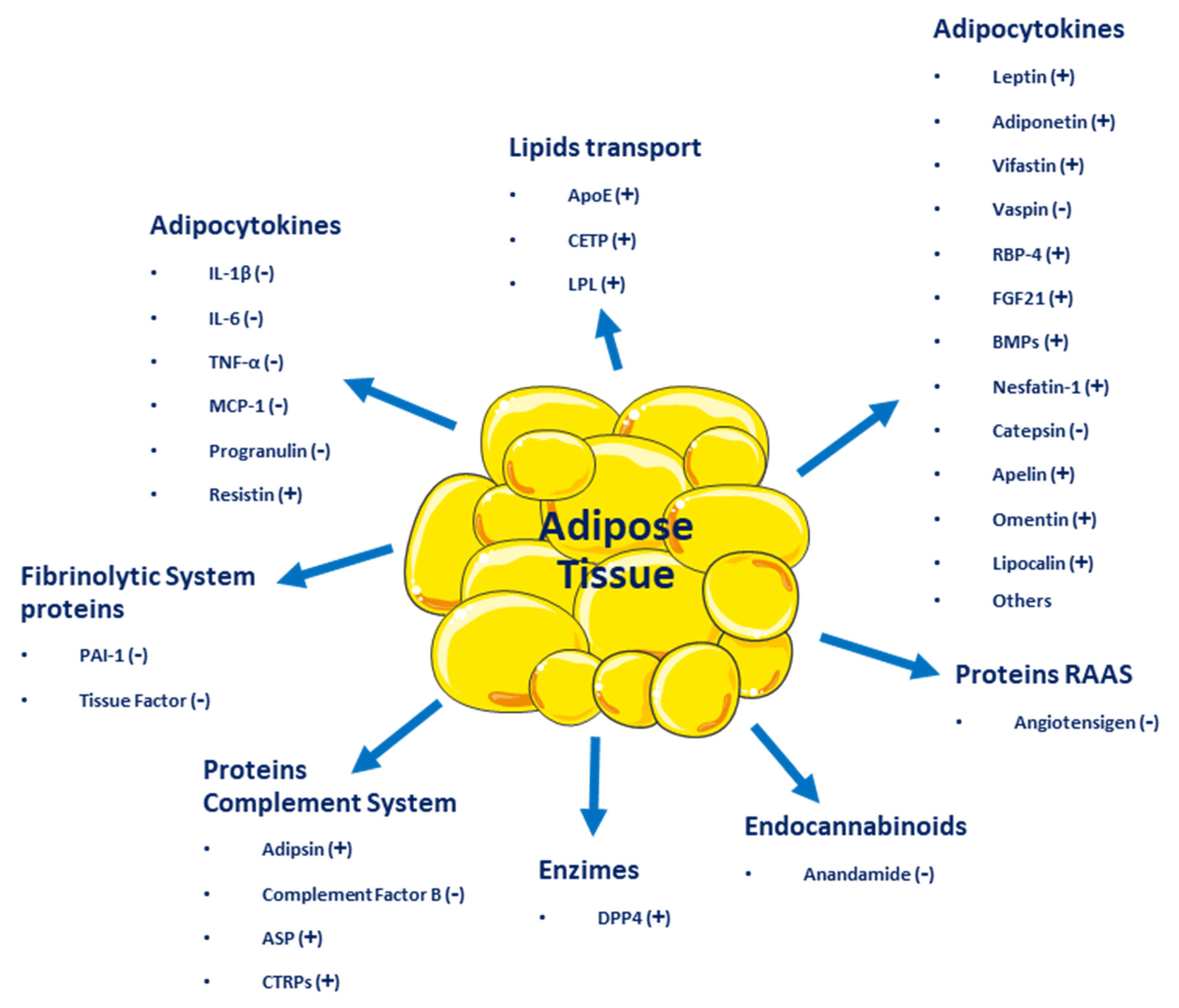
2. Adipose Tissue as an Endocrine Organ
3. Definition and Incidence of Overweight and Obesity
4. Causes or Mechanisms of Obesity
4.1. Genetic Factors
4.2. Fat Cells
4.3. Dysregulation of Energy Balance
4.4. Metabolic and Physiological Effects
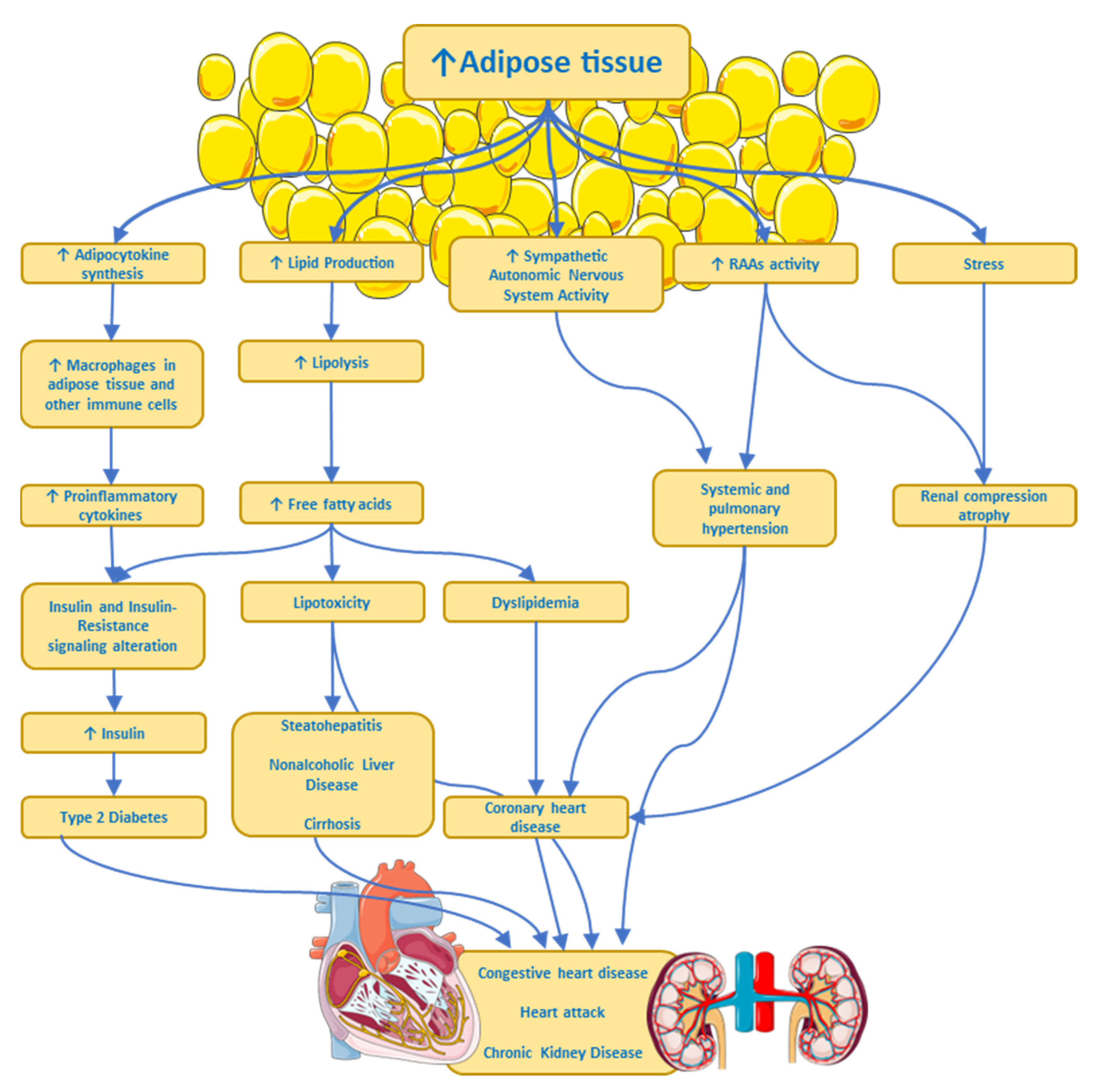
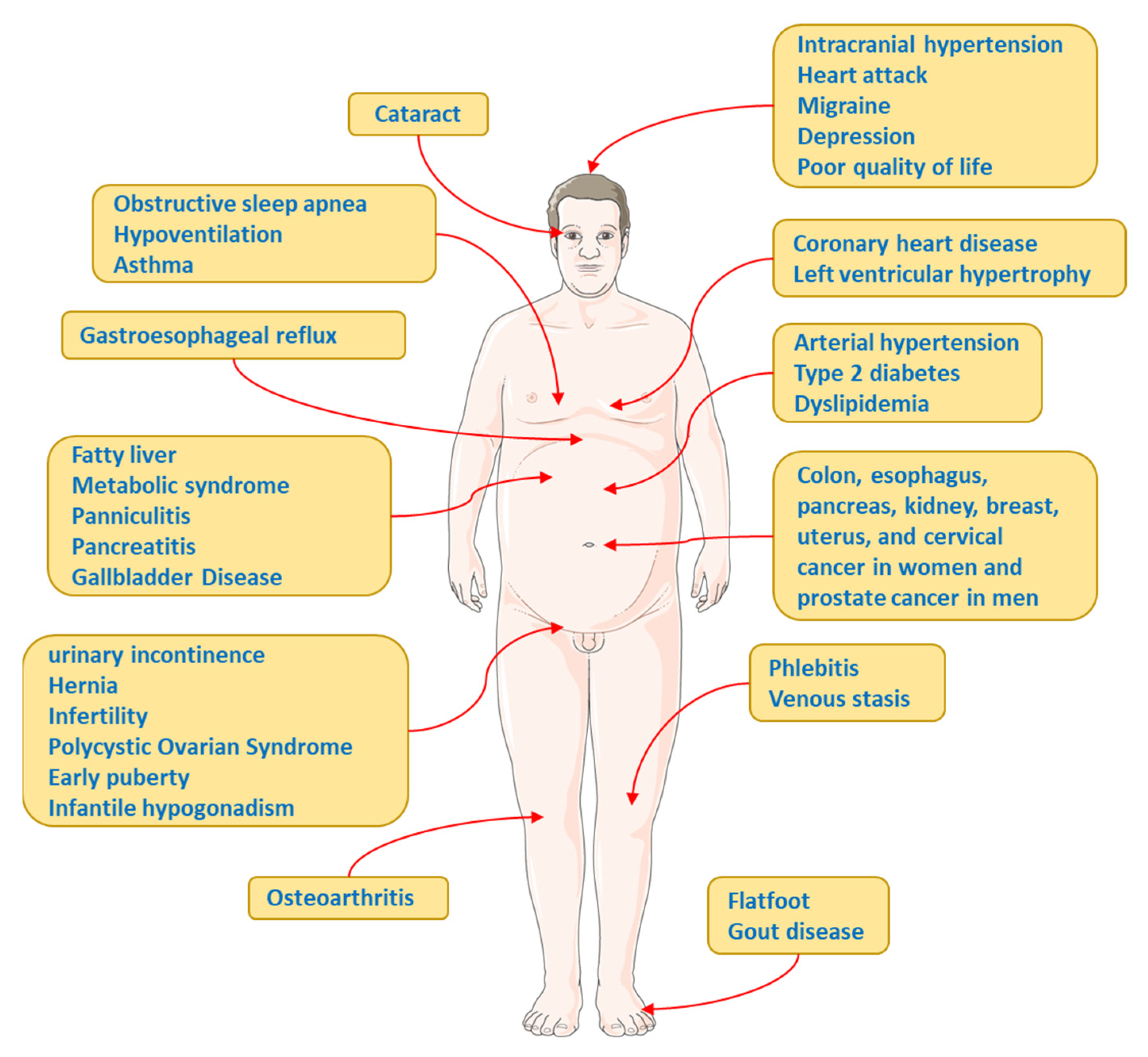
5. Complications and Comorbidities Associated with Obesity
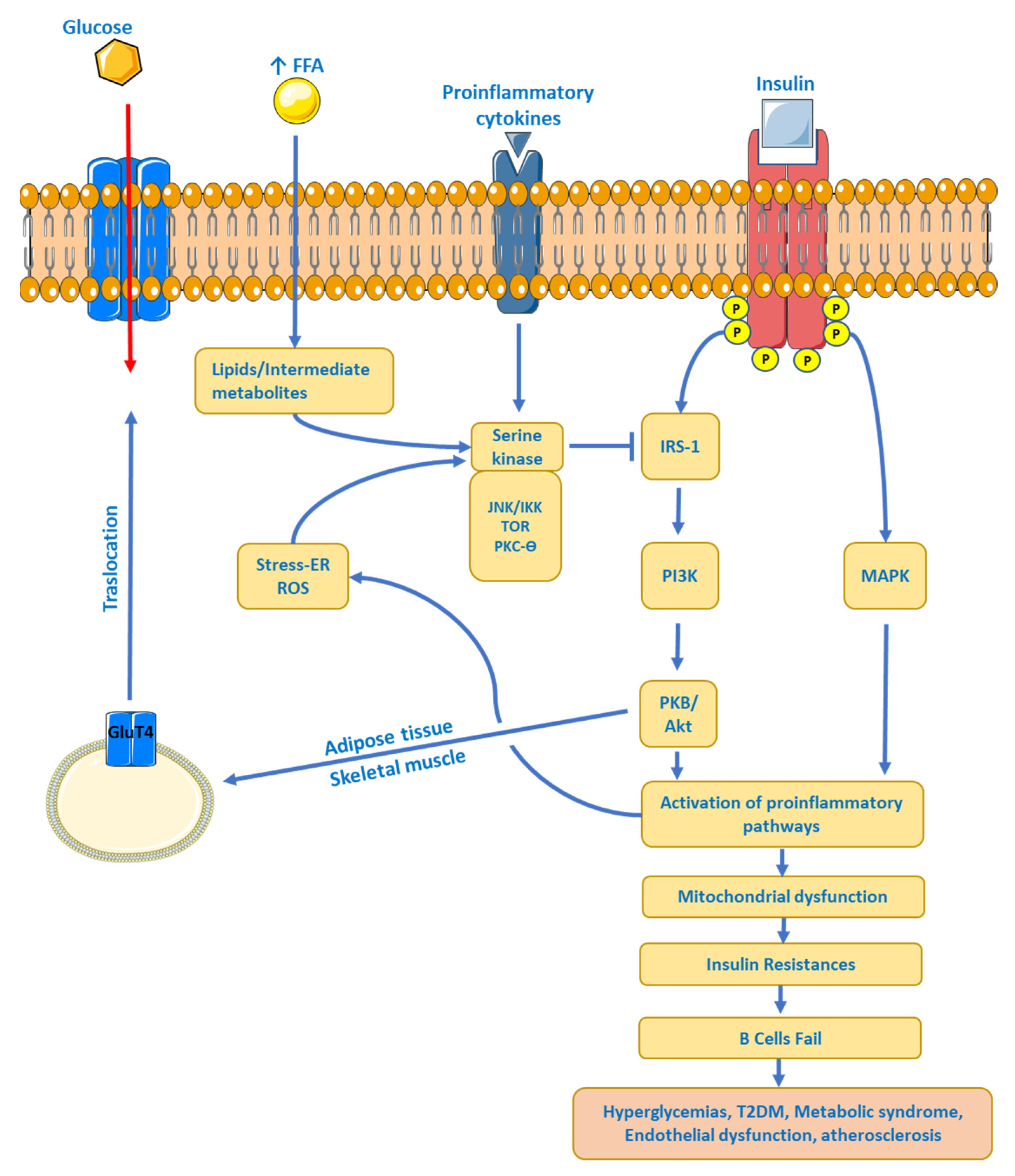
5.1. Insulin Resistance (IR)
5.2. Type 2 Diabetes
5.3. Dyslipidaemia
5.4. Hypertension
5.5. Other Complications and Comorbidities
6. Management of the Overweight-Obesity Patient
6.1. Lifestyle Modification
6.2. Pharmacotherapy: Who Are Candidates to Receive Anti-Obesity Drugs?
6.3. Orlistat
6.4. Liraglutide
6.5. Phentermine
6.6. Naltrexone/Bupropion
6.7. Phentermine/Topiramate
6.8. Diethylpropion
7. Physical Exercise as Treatment
8. Hypocaloric Diets as the Most Effective Approach
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Booth, A.; Magnuson, A.; Fouts, J.; Foster, M.T. Adipose tissue: An endocrine organ playing a role in metabolic regulation. Horm. Mol. Biol. Clin. Investig. 2016, 26, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, R.E.; Klaman, L.D. Adipose tissue as an active endocrine organ: Recent advances. Curr. Opin. Pharmacol. 2005, 5, 122–128. [Google Scholar] [CrossRef]
- Zwick, R.K.; Guerrero-Juarez, C.F.; Horsley, V.; Plikus, M.V. Anatomical, Physiological, and Functional Diversity of Adipose Tissue. Cell Metab. 2018, 27, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; García-Cañaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866.e26. [Google Scholar] [CrossRef]
- Cinti, S. The adipose organ at a glance. Dis. Model Mech. 2012, 5, 588–594. [Google Scholar] [CrossRef]
- Wang, F.; Vihma, V.; Soronen, J.; Turpeinen, U.; Hämäläinen, E.; Savolainen-Peltonen, H.; Mikkola, T.S.; Naukkarinen, J.; Pietiläinen, K.H.; Jauhiainen, M.; et al. 17β estradiol and estradiol fatty acyl esters and estrogen-converting enzyme expression in adipose tissue in obese men and women. J. Clin. Endochrinol. Metab. 2013, 98, 4923–4931. [Google Scholar] [CrossRef]
- Kajimura, S.; Spiegelman, B.M.; Seale, P. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 2015, 22, 546–559. [Google Scholar] [CrossRef]
- Vega-Robledo, G.; Rico-Rosillo, M.G. Adipose tissue: Immune function and alterations caused by obesity. Rev. Alerg. Mex. 2016, 66, 340–353. [Google Scholar] [CrossRef]
- Svensson, K.J.; Long, J.Z.; Jedrychowski, M.P.; Cohen, P.; Lo, J.C.; Serag, S.; Kir, S.; Shinoda, K.; Tartaglia, J.A.; Rao, R.R.; et al. A secreted Slit2 fragment regulates adipose tissue thermogenesis and metabolic function. Cell Metab. 2016, 23, 454–466. [Google Scholar] [CrossRef]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef]
- O’Neill, S.; O’Driscoll, L. Metabolic syndrome: A closer look at the growing epidemic and its associated pathologies. Obes. Rev. 2015, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C. Obesity, Immunity, and Cancer. N. Engl. J. Med. 2021, 384, 1160–1162. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef]
- Szablewski, L. Introductory Chapter: Adipose. In Tissue; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Atzmon, G.; Yang, X.M.; Muzumdar, R.; Ma, X.H.; Gabriely, I.; Barzilai, N. Differential gene expression between visceral and subcutaneous fat depots. Horm. Metab. Res. 2002, 34, 622–628. [Google Scholar] [CrossRef]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef]
- Choi, H.M.; Doss, H.M.; Kim, K.S. Multifaceted Physiological Roles of Adiponectin in Inflammation and Diseases. Int. J. Mol. Sci. 2020, 21, 1219. [Google Scholar] [CrossRef]
- Aida-Souki, A.-R.; Prieto-Fuenmayor, C.; Cano-Ponce, C. Aspectos Básicos en Obesidad; Ediciones Universidad Simón Bolívar: Barranquilla, Colombia, 2018; pp. 1–44. [Google Scholar]
- Hida, K.; Wada, J.; Eguchi, J.; Zhang, H.; Baba, M.; Seida, A.; Hashimoto, I.; Okada, T.; Yasuhara, A.; Nakatsuka, A.; et al. Visceral adipose tissue-derived serine protease inhibitor: A unique insulin-sensitizing adipocytokine in obesity. Proc. Natl. Acad. Sci. USA 2005, 102, 10610–10615. [Google Scholar] [CrossRef]
- Ulbricht, D.; Pippel, J.; Schultz, S.; Meier, R.; Sträter, N.; Heiker, J.T. A unique serpin P1′ glutamate and a conserved β-sheet C arginine are key residues for activity, protease recognition and stability of serpinA12 (vaspin). Biochem. J. 2015, 470, 357–367. [Google Scholar] [CrossRef]
- Pilarski, Ł.; Pelczyńska, M.; Koperska, A.; Seraszek-Jaros, A.; Szulińska, M.; Bogdański, P. Association of Serum Vaspin Concentration with Metabolic Disorders in Obese Individuals. Biomolecules 2023, 13, 508. [Google Scholar] [CrossRef]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; Matsuki, Y.; Murakami, M.; Ichisaka, T.; Murakami, H.; et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005, 307, 426–430. [Google Scholar] [CrossRef] [PubMed]
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar] [CrossRef] [PubMed]
- WHO. Obesity: Preventing and Managing the Global Epidemic. Report of a WHO Consultation; World Health Organization Technical Report Series 894; WHO Consultation on Obesity: Geneva, Switzerland, 2000; 253p. [Google Scholar]
- Poirier, P.; Giles, T.D.; Bray, G.A.; Hong, Y.; Stern, J.S.; Pi-Sunyer, F.X.; Eckel, R.H. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 968–976. [Google Scholar] [CrossRef]
- Apovian, C.M. Obesity: Definition, comorbidities, causes, and burden. Am. J. Manag. Care 2016, 22, s176–s185. [Google Scholar]
- Bray, G.A.; Kim, K.K.; Wilding, J.P.H.; Federation, W.O. Obesity: A chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes. Rev. 2017, 18, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Swinburn, B.A.; Sacks, G.; Hall, K.D.; McPherson, K.; Finegood, D.T.; Moodie, M.L.; Gortmaker, S.L. The global obesity pandemic: Shaped by global drivers and local environments. Lancet 2011, 378, 804–814. [Google Scholar] [CrossRef]
- Heymsfield, S.B.; Wadden, T.A. Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 2017, 376, 1492. [Google Scholar] [CrossRef]
- Wu, Y.; Duan, H.; Tian, X.; Xu, C.; Wang, W.; Jiang, W.; Pang, Z.; Zhang, D.; Tan, Q. Genetics of Obesity Traits: A Bivariate Genome-Wide Association Analysis. Front. Genet. 2018, 9, 179. [Google Scholar] [CrossRef]
- Shungin, D.; Winkler, T.W.; Croteau-Chonka, D.C.; Ferreira, T.; Locke, A.E.; Mägi, R.; Strawbridge, R.J.; Pers, T.H.; Fischer, K.; Justice, A.E.; et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015, 518, 187–196. [Google Scholar] [CrossRef]
- Thaker, V.V. Genetic and Epigenetic Causes of Obesity. Adolesc. Med. State Art Rev. 2017, 28, 379–405. [Google Scholar]
- Huvenne, H.; Dubern, B.; Clément, K.; Poitou, C. Rare Genetic Forms of Obesity: Clinical Approach and Current Treatments in 2016. Obes. Facts 2016, 9, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Koochakpour, G.; Esfandiar, Z.; Hosseini-Esfahani, F.; Mirmiran, P.; Daneshpour, M.S.; Sedaghati-Khayat, B.; Azizi, F. Evaluating the interaction of common FTO genetic variants, added sugar, and trans-fatty acid intakes in altering obesity phenotypes. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 474–480. [Google Scholar] [CrossRef]
- Gupta, N.; Jain, V. Prader Willi Syndrome—A Common Epigenetic Cause of Syndromic Obesity. Indian J. Pediatr. 2017, 84, 809–810. [Google Scholar] [CrossRef] [PubMed]
- Cena, H.; Chiovato, L.; Nappi, R.E. Obesity, Polycystic Ovary Syndrome, and Infertility: A New Avenue for GLP-1 Receptor Agonists. J. Clin. Endocrinol. Metab. 2020, 105, e2695–e2709. [Google Scholar] [CrossRef]
- D’Angelo, C.S.; Koiffmann, C.P. Copy number variants in obesity-related syndromes: Review and perspectives on novel molecular approaches. J. Obes. 2012, 2012, 845480. [Google Scholar] [CrossRef]
- Lin, X.; Li, H. Obesity: Epidemiology, Pathophysiology, and Therapeutics. Front. Endocrinol. 2021, 12, 706978. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Knight, R.; Leibel, R.L. The gut microbiota in human energy homeostasis and obesity. Trends Endocrinol. Metab. 2015, 26, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Wernstedt Asterholm, I.; Tao, C.; Morley, T.S.; Wang, Q.A.; Delgado-Lopez, F.; Wang, Z.V.; Scherer, P.E. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014, 20, 103–118. [Google Scholar] [CrossRef]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef]
- Pigeyre, M.; Yazdi, F.T.; Kaur, Y.; Meyre, D. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin. Sci. 2016, 130, 943–986. [Google Scholar] [CrossRef]
- van der Klaauw, A.A.; Farooqi, I.S. The hunger genes: Pathways to obesity. Cell 2015, 161, 119–132. [Google Scholar] [CrossRef] [PubMed]
- MacLean, P.S.; Higgins, J.A.; Giles, E.D.; Sherk, V.D.; Jackman, M.R. The role for adipose tissue in weight regain after weight loss. Obes. Rev. 2015, 16 (Suppl. S1), 45–54. [Google Scholar] [CrossRef]
- Ochner, C.N.; Tsai, A.G.; Kushner, R.F.; Wadden, T.A. Treating obesity seriously: When recommendations for lifestyle change confront biological adaptations. Lancet Diabetes Endocrinol. 2015, 3, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Thomou, T.; Zhu, Y.; Karagiannides, I.; Pothoulakis, C.; Jensen, M.D.; Kirkland, J.L. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. 2013, 17, 644–656. [Google Scholar] [CrossRef]
- Heymsfield, S.B.; Hu, H.H.; Shen, W.; Carmichael, O. Emerging Technologies and their Applications in Lipid Compartment Measurement. Trends Endocrinol. Metab. 2015, 26, 688–698. [Google Scholar] [CrossRef]
- McCullough, A.J. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin. Liver Dis. 2004, 8, 521–533. [Google Scholar] [CrossRef]
- Kaur, J. A comprehensive review on metabolic syndrome. Cardiol. Res. Pract. 2014, 2014, 943162. [Google Scholar] [CrossRef]
- Schéle, E.; Grahnemo, L.; Anesten, F.; Hallén, A.; Bäckhed, F.; Jansson, J.O. The gut microbiota reduces leptin sensitivity and the expression of the obesity-suppressing neuropeptides proglucagon (Gcg) and brain-derived neurotrophic factor (Bdnf) in the central nervous system. Endocrinology 2013, 154, 3643–3651. [Google Scholar] [CrossRef]
- Jiang, L.; Su, H.; Wu, X.; Shen, H.; Kim, M.H.; Li, Y.; Myers, M.G.; Owyang, C.; Rui, L. Leptin receptor-expressing neuron Sh2b1 supports sympathetic nervous system and protects against obesity and metabolic disease. Nat. Commun. 2020, 11, 1517. [Google Scholar] [CrossRef]
- Ochoa-Repáraz, J.; Kasper, L.H. The Second Brain: Is the Gut Microbiota a Link Between Obesity and Central Nervous System Disorders? Curr. Obes. Rep. 2016, 5, 51–64. [Google Scholar] [CrossRef]
- Di Vincenzo, F.; Del Gaudio, A.; Petito, V.; Lopetuso, L.R.; Scaldaferri, F. Gut microbiota, intestinal permeability, and systemic inflammation: A narrative review. Intern. Emerg. Med. 2023. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Rowe, D.C.; Barnes, B.J.; Caffrey, D.R.; Visintin, A.; Latz, E.; Monks, B.; Pitha, P.M.; Golenbock, D.T. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 2003, 198, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Munford, R.S. Murine responses to endotoxin: Another dirty little secret? J. Infect. Dis. 2010, 201, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; da Silva, A.A.; do Carmo, J.M.; Dubinion, J.; Hamza, S.; Munusamy, S.; Smith, G.; Stec, D.E. Obesity-induced hypertension: Role of sympathetic nervous system, leptin, and melanocortins. J. Biol. Chem. 2010, 285, 17271–17276. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, G.; Lewington, S.; Sherliker, P.; Clarke, R.; Emberson, J.; Halsey, J.; Qizilbash, N.; Collins, R.; Peto, R.; Collaboration, P.S. Body-mass index and cause-specific mortality in 900 000 adults: Collaborative analyses of 57 prospective studies. Lancet 2009, 373, 1083–1096. [Google Scholar] [CrossRef]
- Ortiz-Martínez, M.; González-González, M.; Martagón, A.J.; Hlavinka, V.; Willson, R.C.; Rito-Palomares, M. Recent Developments in Biomarkers for Diagnosis and Screening of Type 2 Diabetes Mellitus. Curr. Diabetes Rep. 2022, 22, 95–115. [Google Scholar] [CrossRef]
- Upadhyay, J.; Farr, O.; Perakakis, N.; Ghaly, W.; Mantzoros, C. Obesity as a Disease. Med. Clin. N. Am. 2018, 102, 13–33. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, T.; Zhang, S.; Zhou, L. Associations of Different Adipose Tissue Depots with Insulin Resistance: A Systematic Review and Meta-analysis of Observational Studies. Sci. Rep. 2015, 5, 18495. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef]
- Wondmkun, Y.T. Obesity, Insulin Resistance, and Type 2 Diabetes: Associations and Therapeutic Implications. Diabetes Metab. Syndr. Obes. 2020, 13, 3611–3616. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Ota, T. Obesity-induced inflammation and insulin resistance. Front. Endocrinol. 2014, 5, 204. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Krssak, M.; Brehm, A.; Bernroider, E.; Anderwald, C.; Nowotny, P.; Dalla Man, C.; Cobelli, C.; Cline, G.W.; Shulman, G.I.; Waldhäusl, W.; et al. Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes 2004, 53, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. S2), S157–S163. [Google Scholar] [CrossRef]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Spijker, H.S.; Song, H.; Ellenbroek, J.H.; Roefs, M.M.; Engelse, M.A.; Bos, E.; Koster, A.J.; Rabelink, T.J.; Hansen, B.C.; Clark, A.; et al. Loss of β-Cell Identity Occurs in Type 2 Diabetes and Is Associated With Islet Amyloid Deposits. Diabetes 2015, 64, 2928–2938. [Google Scholar] [CrossRef]
- Pendergrass, M.; Bertoldo, A.; Bonadonna, R.; Nucci, G.; Mandarino, L.; Cobelli, C.; Defronzo, R.A. Muscle glucose transport and phosphorylation in type 2 diabetic, obese nondiabetic, and genetically predisposed individuals. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E92–E100. [Google Scholar] [CrossRef]
- Edgerton, D.S.; Kraft, G.; Smith, M.; Farmer, B.; Williams, P.E.; Coate, K.C.; Printz, R.L.; O’Brien, R.M.; Cherrington, A.D. Insulin’s direct hepatic effect explains the inhibition of glucose production caused by insulin secretion. JCI Insight 2017, 2, e91863. [Google Scholar] [CrossRef]
- Lewis, G.F.; Carpentier, A.C.; Pereira, S.; Hahn, M.; Giacca, A. Direct and indirect control of hepatic glucose production by insulin. Cell Metab. 2021, 33, 709–720. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, G.; Duval, S.; Jacobs, D.R.; Silventoinen, K. Comparison of body mass index, waist circumference, and waist/hip ratio in predicting incident diabetes: A meta-analysis. Epidemiol. Rev. 2007, 29, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Thapa, S.D.K.S.R.; Gautam, S.; Gyawali, D. Dyslipidemia in Type 2 Diabetes mellitus. J. Pathol. 2017, 7, 1149–1154. [Google Scholar] [CrossRef]
- Chan, D.C.; Barrett, H.P.; Watts, G.F. Dyslipidemia in visceral obesity: Mechanisms, implications, and therapy. Am. J. Cardiovasc. Drugs 2004, 4, 227–246. [Google Scholar] [CrossRef]
- Beers, A.; Haas, M.J.; Wong, N.C.; Mooradian, A.D. Inhibition of apolipoprotein AI gene expression by tumor necrosis factor alpha: Roles for MEK/ERK and JNK signaling. Biochemistry 2006, 45, 2408–2413. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Haas, M.J.; Wehmeier, K.R.; Wong, N.C. Obesity-related changes in high-density lipoprotein metabolism. Obesity 2008, 16, 1152–1160. [Google Scholar] [CrossRef]
- Gupta-Malhotra, M.; Hashmi, S.S.; Poffenbarger, T.; McNiece-Redwine, K. Left Ventricular Hypertrophy Phenotype in Childhood-Onset Essential Hypertension. J. Clin. Hypertens. 2016, 18, 449–455. [Google Scholar] [CrossRef]
- Alpert, M.A.; Lambert, C.R.; Panayiotou, H.; Terry, B.E.; Cohen, M.V.; Massey, C.V.; Hashimi, M.W.; Mukerji, V. Relation of duration of morbid obesity to left ventricular mass, systolic function, and diastolic filling, and effect of weight loss. Am. J. Cardiol. 1995, 76, 1194–1197. [Google Scholar] [CrossRef]
- Avelar, E.; Cloward, T.V.; Walker, J.M.; Farney, R.J.; Strong, M.; Pendleton, R.C.; Segerson, N.; Adams, T.D.; Gress, R.E.; Hunt, S.C.; et al. Left ventricular hypertrophy in severe obesity: Interactions among blood pressure, nocturnal hypoxemia, and body mass. Hypertension 2007, 49, 34–39. [Google Scholar] [CrossRef]
- Alpert, M.A.; Omran, J.; Bostick, B.P. Effects of Obesity on Cardiovascular Hemodynamics, Cardiac Morphology, and Ventricular Function. Curr. Obes. Rep. 2016, 5, 424–434. [Google Scholar] [CrossRef]
- Mente, A.; O’Donnell, M.J.; Rangarajan, S.; McQueen, M.J.; Poirier, P.; Wielgosz, A.; Morrison, H.; Li, W.; Wang, X.; Di, C.; et al. Association of urinary sodium and potassium excretion with blood pressure. N. Engl. J. Med. 2014, 371, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Grillo, A.; Salvi, L.; Coruzzi, P.; Salvi, P.; Parati, G. Sodium Intake and Hypertension. Nutrients 2019, 11, 1970. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.E.; do Carmo, J.M.; da Silva, A.A.; Juncos, L.A.; Wang, Z.; Hall, J.E. Obesity, hypertension, and chronic kidney disease. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Yu, Y.; Zhang, Z.; Guo, F.; Beltz, T.G.; Thunhorst, R.L.; Felder, R.B.; Johnson, A.K. Leptin Mediates High-Fat Diet Sensitization of Angiotensin II-Elicited Hypertension by Upregulating the Brain Renin-Angiotensin System and Inflammation. Hypertension 2016, 67, 970–976. [Google Scholar] [CrossRef]
- Lohmeier, T.E.; Iliescu, R. The sympathetic nervous system in obesity hypertension. Curr. Hypertens. Rep. 2013, 15, 409–416. [Google Scholar] [CrossRef]
- Susic, D.; Varagic, J. Obesity: A Perspective from Hypertension. Med. Clin. N. Am. 2017, 101, 139–157. [Google Scholar] [CrossRef]
- Muñoz, M.; López-Oliva, M.E.; Rodríguez, C.; Martínez, M.P.; Sáenz-Medina, J.; Sánchez, A.; Climent, B.; Benedito, S.; García-Sacristán, A.; Rivera, L.; et al. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol. 2020, 28, 101330. [Google Scholar] [CrossRef]
- Virdis, A.; Taddei, S. Endothelial Dysfunction in Resistance Arteries of Hypertensive Humans: Old and New Conspirators. J. Cardiovasc. Pharmacol. 2016, 67, 451–457. [Google Scholar] [CrossRef]
- Marchesi, C.; Ebrahimian, T.; Angulo, O.; Paradis, P.; Schiffrin, E.L. Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertension 2009, 54, 1384–1392. [Google Scholar] [CrossRef]
- Lu, Y.; Hajifathalian, K.; Ezzati, M.; Woodward, M.; Rimm, E.B.; Danaei, G.; D’Este, C. Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: A pooled analysis of 97 prospective cohorts with 1·8 million participants. Lancet 2014, 383, 970–983. [Google Scholar] [CrossRef]
- Barber, T.M.; Franks, S. Obesity and polycystic ovary syndrome. Clin. Endocrinol. 2021, 95, 531–541. [Google Scholar] [CrossRef]
- Barber, T.M.; McCarthy, M.I.; Wass, J.A.; Franks, S. Obesity and polycystic ovary syndrome. Clin. Endocrinol. 2006, 65, 137–145. [Google Scholar] [CrossRef]
- Purwar, A.; Nagpure, S. Insulin Resistance in Polycystic Ovarian Syndrome. Cureus 2022, 14, e30351. [Google Scholar] [CrossRef]
- Kositanurit, W.; Muntham, D.; Udomsawaengsup, S.; Chirakalwasan, N. Prevalence and associated factors of obstructive sleep apnea in morbidly obese patients undergoing bariatric surgery. Sleep Breath 2018, 22, 251–256. [Google Scholar] [CrossRef]
- Newman, A.B.; Foster, G.; Givelber, R.; Nieto, F.J.; Redline, S.; Young, T. Progression and regression of sleep-disordered breathing with changes in weight: The Sleep Heart Health Study. Arch. Intern. Med. 2005, 165, 2408–2413. [Google Scholar] [CrossRef] [PubMed]
- Jehan, S.; Farag, M.; Zizi, F.; Pandi-Perumal, S.R.; Chung, A.; Truong, A.; Jean-Louis, G.; Tello, D.; McFarlane, S.I. Obstructive sleep apnea and stroke. Sleep Med. Disord. 2018, 2, 120–125. [Google Scholar]
- Hamilton, G.S.; Joosten, S.A. Obstructive sleep apnoea and obesity. Aust. Fam. Physician 2017, 46, 460–463. [Google Scholar]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Park, J.; Euhus, D.M.; Scherer, P.E. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr. Rev. 2011, 32, 550–570. [Google Scholar] [CrossRef]
- Gadde, K.M.; Apolzan, J.W.; Berthoud, H.R. Pharmacotherapy for Patients with Obesity. Clin. Chem. 2018, 64, 118–129. [Google Scholar] [CrossRef]
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Ard, J.D.; Comuzzie, A.G.; Donato, K.A.; Hu, F.B.; Hubbard, V.S.; Jakicic, J.M.; Kushner, R.F.; et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J. Am. Coll. Cardiol. 2014, 63, 2985–3023. [Google Scholar] [CrossRef] [PubMed]
- Preiss Contreras, Y.; Ramos Salas, X.; Ávila Oliver, C.; Saquimux Contreras, M.A.; Muñoz Claro, R.; Canales Ferrada, C.; Obesidad, C.C.p.e.E.d.l. Obesity in adults: Clinical practice guideline adapted for Chile. Medwave 2022, 22, e2649. [Google Scholar] [CrossRef] [PubMed]
- Gadde, K.M.; Martin, C.K.; Berthoud, H.R.; Heymsfield, S.B. Obesity: Pathophysiology and Management. J. Am. Coll. Cardiol. 2018, 71, 69–84. [Google Scholar] [CrossRef]
- Wing, R.R.; Tate, D.F.; Gorin, A.A.; Raynor, H.A.; Fava, J.L. A self-regulation program for maintenance of weight loss. N. Engl. J. Med. 2006, 355, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Rock, C.L.; Flatt, S.W.; Sherwood, N.E.; Karanja, N.; Pakiz, B.; Thomson, C.A. Effect of a free prepared meal and incentivized weight loss program on weight loss and weight loss maintenance in obese and overweight women: A randomized controlled trial. JAMA 2010, 304, 1803–1810. [Google Scholar] [CrossRef]
- Lee, E.Y.; Yoon, K.H. Epidemic obesity in children and adolescents: Risk factors and prevention. Front. Med. 2018, 12, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.; Clements, J. Obesity management among patients with type 2 diabetes and prediabetes: A focus on lifestyle modifications and evidence of antiobesity medications. Expert Rev. Endocrinol. Metab. 2017, 12, 303–313. [Google Scholar] [CrossRef]
- Pedersen, S.D.M.P.; Wharton, S. Canadian Adult Obesity Clinical Practice Guidelines: Pharmacotherapy in Obesity Management; Obesity Canada: Edmonton, AB, Canada, 2020. [Google Scholar]
- Leblanc, E.S.; O’Connor, E.; Whitlock, E.P.; Patnode, C.D.; Kapka, T. Effectiveness of primary care-relevant treatments for obesity in adults: A systematic evidence review for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2011, 155, 434–447. [Google Scholar] [CrossRef]
- Rucker, D.; Padwal, R.; Li, S.K.; Curioni, C.; Lau, D.C. Long term pharmacotherapy for obesity and overweight: Updated meta-analysis. BMJ 2007, 335, 1194–1199. [Google Scholar] [CrossRef]
- Secher, A.; Jelsing, J.; Baquero, A.F.; Hecksher-Sørensen, J.; Cowley, M.A.; Dalbøge, L.S.; Hansen, G.; Grove, K.L.; Pyke, C.; Raun, K.; et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J. Clin. Investig. 2014, 124, 4473–4488. [Google Scholar] [CrossRef]
- Sisley, S.; Gutierrez-Aguilar, R.; Scott, M.; D’Alessio, D.A.; Sandoval, D.A.; Seeley, R.J. Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effect. J. Clin. Investig. 2014, 124, 2456–2463. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, L.V.; Flint, A.; Olsen, A.K.; Ingwersen, S.H. Liraglutide in Type 2 Diabetes Mellitus: Clinical Pharmacokinetics and Pharmacodynamics. Clin. Pharmacokinet. 2016, 55, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Novo Nordisk Canada Inc. SAXENDA® Liraglutide; Novo Nordisk Canada Inc.: Mississauga, ON, Canada, 2017. [Google Scholar]
- Allison, D.B.; Gadde, K.M.; Garvey, W.T.; Peterson, C.A.; Schwiers, M.L.; Najarian, T.; Tam, P.Y.; Troupin, B.; Day, W.W. Controlled-release phentermine/topiramate in severely obese adults: A randomized controlled trial (EQUIP). Obesity 2012, 20, 330–342. [Google Scholar] [CrossRef]
- Rothman, R.B.; Hendricks, E.J. Phentermine cardiovascular safety. Am. J. Emerg. Med. 2009, 27, 1010–1013. [Google Scholar] [CrossRef]
- Steidl, K.E.; Darko, W.; Probst, L.A.; Noviasky, J.A.; Nasser, S. Rhabdomyolysis associated with phentermine. Am. J. Health Syst. Pharm. 2010, 67, 1929–1932. [Google Scholar] [CrossRef]
- Gadde, K.M.; Xiong, G.L. Bupropion for weight reduction. Expert Rev. Neurother. 2007, 7, 17–24. [Google Scholar] [CrossRef]
- Smith, S.R.; Fujioka, K.; Gupta, A.K.; Billes, S.K.; Burns, C.; Kim, D.; Dunayevich, E.; Greenway, F.L. Combination therapy with naltrexone and bupropion for obesity reduces total and visceral adiposity. Diabetes Obes. Metab. 2013, 15, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Rothman, R.B.; Baumann, M.H. Appetite suppressants, cardiac valve disease and combination pharmacotherapy. Am. J. Ther. 2009, 16, 354–364. [Google Scholar] [CrossRef]
- Scudeler, M.A.; Morreale, S.; Doretto-Silva, L.; Petri, G.; Santos, J.F.R.D.; Nassis, C.; Correa, O.M.T.; Veridiano, J.M. Effects of topiramate, bupropion and naltrexone isolated or combined on subcutaneous adipose tissue in obese rats. Einstein 2022, 20, eAO5587. [Google Scholar] [CrossRef]
- Food and Drug Administration. Advisory Committee Meeting for Phentermine/Topiramate (Qnexa); Division of Metabolism and Endocrinology Products (DMEP), Office of Drug Evaluation II, Center for Drug Evaluation and Research: Silver Spring, MD, USA, 2010.
- Arias, H.R.; Santamaría, A.; Ali, S.F. Pharmacological and neurotoxicological actions mediated by bupropion and diethylpropion. Int. Rev. Neurobiol. 2009, 88, 223–255. [Google Scholar] [CrossRef]
- Jones, H.S. Diethylpropion dependence. Med. J. Aust. 1968, 1, 267. [Google Scholar] [CrossRef] [PubMed]
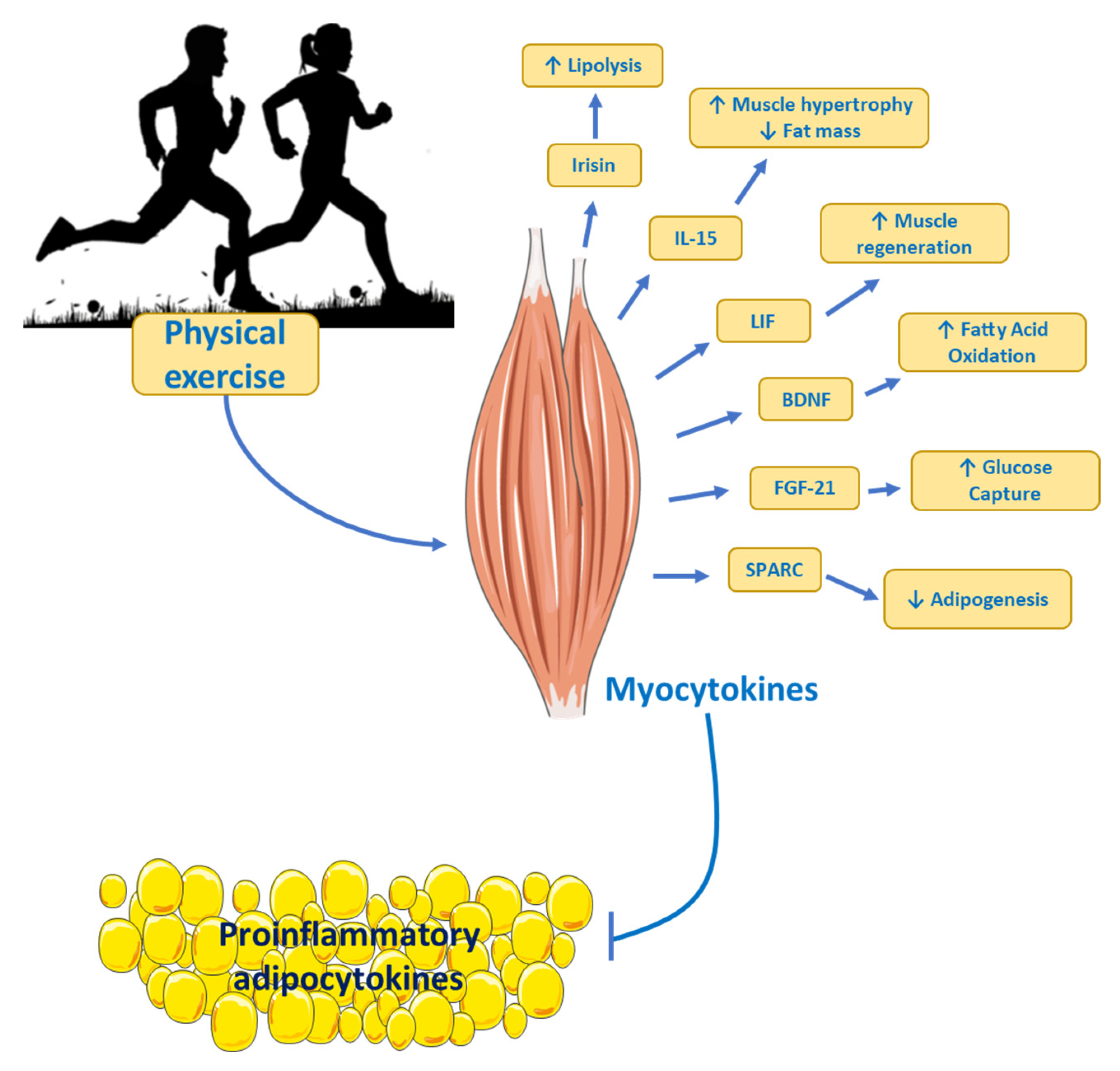
- So, B.; Kim, H.J.; Kim, J.; Song, W. Exercise-induced myokines in health and metabolic diseases. Integr. Med. Res. 2014, 3, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef]
- Scherer, P.E. Adipose tissue: From lipid storage compartment to endocrine organ. Diabetes 2006, 55, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. Muscles and their myokines. J. Exp. Biol. 2011, 214, 337–346. [Google Scholar] [CrossRef]
- Leal, L.G.; Lopes, M.A.; Batista, M.L. Physical Exercise-Induced Myokines and Muscle-Adipose Tissue Crosstalk: A Review of Current Knowledge and the Implications for Health and Metabolic Diseases. Front. Physiol. 2018, 9, 1307. [Google Scholar] [CrossRef]
- Petersen, A.M.; Pedersen, B.K. The anti-inflammatory effect of exercise. J. Appl. Physiol. 2005, 98, 1154–1162. [Google Scholar] [CrossRef]
- Ng, S.W.; Popkin, B.M. Time use and physical activity: A shift away from movement across the globe. Obes. Rev. 2012, 13, 659–680. [Google Scholar] [CrossRef]
- Merritt, E.K. Why is it so hard to lose fat? Because it has to get out through your nose! An exercise physiology laboratory on oxygen consumption, metabolism, and weight loss. Adv. Physiol. Educ. 2021, 45, 599–606. [Google Scholar] [CrossRef]
- Popson, M.S.D.M.; Borger, J. Biochemistry, Heat and Calories; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Merry, B.J. Molecular mechanisms linking calorie restriction and longevity. Int. J. Biochem. Cell Biol. 2002, 34, 1340–1354. [Google Scholar] [CrossRef]
- Asami, D.K.; McDonald, R.B.; Hagopian, K.; Horwitz, B.A.; Warman, D.; Hsiao, A.; Warden, C.; Ramsey, J.J. Effect of aging, caloric restriction, and uncoupling protein 3 (UCP3) on mitochondrial proton leak in mice. Exp. Gerontol. 2008, 43, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Poirier, P.; Després, J.P. Exercise in weight management of obesity. Cardiol. Clin. 2001, 19, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; Barrea, L.; Laudisio, D.; Pugliese, G.; Salzano, C.; Savastano, S.; Colao, A. The management of very low-calorie ketogenic diet in obesity outpatient clinic: A practical guide. J. Transl. Med. 2019, 17, 356. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monsalve, F.A.; Delgado-López, F.; Fernández-Tapia, B.; González, D.R. Adipose Tissue, Non-Communicable Diseases, and Physical Exercise: An Imperfect Triangle. Int. J. Mol. Sci. 2023, 24, 17168. https://doi.org/10.3390/ijms242417168
Monsalve FA, Delgado-López F, Fernández-Tapia B, González DR. Adipose Tissue, Non-Communicable Diseases, and Physical Exercise: An Imperfect Triangle. International Journal of Molecular Sciences. 2023; 24(24):17168. https://doi.org/10.3390/ijms242417168
Chicago/Turabian StyleMonsalve, Francisco A., Fernando Delgado-López, Barbra Fernández-Tapia, and Daniel R. González. 2023. "Adipose Tissue, Non-Communicable Diseases, and Physical Exercise: An Imperfect Triangle" International Journal of Molecular Sciences 24, no. 24: 17168. https://doi.org/10.3390/ijms242417168
APA StyleMonsalve, F. A., Delgado-López, F., Fernández-Tapia, B., & González, D. R. (2023). Adipose Tissue, Non-Communicable Diseases, and Physical Exercise: An Imperfect Triangle. International Journal of Molecular Sciences, 24(24), 17168. https://doi.org/10.3390/ijms242417168