
The Structure and Nucleotide-Binding Characteristics of Regulated Cystathionine β-Synthase Domain-Containing Pyrophosphatase without One Catalytic Domain

 , , and
, , and
Abstract
:
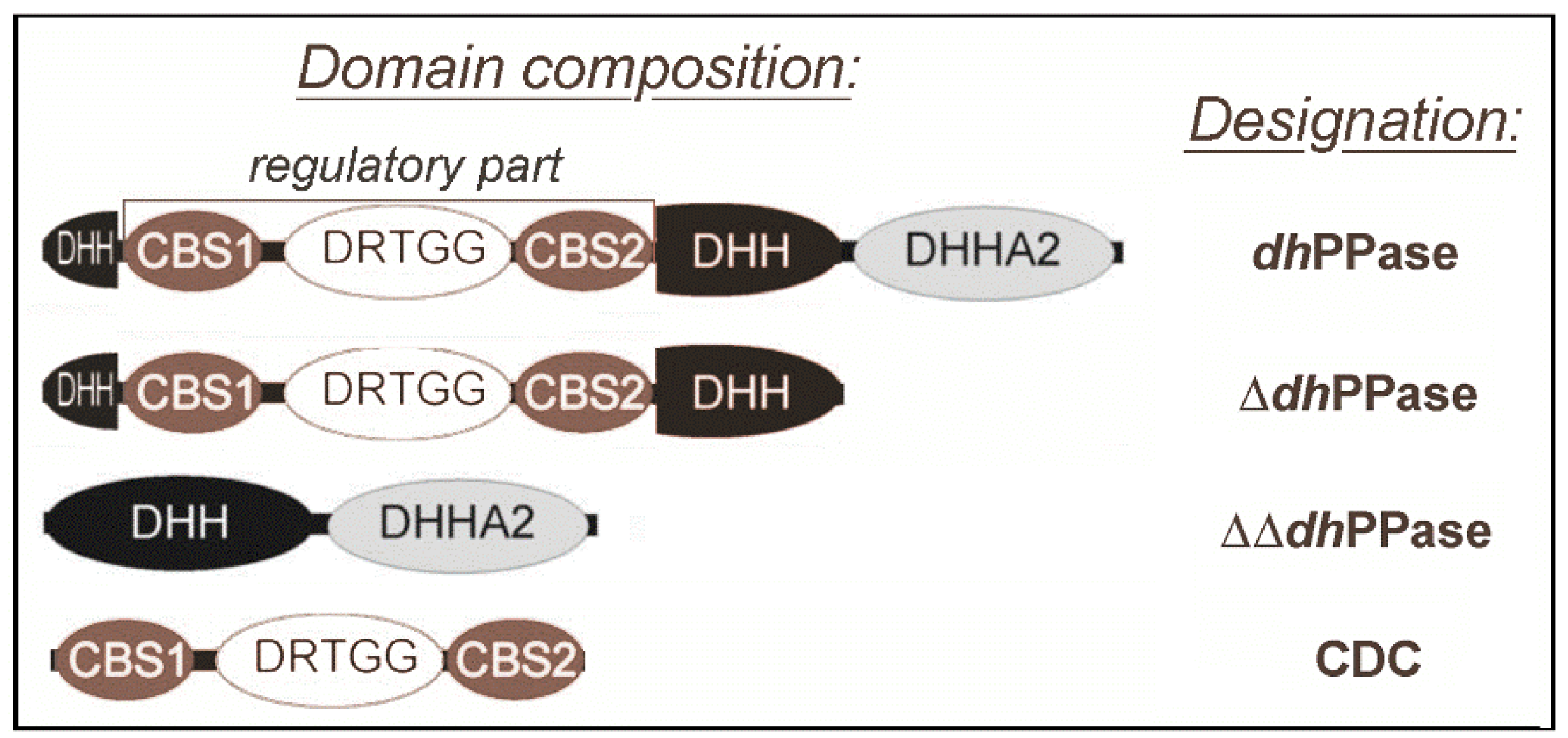
1. Introduction
2. Results
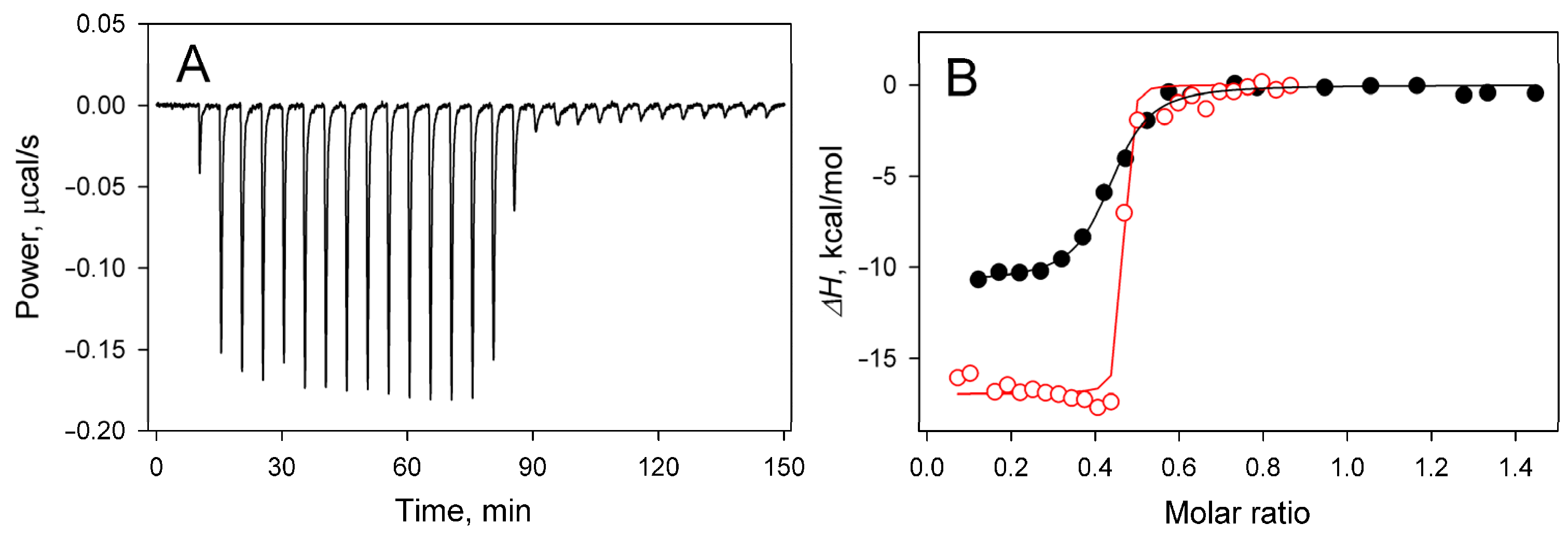
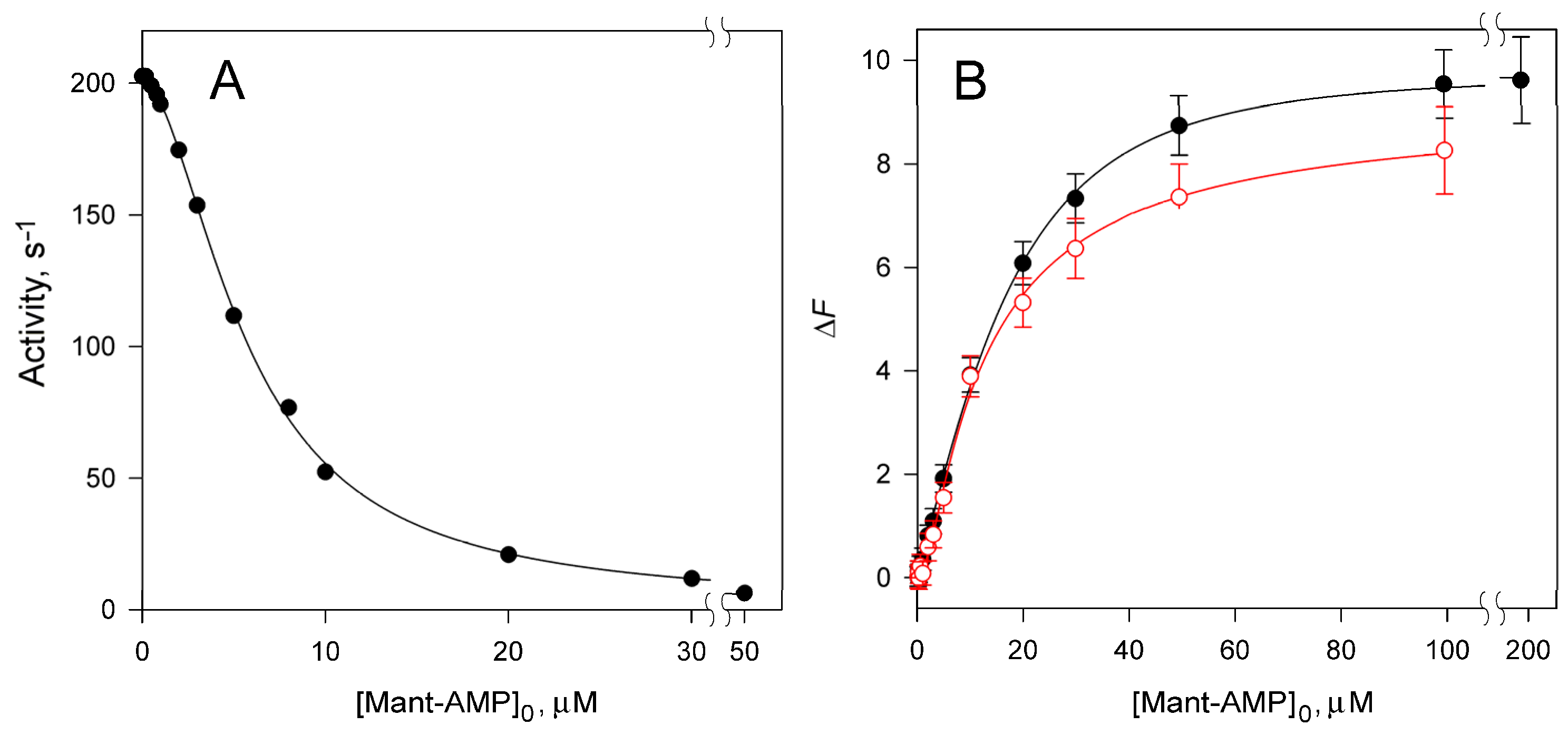
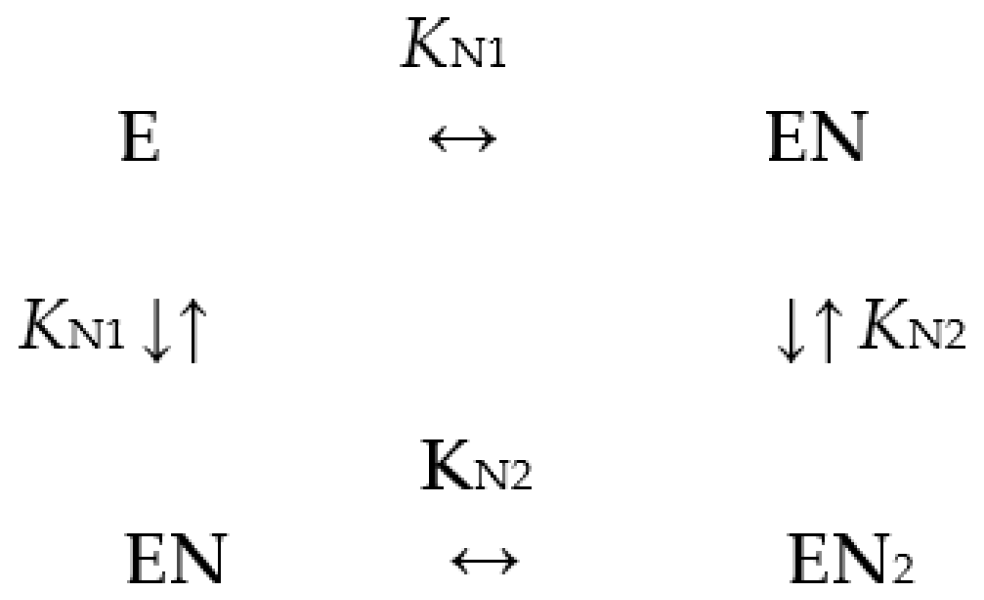
2.1. Nucleoside Phosphate (Ap4A and AMP) Binding
2.2. Effects of Deletion and Nucleotide Binding on Protein Thermal Stability
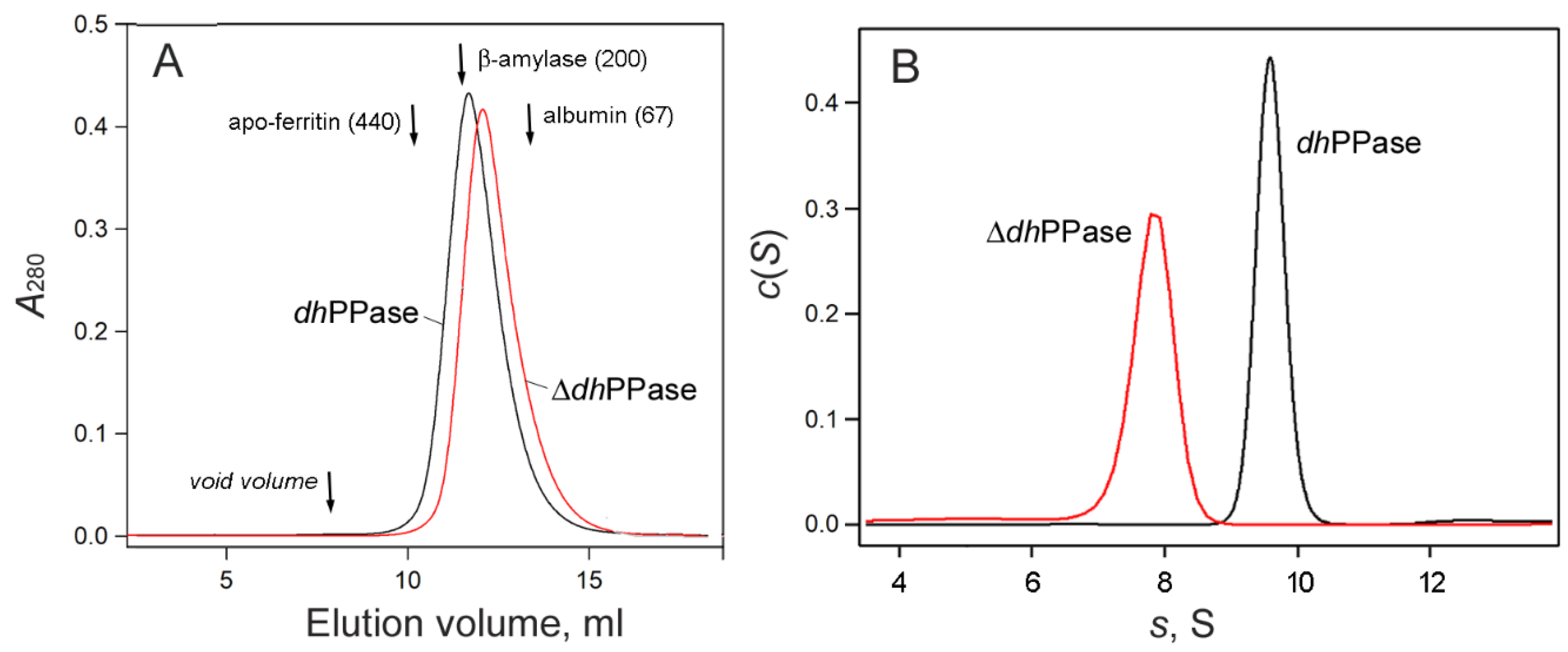
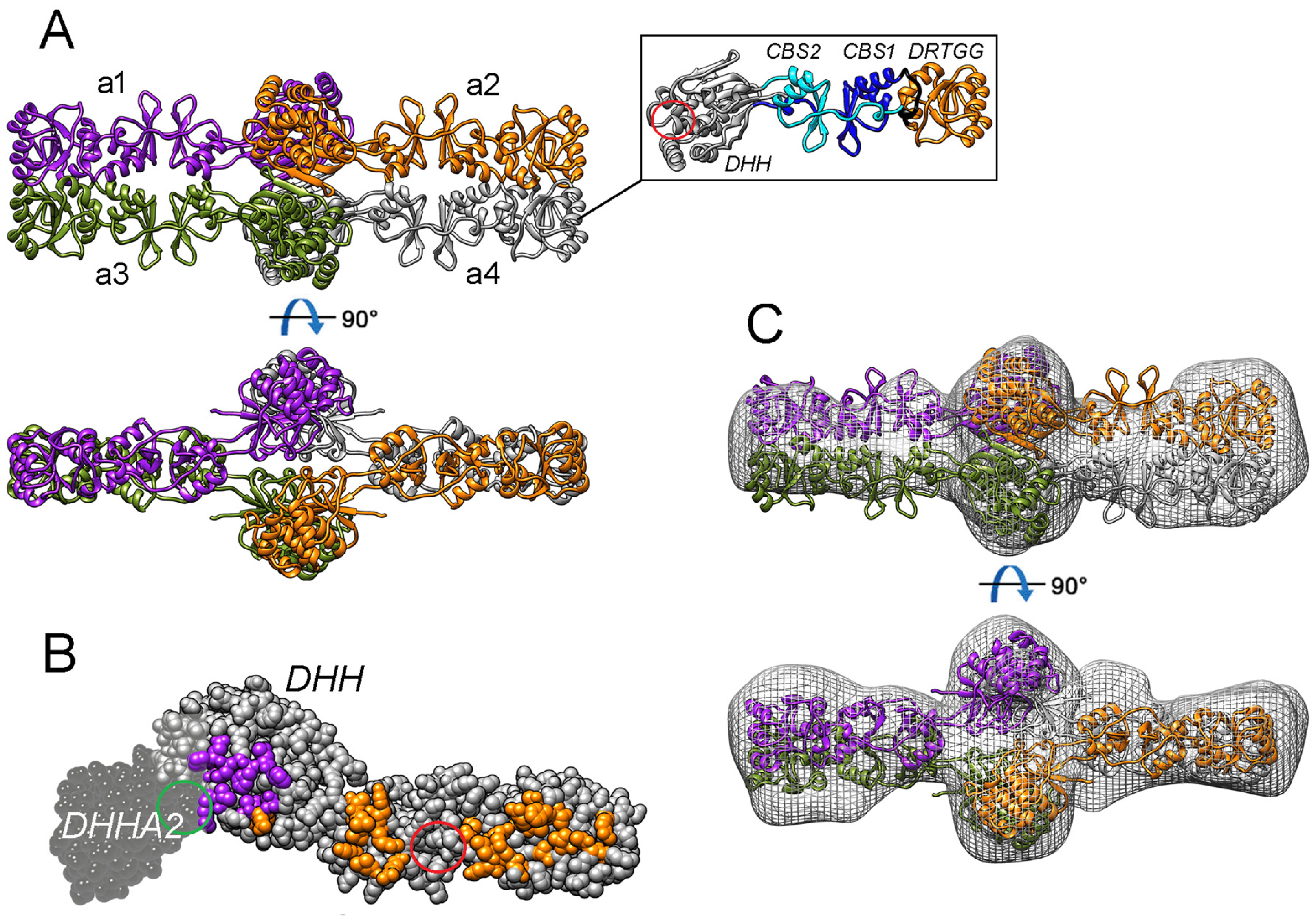
2.3. Oligomeric Structure of ΔdhPPase
2.4. Single-Particle Electron Microscopy of ΔdhPPase
2.5. The Modeled 3D Structure of ΔdhPPase
3. Discussion
3.1. 3D Structure of dhPPase
3.2. Mechanism of Regulation
4. Materials and Methods
4.1. Materials
4.2. Enzyme Activity Assay
4.3. Isothermal Titration Calorimetry (ITC)
4.4. Förster Resonance Energy Transfer (FRET) Measurements
4.5. Binding Data Treatment
4.6. Protein Thermal Stability Measurements (ThermoFluor)
4.7. Size-Exclusion Chromatography
4.8. Sedimentation Velocity Analysis
4.9. Structure Modeling and Refinement
4.10. Single-Particle Electron Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Heinonen, J.K. Biological Role of Inorganic Pyrophosphate; Kluwer Academic Publishers: London, UK, 2001. [Google Scholar]
- Kajander, T.K.; Kellosalo, J.; Goldman, A. Inorganic pyrophosphatases: One substrate, three mechanisms. FEBS Lett. 2013, 587, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Baykov, A.A.; Anashkin, V.A.; Salminen, A.; Lahti, R. Inorganic pyrophosphatases of Family II—Two decades after their discovery. FEBS Lett. 2017, 591, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Patskovsky, Y.; Toro, R.; Farelli, J.D.; Pandya, C.; Almo, S.C.; Allen, K.N.; Dunaway-Mariano, D. Divergence of structure and function in the haloacid dehalogenase enzyme superfamily: Bacteroides thetaiotaomicron BT2127 is an inorganic pyrophosphatase. Biochemistry 2011, 50, 8937–8949. [Google Scholar] [CrossRef] [PubMed]
- Jämsen, J.; Tuominen, H.; Salminen, A.; Belogurov, G.A.; Magretova, N.N.; Baykov, A.A.; Lahti, R. A CBS domain-containing pyrophosphatase of Moorella thermoacetica is regulated by adenine nucleotides. Biochem. J. 2007, 408, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Anashkin, V.A.; Salminen, A.; Tuominen, H.K.; Orlov, V.N.; Lahti, R.; Baykov, A.A. Cystathionine β-synthase (CBS) domain-containing pyrophosphatase as a target for diadenosine polyphosphates in bacteria. J. Biol. Chem. 2015, 290, 27594–27603. [Google Scholar] [CrossRef]
- Varshavsky, A. Diadenosine 5′,5′′′-P1,P4-tetraphosphate: A pleiotypically acting alarmone. Cell 1983, 34, 711–712. [Google Scholar] [CrossRef]
- Ereno-Orbea, J.; Oyenarte, I.; Martìnez-Cruz, L.A. CBS domains: Ligand binding sites and conformational variability. Arch. Biochem. Biophys. 2013, 540, 70–81. [Google Scholar] [CrossRef]
- Bateman, A. The structure of a domain common to Archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 1997, 22, 12–13. [Google Scholar] [CrossRef]
- Baykov, A.A.; Tuominen, H.K.; Lahti, R. The CBS domain: A protein module with an emerging prominent role in regulation. ACS Chem. Biol. 2011, 6, 1156–1163. [Google Scholar] [CrossRef]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Investig. 2004, 113, 274–284. [Google Scholar] [CrossRef]
- Kery, V.; Poneleit, L.; Kraus, J. Trypsin cleavage of human cystathionine β-synthase into an evolutionarily conserved active core: Structural and functional consequences. Arch. Biochem. Biophys. 1998, 355, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Ereño-Orbea, J.; Majtan, T.; Oyenarte, I.; Kraus, J.P.; Martίnez-Cruz, L.A. Structural basis of regulation and oligomerization of human cystathionine β-synthase, the central enzyme of transsulfuration. Proc. Natl. Acad. Sci. USA 2013, 110, 3790–3799. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Chen, Z.P.; Van Denderen, B.J.; Morton, C.J.; Parker, M.W.; Witters, L.A.; Stapleton, D.; Kemp, B.E. Intrasteric control of AMPK via the c1 subunit AMP allosteric regulatory site. Protein Sci. 2004, 13, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Anashkin, V.A.; Lahti, M.; Tuominen, H.K.; Lahti, R.; Baykov, A.A. Cystathionine β-synthase (CBS) domains confer multiple forms of Mg2+-dependent cooperativity to family II pyrophosphatases. J. Biol. Chem. 2014, 289, 22865–22876. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Milner, A.J.; Fütterer, K.; Konopka, M.; Ilias, M.; Young, T.W.; White, S.A. The “open” and “closed” structures of the type-C inorganic pyrophosphatases from Bacillus subtilis and Streptococcus gordonii. J. Mol. Biol. 2001, 313, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Fabrichniy, I.P.; Lehtio, L.; Salminen, A.; Zyryanov, A.B.; Baykov, A.A.; Lahti, R.; Goldman, A. Structural studies of metal ions in Family II pyrophosphatases: The requirement for a Janus ion. Biochemistry 2004, 43, 14403–14411. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, H.; Salminen, A.; Oksanen, E.; Jämsen, J.; Heikkilä, O.; Lehtio, L.; Magretova, N.N.; Goldman, A.; Baykov, A.A.; Lahti, R. Crystal structures of the CBS and DRTGG domains of the regulatory region of Clostridium perfringens pyrophosphatase complexed with the inhibitor, AMP, and activator, diadenosine tetraphosphate. J. Mol. Biol. 2010, 398, 400–413. [Google Scholar] [CrossRef]
- Anashkin, V.A.; Salminen, A.; Orlov, V.N.; Lahti, R.; Baykov, A.A. The tetrameric structure of nucleotide-regulated pyrophosphatase and its modulation by deletion mutagenesis and ligand binding. Arch. Biochem. Biophys. 2022, 692, 108537. [Google Scholar] [CrossRef]
- Dadinova, L.A.; Soshinskaia, E.Y.; Jeffries, C.M.; Svergun, D.A.; Shtykova, E.V. Tetrameric structures of inorganic CBS-pyrophosphatases from various bacterial species revealed by small-angle X-ray scattering in solution. Biomolecules 2020, 10, 564. [Google Scholar] [CrossRef]
- Zhang, R.; Monsma, F. Fluorescence-based thermal shift assays. Curr. Opin. Drug Discov. Dev. 2010, 13, 389–402. [Google Scholar]
- Parfenyev, A.N.; Salminen, A.; Halonen, P. Quaternary structure and metal-ion requirement of family II pyrophosphatases from Bacillus subtilis, Streptococcus gordonii and Streptococcus mutans. J. Biol. Chem. 2001, 276, 24511–24518. [Google Scholar] [CrossRef] [PubMed]
- Schuck, P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000, 78, 1606–1619. [Google Scholar] [CrossRef] [PubMed]
- Dadinova, L.A.; Soshinskaya, E.Y.; Chesnokov, Y.M.; Kamyshinsky, R.A.; Vasilieva, A.L.; Shtykova, E.V. Formation of high-order structures in solution by CBS-pyrophosphatase from D. hafniense. Crystallogr. Rep. 2021, 66, 833–839. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Gronenborn, A.M. Domain Swapping. In Encyclopedia of Biophysics; Roberts, G.C.K., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 498–503. [Google Scholar]
- Bandyopadhyay, P.; Pramanick, I.; Biswas, R.; PS, S.; Sreedharan, S.; Singh, S.; Rajmani, R.S.; Laxman, S.; Dutta, S.; Singh, A. S-Adenosylmethionine-responsive cystathionine β-synthase modulates sulfur metabolism and redox balance in Mycobacterium tuberculosis. Sci. Adv. 2022, 8, eabo0097. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, M.; Shibata, N.; Ishido-Nakai, E.; Kanagawa, M.; Iio, Y.; Komori, H.; Ueda, Y.; Nakagawa, N.; Kuramitsu, S.; Higuchi, Y. Crystal structure of a hypothetical protein, TTHA0829 from Thermus thermophilus HB8, composed of cystathionine-β-synthase (CBS) and aspartate-kinase chorismate-mutase tyrA (ACT) domains. Extremophiles 2016, 20, 275–282. [Google Scholar] [CrossRef]
- Zhang, R.; Evans, G.; Rotella, F.J.; Westbrook, E.M.; Beno, D.; Huberman, E.; Joachimiak, A.; Collart, F.R. Characteristics and crystal structure of bacterial inosine-5′-monophosphate dehydrogenase. Biochemistry 1999, 38, 4691–4700. [Google Scholar] [CrossRef]
- Labesse, G.; Alexandre, T.; Vaupre, L.; Salard-Arnaud, I.; Him, J.L.; Raynal, B.; Bron, P.; Munier-Lehmann, H. MgATP regulates allostery and fiber formation in IMPDHs. Structure 2013, 21, 975–985. [Google Scholar] [CrossRef]
- Giammarinaro, P.I.; Young, M.K.M.; Steinchen, W.; Mais, C.N.; Hochberg, G.; Yang, J.; Stevenson, D.M.; Amador-Noguez, D.; Paulus, A.; Wang, J.D.; et al. Diadenosine tetraphosphate regulates biosynthesis of GTP in Bacillus subtilis. Nat. Microbiol. 2022, 7, 1442–1452. [Google Scholar] [CrossRef]
- Fernandez-Justel, D.; Marcos-Alcalde, I.; Abascal, F.; Vidana, N.; Gomez-Puertas, P.; Jimenez, A.; Revuelta, J.L.; Buey, R.M. Diversity of mechanisms to control bacterial GTP homeostasis by the mutually exclusive binding of adenine and guanine nucleotides to IMP dehydrogenase. Protein Sci. 2022, 31, e4314. [Google Scholar] [CrossRef] [PubMed]
- Imamura, A.; Okada, T.; Mase, H.; Otani, T.; Kobayashi, T.; Tamura, M.; Kubata, B.K.; Inoue, K.; Rambo, R.P.; Uchiyama, S.; et al. Allosteric regulation accompanied by oligomeric state changes of Trypanosoma brucei GMP reductase through cystathionine-β-synthase domain. Nat. Commun. 2020, 11, 1837. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; King, N.P.; Sawaya, M.R.; Cascio, D.; Yeates, T.O. Structures and functional implications of an AMP-binding cystathionine β-synthase domain protein from a hyperthermophilic archaeon. J. Mol. Biol. 2008, 380, 181–192. [Google Scholar]
- Anashkin, V.A.; Orlov, V.N.; Lahti, R.; Baykov, A.A. An arginine residue involved in allosteric regulation of cystathionine β-synthase (CBS) domain-containing pyrophosphatase. Arch. Biochem. Biophys. 2019, 662, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Anashkin, V.A.; Anu, S.; Osipova, E.; Kurilova, S.A.; Deltsov, I.D.; Reijo, L.; Baykov, A.A. Residue network involved in the allosteric regulation of cystathionine β-synthase domain-containing pyrophosphatase by adenine nucleotides. ACS Omega 2019, 4, 15549–15559. [Google Scholar] [CrossRef] [PubMed]
- Jämsen, J.; Tuominen, H.; Baykov, A.A.; Lahti, R. Mutational analysis of residues in the regulatory CBS domains of Moorella thermoacetica pyrophosphatase corresponding to disease-related residues of human proteins. Biochem. J. 2011, 433, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Baykov, A.A.; Avaeva, S.M. A simple and sensitive apparatus for continuous monitoring of orthophosphate in the presence of acid-labile compounds. Anal. Biochem. 1981, 116, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Resch, G.P.; Brandstetter, M.; Konigsmaier, L.; Urban, E.; Pickl-Herk, A.M. Immersion freezing of suspended particles and cells for cryo-electron microscopy. Cold Spring Harb. Protoc. 2011, 2011, 803–814. [Google Scholar] [CrossRef]
- Mastronarde, D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005, 152, 36–51. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Variant | Nucleotide | ΔH, kcal/mol | n | KN, µM |
|---|---|---|---|---|
| ΔdhPPase | Ap4A | −17.0 ± 0.3 | 0.45 ± 0.01 | <<0.01 |
| dhPPase | Ap4A | −10.8 ± 0.2 (−10.4 ± 0.3) a | 0.42 ± 0.01 (0.41 ± 0.01) a | 0.01–0.1 |
| dhPPase a | AMP | −5.6 ± 0.5 | 0.79 ± 0.05 | 0.8 ± 0.3 |
| Nucleotide | Residual Activity, % | h | Ki1, µM | Ki2, µM |
|---|---|---|---|---|
| Mant-AMP | <1 | 1.75 ± 0.04 | 41 ± 7 | 0.8 ± 0.1 |
| AMP a | 3.7 ± 0.1 | 1.68 ± 0.04 | 9.6 ± 1.4 | 0.21 ± 0.08 |
| Enzyme Variant | ΔFmax | KN1, µM | KN2, µM |
|---|---|---|---|
| dhPPase | 10.3 ± 0.4 | 22 ± 2 | 6.5 ± 1 |
| ΔdhPPase | 8.8 ± 0.4 | 21 ± 6 | 6 ± 2 |
| Nucleotide | Tm, °C | |||
|---|---|---|---|---|
| dhPPase | ΔdhPPase | CDC | ΔΔdhPPase | |
| None | 57.0, 64.0, 77.5 | 68.3, 83.5 | 58.0 | 56.5, 77.5 |
| AMP | 57.1, 67.4, 80.8 | 68.6, 85.5 | 66.5 | |
| Ap4A | 52.9, 68.8, 79.7 | 71.0, 86.5 | 61.5 | |
| Enzyme Variant | Molecular Mass, kDa | ||
|---|---|---|---|
| Sedimentation a | SEC | Theory b | |
| ΔdhPPase | 171–192 | ~176 | 186 |
| dhPPase c | 203–234 | ~214 | 241 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamakhov, I.M.; Anashkin, V.A.; Moiseenko, A.V.; Orlov, V.N.; Vorobyeva, N.N.; Sokolova, O.S.; Baykov, A.A. The Structure and Nucleotide-Binding Characteristics of Regulated Cystathionine β-Synthase Domain-Containing Pyrophosphatase without One Catalytic Domain. Int. J. Mol. Sci. 2023, 24, 17160. https://doi.org/10.3390/ijms242417160
Zamakhov IM, Anashkin VA, Moiseenko AV, Orlov VN, Vorobyeva NN, Sokolova OS, Baykov AA. The Structure and Nucleotide-Binding Characteristics of Regulated Cystathionine β-Synthase Domain-Containing Pyrophosphatase without One Catalytic Domain. International Journal of Molecular Sciences. 2023; 24(24):17160. https://doi.org/10.3390/ijms242417160
Chicago/Turabian StyleZamakhov, Ilya M., Viktor A. Anashkin, Andrey V. Moiseenko, Victor N. Orlov, Natalia N. Vorobyeva, Olga S. Sokolova, and Alexander A. Baykov. 2023. "The Structure and Nucleotide-Binding Characteristics of Regulated Cystathionine β-Synthase Domain-Containing Pyrophosphatase without One Catalytic Domain" International Journal of Molecular Sciences 24, no. 24: 17160. https://doi.org/10.3390/ijms242417160
APA StyleZamakhov, I. M., Anashkin, V. A., Moiseenko, A. V., Orlov, V. N., Vorobyeva, N. N., Sokolova, O. S., & Baykov, A. A. (2023). The Structure and Nucleotide-Binding Characteristics of Regulated Cystathionine β-Synthase Domain-Containing Pyrophosphatase without One Catalytic Domain. International Journal of Molecular Sciences, 24(24), 17160. https://doi.org/10.3390/ijms242417160