
Transient Receptor Potential Vanilloid 4-Dependent Microglial Function in Myelin Injury and Repair
 , ,
, ,
Abstract
:1. Introduction
2. Results
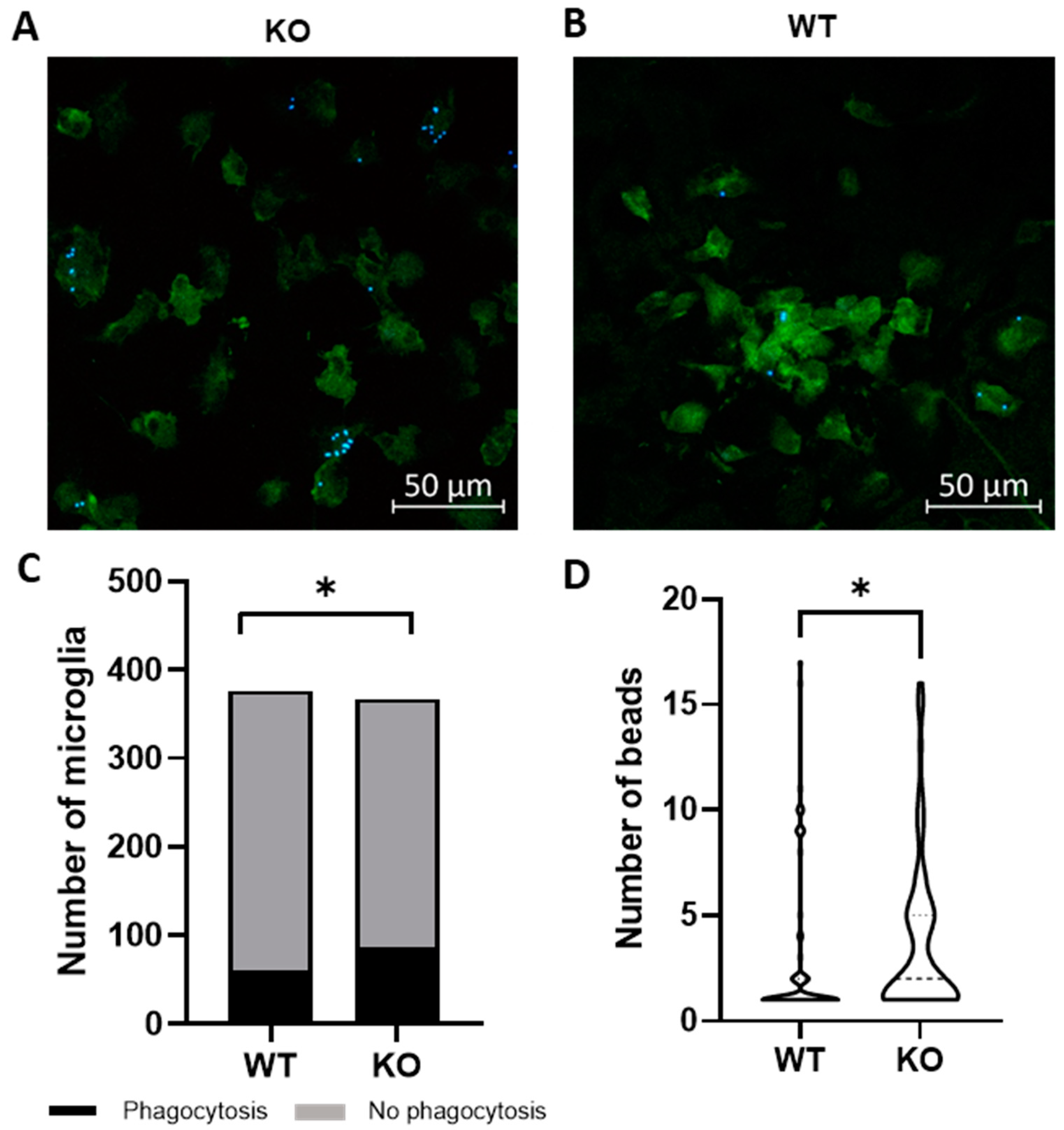
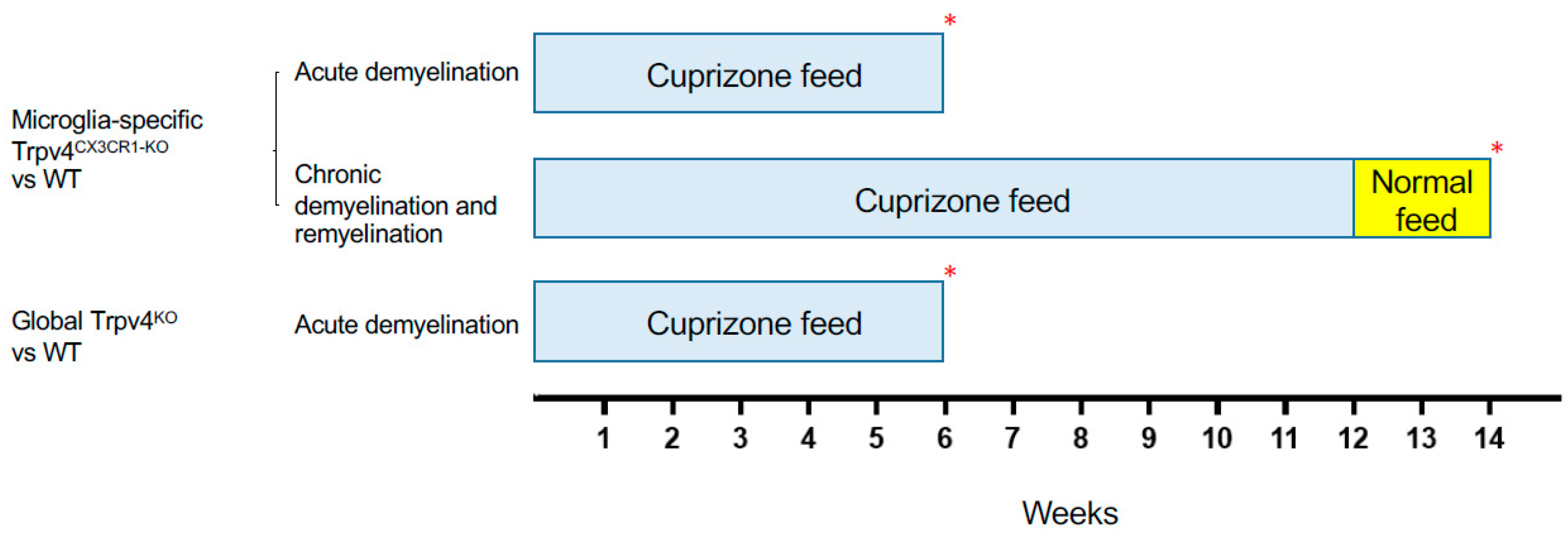
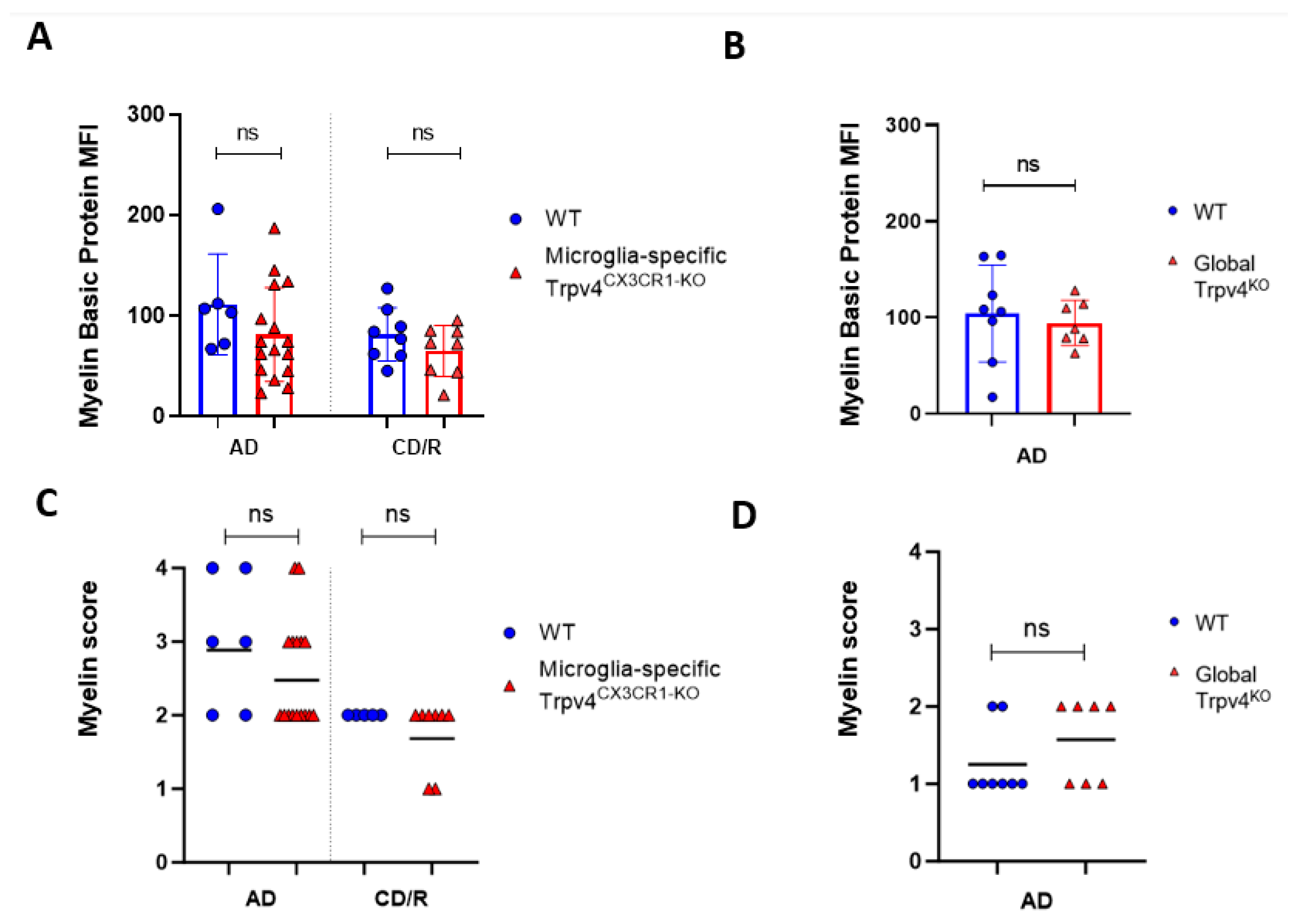
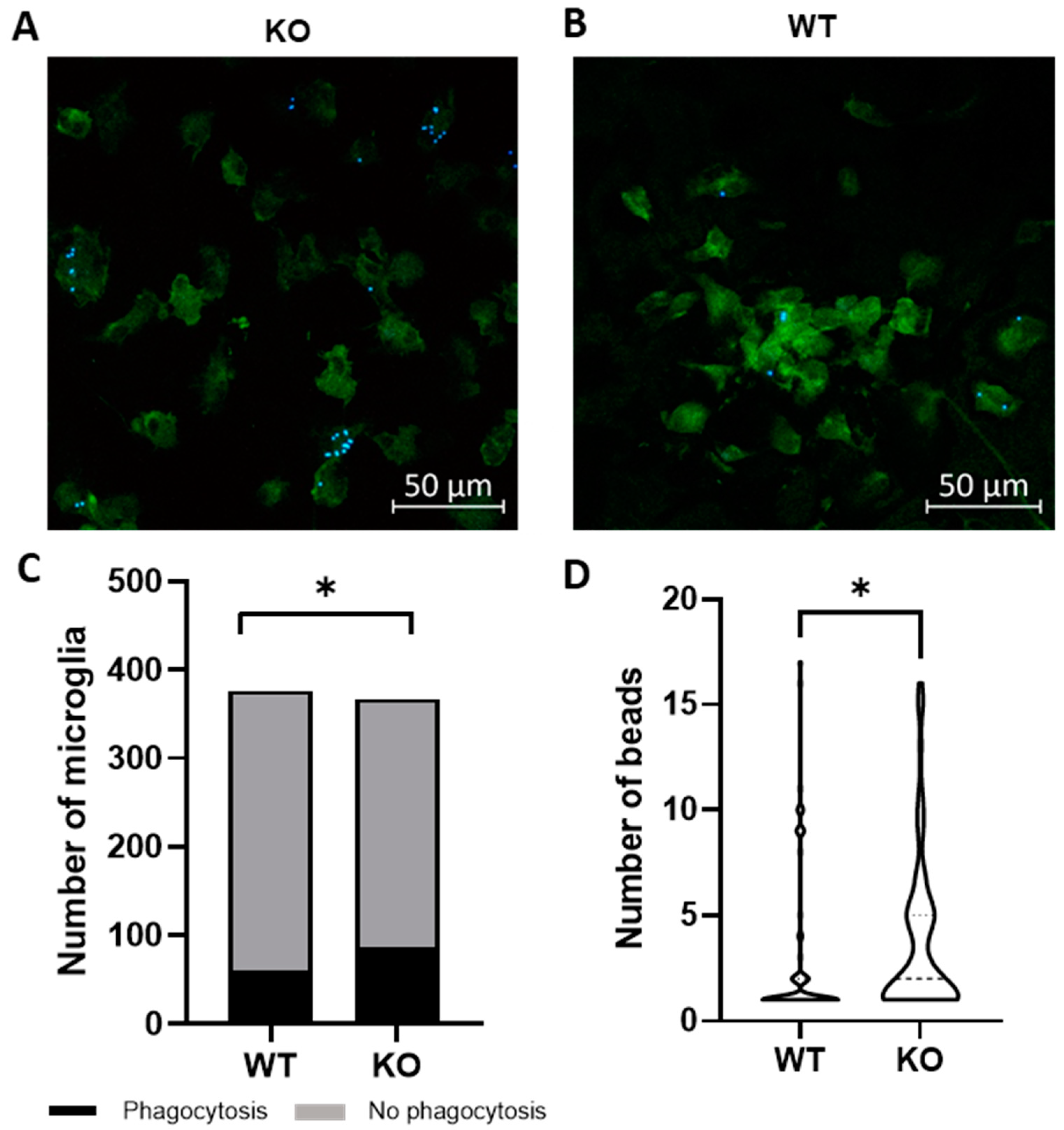
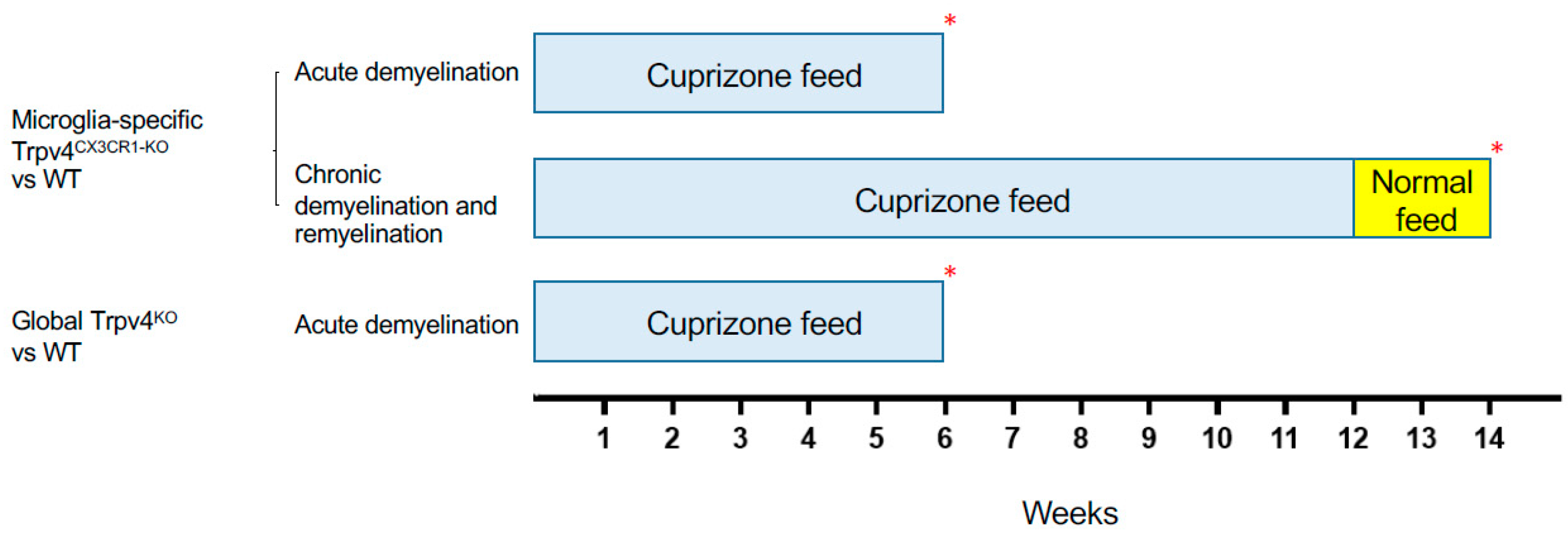
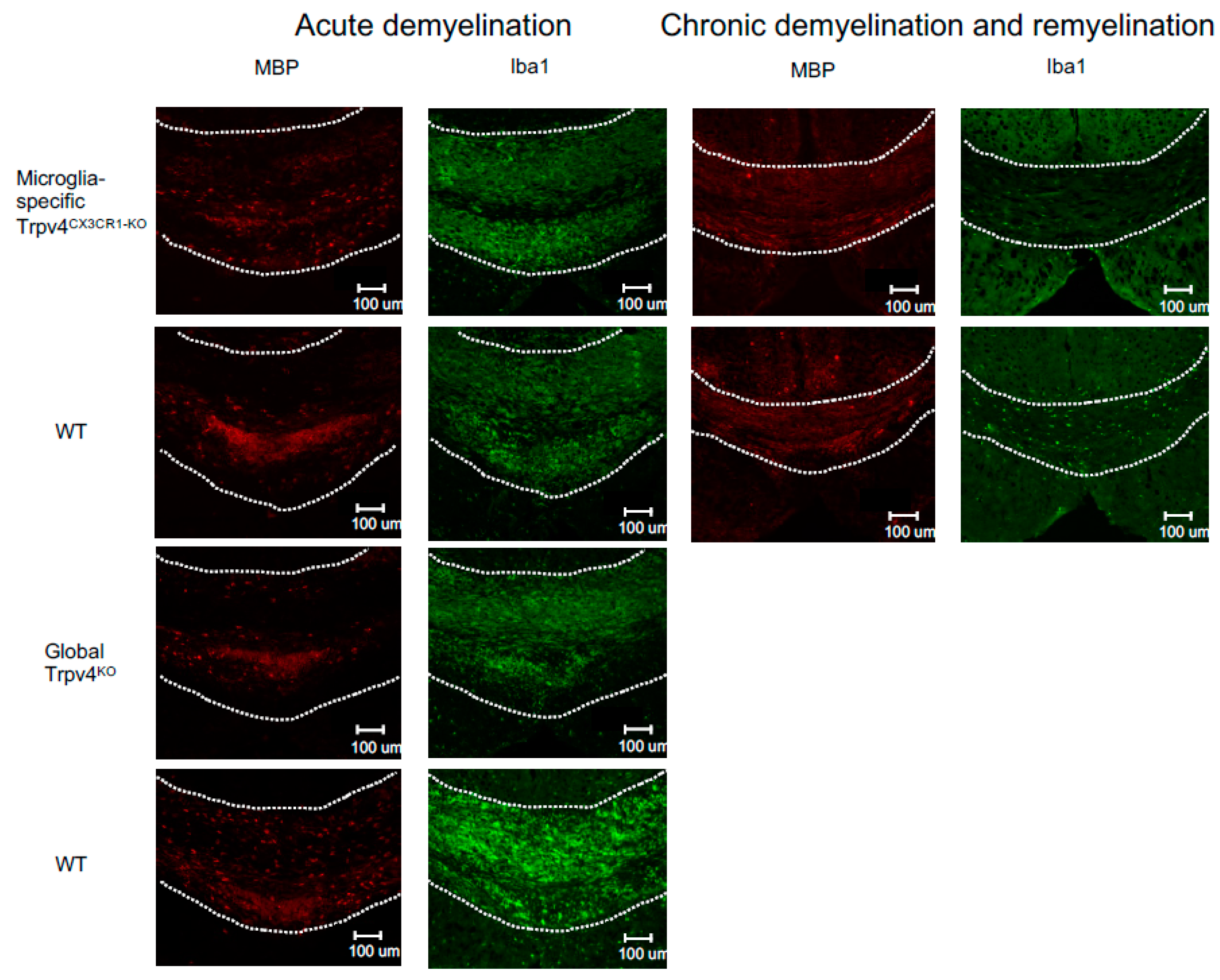
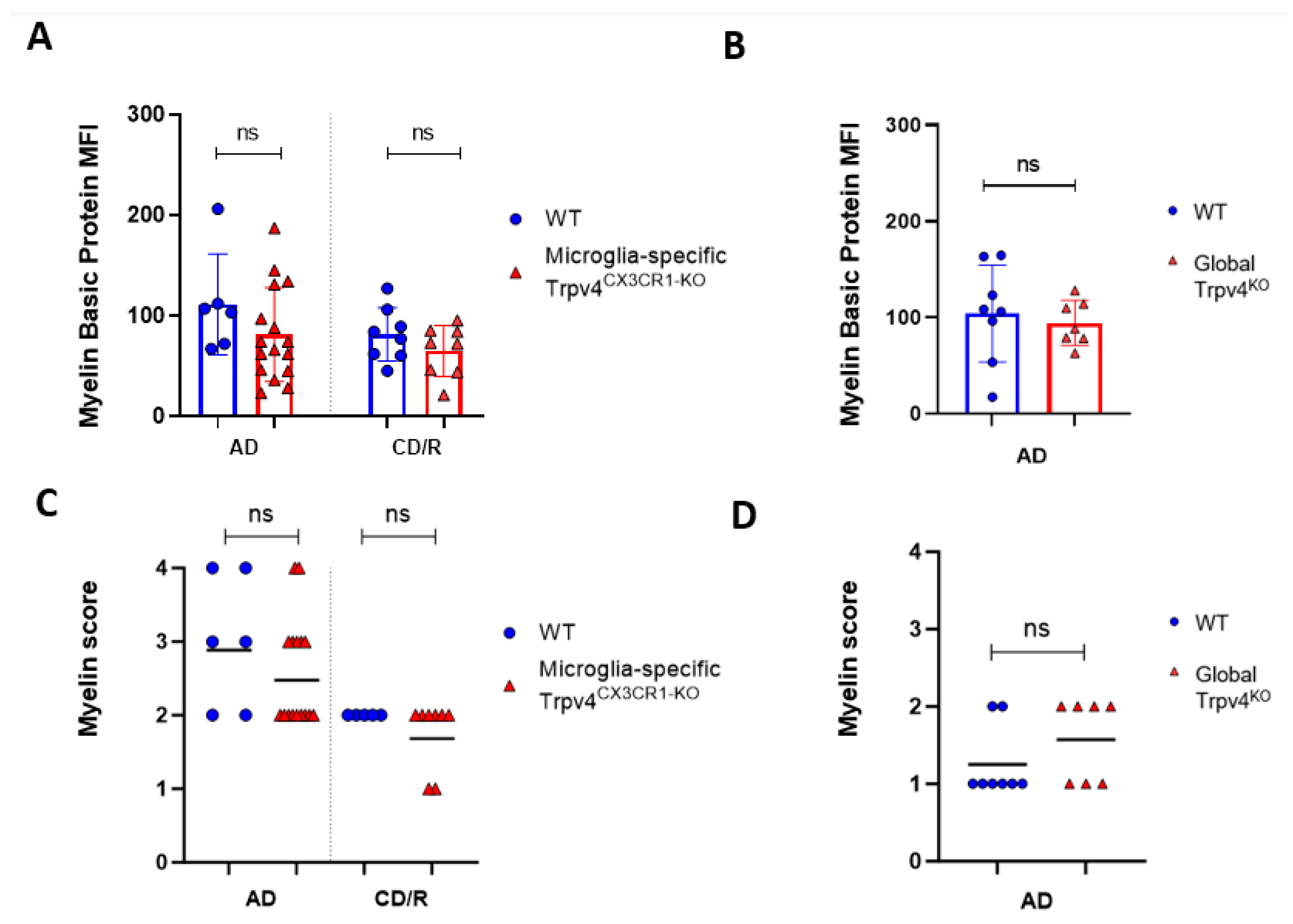
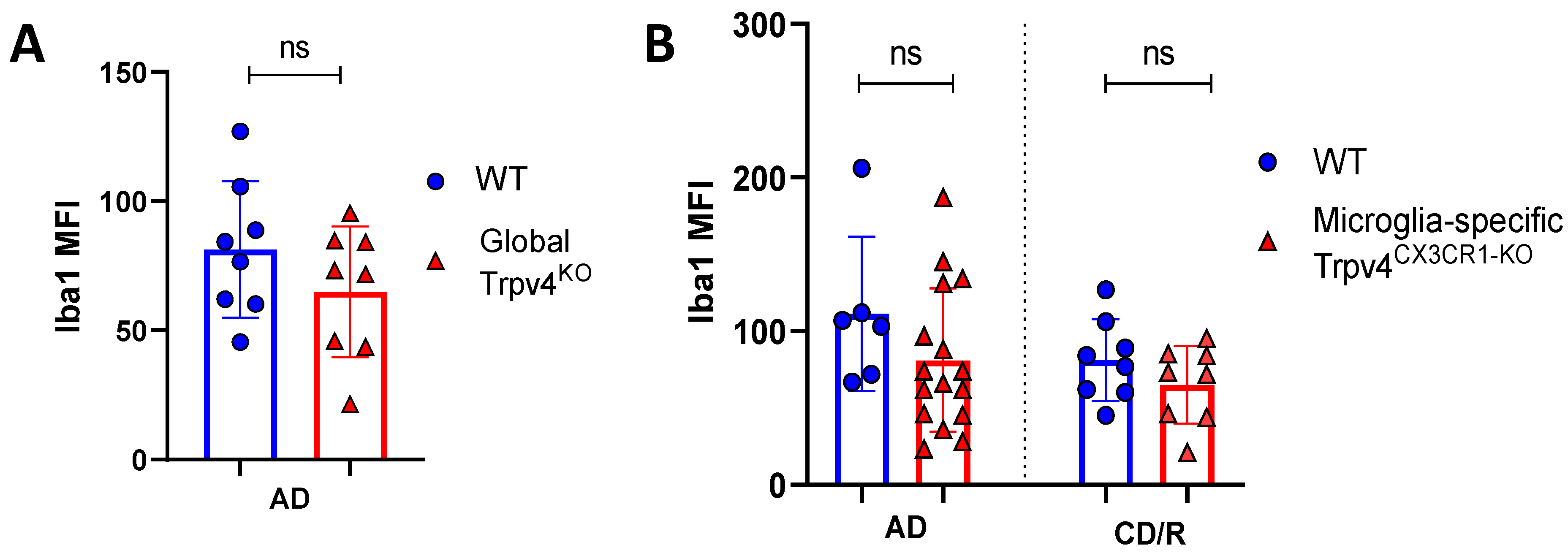
2.1. Trpv4 Deletion in Microglia Leads to Increased In Vitro Phagocytosis but Does Not Alter the Degree of Demyelination, Remyelination, or Microgliosis following Cuprizone Treatment
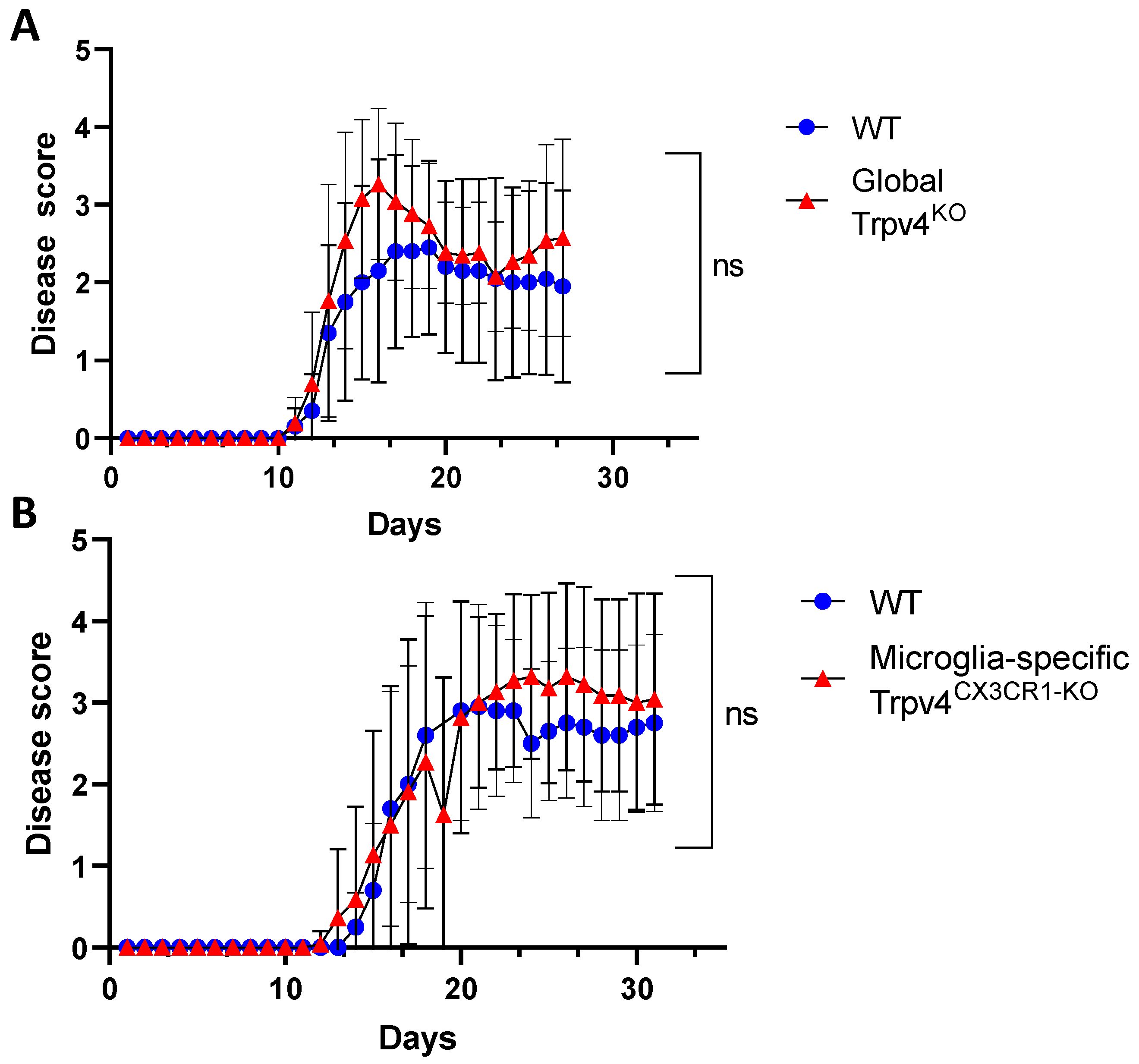
2.2. Global and Microglia-Specific Trpv4 Deletion Does Not Impact EAE
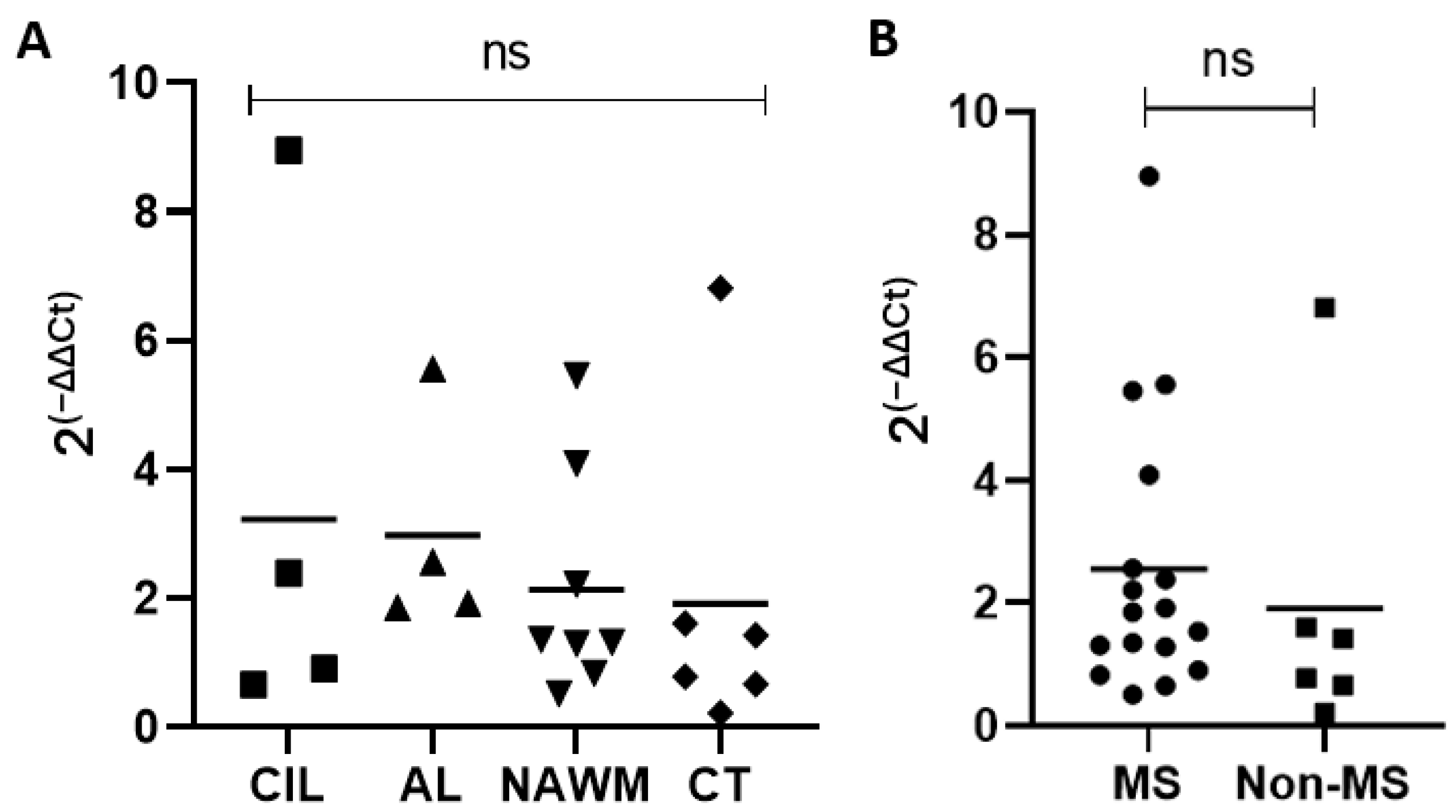
2.3. Expression of TRPV4 in MS Lesions, MS Normal-Appearing White Matter, and Control Tissue
3. Discussion
4. Materials and Methods
4.1. Microglia Cultures
4.2. Microglia Phagocytosis Assay
4.3. Mice
4.4. EAE Induction and Clinical Score Assessment
4.5. Cuprizone-Induced Acute Demyelination and Chronic Demyelination/Remyelination Models
4.6. Immunohistochemistry
4.7. Myelin Content Quantification
4.8. Solochrome Cyanine Staining
4.9. Quantification of TRPV4 Gene Expression in MS Brain Tissue
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; Van Der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple sclerosis: Pathogenesis, symptoms, diagnoses and cell-based therapy. Cell J. 2017, 19, 1. [Google Scholar] [PubMed]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef]
- Baker, D.; O’Neill, J.; Turk, J. Cytokines in the central nervous system of mice during chronic relapsing experimental allergic encephalomyelitis. Cell. Immunol. 1991, 134, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Sullivan, B.; Szabo, S.J.; Sobel, R.A.; Glimcher, L.H.; Kuchroo, V.K. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J. Exp. Med. 2004, 200, 79–87. [Google Scholar] [CrossRef]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Höftberger, R.; Berger, T.; Auff, E.; Leutmezer, F.; Trattnig, S.; Lassmann, H.; et al. Slow expansion of multiple sclerosis iron rim lesions: Pathology and 7 T magnetic resonance imaging. Acta Neuropathol. 2017, 133, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.; McGeer, P. Cellular immune response in multiple sclerosis plaques. Am. J. Pathol. 1990, 137, 575. [Google Scholar]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Singh, S.; Metz, I.; Amor, S.; van der Valk, P.; Stadelmann, C.; Brück, W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol. 2013, 125, 595–608. [Google Scholar] [CrossRef]
- Ramaglia, V.; Hughes, T.R.; Donev, R.M.; Ruseva, M.M.; Wu, X.; Huitinga, I.; Baas, F.; Neal, J.W.; Morgan, B.P. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 965–970. [Google Scholar] [CrossRef]
- Neumann, H.; Kotter, M.; Franklin, R. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2009, 132, 288–295. [Google Scholar] [CrossRef]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.-M.; Sánchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef]
- Kanju, P.; Liedtke, W. Pleiotropic function of TRPV4 ion channels in the central nervous system. Exp. Physiol. 2016, 101, 1472–1476. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Cibelli, M.; Urban, L.; Nilius, B.; McGeown, J.G.; Nagy, I. TRPV4: Molecular conductor of a diverse orchestra. Physiol. Rev. 2016, 96, 911–973. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, P.; Poole, D.P.; Veldhuis, N.A. Role of nonneuronal TRPV4 signaling in inflammatory processes. Adv. Pharmacol. 2017, 79, 117–139. [Google Scholar] [PubMed]
- Rosenkranz, S.C.; Shaposhnykov, A.; Schnapauff, O.; Epping, L.; Vieira, V.; Heidermann, K.; Schattling, B.; Tsvilovskyy, V.; Liedtke, W.; Meuth, S.G.; et al. TRPV4-mediated regulation of the blood brain barrier is abolished during inflammation. Front. Cell Dev. Biol. 2020, 8, 849. [Google Scholar] [CrossRef]
- Liedtke, W. TRPV4 plays an evolutionary conserved role in the transduction of osmotic and mechanical stimuli in live animals. J. Physiol. 2005, 567, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Stampanoni Bassi, M.; Gentile, A.; Iezzi, E.; Zagaglia, S.; Musella, A.; Simonelli, I.; Gilio, L.; Furlan, R.; Finardi, A.; Marfia, G.A.; et al. Transient receptor potential vanilloid 1 modulates central inflammation in multiple sclerosis. Front. Neurol. 2019, 10, 30. [Google Scholar] [CrossRef]
- Redmon, S.N.; Yarishkin, O.; Lakk, M.; Jo, A.; Mustafić, E.; Tvrdik, P.; Križaj, D. TRPV4 channels mediate the mechanoresponse in retinal microglia. Glia 2021, 69, 1563–1582. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, L.; An, D.; Xu, W.; Wu, C.; Sha, S.; Li, Y.; Zhu, Y.; Chen, A.; Du, Y.; et al. TRPV4-induced inflammatory response is involved in neuronal death in pilocarpine model of temporal lobe epilepsy in mice. Cell Death Dis. 2019, 10, 386. [Google Scholar] [CrossRef]
- Luo, J.; Feng, J.; Yu, G.; Yang, P.; Mack, M.R.; Du, J.; Yu, W.; Qian, A.; Zhang, Y.; Liu, S.; et al. Transient receptor potential vanilloid 4–expressing macrophages and keratinocytes contribute differentially to allergic and nonallergic chronic itch. J. Allergy Clin. Immunol. 2018, 141, 608–619.e7. [Google Scholar] [CrossRef]
- Liu, M.; Liu, X.; Wang, L.; Wang, Y.; Dong, F.; Wu, J.; Qu, X.; Liu, Y.; Liu, Z.; Fan, H.; et al. TRPV4 inhibition improved myelination and reduced glia reactivity and inflammation in a cuprizone-induced mouse model of demyelination. Front. Cell. Neurosci. 2018, 12, 392. [Google Scholar] [CrossRef]
- Almolda, B.; González, B.; Castellano, B. Activated microglial cells acquire an immature dendritic cell phenotype and may terminate the immune response in an acute model of EAE. J. Neuroimmunol. 2010, 223, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2010, 24, 641–651. [Google Scholar] [CrossRef]
- Konno, M.; Shirakawa, H.; Iida, S.; Sakimoto, S.; Matsutani, I.; Miyake, T.; Kageyama, K.; Nakagawa, T.; Shibasaki, K.; Kaneko, S. Stimulation of transient receptor potential vanilloid 4 channel suppresses abnormal activation of microglia induced by lipopolysaccharide. Glia 2012, 60, 761–770. [Google Scholar] [CrossRef]
- Peferoen, L.A.; Vogel, D.Y.; Ummenthum, K.; Breur, M.; Heijnen, P.D.; Gerritsen, W.H.; Peferoen-Baert, R.M.; van der Valk, P.; Dijkstra, C.D.; Amor, S. Activation status of human microglia is dependent on lesion formation stage and remyelination in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2015, 74, 48–63. [Google Scholar] [CrossRef]
- Harnisch, K.; Teuber-Hanselmann, S.; Macha, N.; Mairinger, F.; Fritsche, L.; Soub, D.; Meinl, E.; Junker, A. Myelination in multiple sclerosis lesions is associated with regulation of bone morphogenetic protein 4 and its antagonist noggin. Int. J. Mol. Sci. 2019, 20, 154. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Beeken, J.; Mertens, M.; Stas, N.; Kessels, S.; Aerts, L.; Janssen, B.; Mussen, F.; Pinto, S.; Vennekens, R.; Rigo, J.; et al. Acute inhibition of transient receptor potential vanilloid-type 4 cation channel halts cytoskeletal dynamism in microglia. Glia 2022, 70, 2157–2168. [Google Scholar] [CrossRef]
- Scheraga, R.G.; Abraham, S.; Niese, K.A.; Southern, B.D.; Grove, L.M.; Hite, R.D.; McDonald, C.; Hamilton, T.A.; Olman, M.A. TRPV4 mechanosensitive ion channel regulates lipopolysaccharide-stimulated macrophage phagocytosis. J. Immunol. 2016, 196, 428–436. [Google Scholar] [CrossRef]
- Lian, H.; Roy, E.; Zheng, H. Protocol for primary microglial culture preparation. Bio-Protocol 2016, 6, e1989. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Mizuno, A.; Kodaira, K.; Imai, M. Impaired pressure sensation in mice lacking TRPV4. J. Biol. Chem. 2003, 278, 22664–22668. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Du, L.; Liu, S.; Lan, Z.; Zang, K.; Feng, J.; Zhao, Y.; Yang, X.; Xie, Z.; Wang, P.L.; et al. A TRPV4-dependent neuroimmune axis in the spinal cord promotes neuropathic pain. J. Clin. Investig. 2023, 133, e161507. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.F.; Shindler, K.S.; Allenspach, E.J.; Stephen, T.L.; Thomas, H.L.; Mikesell, R.J.; Cross, A.H.; Laufer, T.M. Limited sufficiency of antigen presentation by dendritic cells in models of central nervous system autoimmunity. J. Autoimmun. 2011, 36, 56–64. [Google Scholar] [CrossRef]
- Sachs, H.H.; Bercury, K.K.; Popescu, D.C.; Narayanan, S.P.; Macklin, W.B. A new model of cuprizone-mediated demyelination/remyelination. ASN Neuro 2014, 6, 1759091414551955. [Google Scholar] [CrossRef]
- Lindner, M.; Fokuhl, J.; Linsmeier, F.; Trebst, C.; Stangel, M. Chronic toxic demyelination in the central nervous system leads to axonal damage despite remyelination. Neurosci. Lett. 2009, 453, 120–125. [Google Scholar] [CrossRef]
- Steelman, A.J.; Thompson, J.P.; Li, J. Demyelination and remyelination in anatomically distinct regions of the corpus callosum following cuprizone intoxication. Neurosci. Res. 2012, 72, 32–42. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex/Age | MS Subtype | PMI (Hours) | Cause of Death | Lesion Type |
|---|---|---|---|---|
| M/60 | SPMS | 9 | Respiratory failure | Active |
| F/45 | PPMS | 4 | Pneumonia | Chronic active |
| F/50 | SPMS | 20 | Unknown | Active, chronic inactive |
| M/35 | PPMS | 10 | Unknown | Chronic inactive |
| F/54 | SPMS | 8 | Pneumonia | Chronic inactive |
| F/69 | PPMS | 5 | Metastatic colon cancer | Active |
| F/79 | RRMS | 8 | Pulmonary edema | NAWM, chronic inactive |
| F/95 | SPMS | 9 | Pulmonary edema | NAWM |
| F/41 | RRMS | 12 | Complications from T1D | NAWM |
| F/54 | SPMS | 7 | Pneumonia | Chronic inactive |
| F/86 | PPMS | 12 | Cardiac arrest | NAWM |
| F/66 | Unknown | 29 | Unknown | NAWM |
| F/39 | HC | 4 | CNS lymphoma | NA |
| F/56 | HC | 18 | Myocardial infarction | NA |
| F/69 | HC | 43 | Sepsis | NA |
| M/41 | HC | 24 | Heart failure | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holloman, J.P.; Dimas, S.H.; Archambault, A.S.; Filipello, F.; Du, L.; Feng, J.; Zhao, Y.; Bollman, B.; Piccio, L.; Steelman, A.J.; et al. Transient Receptor Potential Vanilloid 4-Dependent Microglial Function in Myelin Injury and Repair. Int. J. Mol. Sci. 2023, 24, 17097. https://doi.org/10.3390/ijms242317097
Holloman JP, Dimas SH, Archambault AS, Filipello F, Du L, Feng J, Zhao Y, Bollman B, Piccio L, Steelman AJ, et al. Transient Receptor Potential Vanilloid 4-Dependent Microglial Function in Myelin Injury and Repair. International Journal of Molecular Sciences. 2023; 24(23):17097. https://doi.org/10.3390/ijms242317097
Chicago/Turabian StyleHolloman, Jameson P., Sophia H. Dimas, Angela S. Archambault, Fabia Filipello, Lixia Du, Jing Feng, Yonghui Zhao, Bryan Bollman, Laura Piccio, Andrew J. Steelman, and et al. 2023. "Transient Receptor Potential Vanilloid 4-Dependent Microglial Function in Myelin Injury and Repair" International Journal of Molecular Sciences 24, no. 23: 17097. https://doi.org/10.3390/ijms242317097
APA StyleHolloman, J. P., Dimas, S. H., Archambault, A. S., Filipello, F., Du, L., Feng, J., Zhao, Y., Bollman, B., Piccio, L., Steelman, A. J., Hu, H., & Wu, G. F. (2023). Transient Receptor Potential Vanilloid 4-Dependent Microglial Function in Myelin Injury and Repair. International Journal of Molecular Sciences, 24(23), 17097. https://doi.org/10.3390/ijms242317097