1. Introduction
Cystinosis is an autosomal recessive disease resulting from mutations in the
ctns gene that encodes for cystinosin [
1], a member of the Lysosomal Cystine Transporter (LCT) family, with significant homology to Microbial Rhodopsins (MRs) [
2]. Cystinosin is a cystine/proton cotransporter that extrudes cystine from lysosomes [
1]. However, cystine efflux from lysosomes declines, as a consequence of a number of the
ctns mutations that alter cystinosin, and as a consequence, intracellular cystine levels increase to such an extent that cystine crystals form [
3]. Cystinosis has three major clinical forms, based on the age of onset as well as the severity of the disease [
3]. They include infantile cystinosis, with an early onset, juvenile cystinosis, with a late onset, and non-nephropathic cystinosis. Infantile cystinosis (also called nephropathic cystinosis), the most severe form of cystinosis, affects 95% of afflicted individuals, being detected as early as 6 months of age, due to the appearance of the Fanconi Syndrome. Excessive levels of such solutes as sodium, phosphate, bicarbonate, amino acids, and glucose appear in the urine of infants with the Fanconi Syndrome due to the defective reabsorption by the Renal Proximal Tubule (RPT). Consequences include rickets, growth retardation, and end-stage renal failure by around 10 years of age [
3]. Subsequently, other organs are affected. Although cystinosis occurs at a low frequency (1 in 100,000 to 200,000 births), the consequences of the disease are devastating, drastically limiting the patient’s lifespan even when administering therapy (which is expensive).
The most common form of therapy is the administration of cysteamine [
4], a drug that reacts with lysosomal cystine, yielding cysteine and a cysteamine-cystine disulfide, both of which leave the lysosomes, thereby reducing lysosomal cystine levels. Although cysteamine increases the lifespan, the effects of cysteamine are only transient (requiring ingestion every 6 h). In addition, the drug is costly, and there are problems with compliance due to complications caused by the drug (including skin rashes, ulcers, blurry vision, and bad breath). Moreover, cysteamine does not prevent the Fanconi syndrome [
5]. Unfortunately, alternative treatment options are not available. Thus, it is imperative that a further understanding of the molecular consequences of elevated lysosomal cystine is obtained.
In vitro studies utilizing tissue culture models of cystinosis are extremely important in these regards. However, initially, in vitro studies were seriously limited, being conducted with undifferentiated, cystinotic fibroblasts [
6]. Although increased cystine levels are observed in all cells in the body in cystinosis, only specific tissues are affected. Thus, the molecular basis of the defects which occur in the specific tissues affected in cystinosis need to be studied using differentiated tissue culture systems. Cell cultures closely resembling cystinotic renal proximal tubules (RPTs) are particularly important in this regard, if an understanding of the molecular basis of the renal Fanconi syndrome in cystinosis is to be obtained [
1]. Previous studies have indicated that cystinotic RPT cells are more sensitive to oxidative stress and are more likely to undergo apoptosis than normal cells [
7]. However, in vitro model systems that more closely resemble cystinotic RPTs are needed to determine the molecular basis for the changes that occur in cystinotic RPTs, as well as how the different
ctns mutations that cause cystinosis affect renal cells, as well as cells in different organs.
An important means towards achieving this goal is to conduct studies with human-induced pluripotent stem cells (hiPSCs). Not only can hiPSCs be readily derived from human fibroblasts obtained from patients with inherited diseases such as cystinosis [
8], but in addition, mutations can be introduced into hiPSCs using CRISPR technology [
9]. Moreover, hiPSCs can be differentiated into different types of organoids that resemble tissues present in a number of different organs, including the kidney [
10]. Of particular importance, hiPSCs can be differentiated into organoids consisting of RPTs [
11]. Thus, impairments in the RPT in the different forms of cystinosis, including infantile cystinosis, can be studied with differentiated hiPSCs.
In the studies described below, hiPSCs were developed from dermal fibroblasts obtained from an individual with the most common mutation in Western Europeans, a 57 kb deletion that enters the
ctns gene [
12,
13], in parallel with developing hiPSCs from a “normal” individual. Our studies indicate that cystinotic hiPSCs are pluripotent, although alterations are apparent when normal and cystinotic-derived hiPSCs are differentiated into organoids consisting of RPTs.
3. Discussion
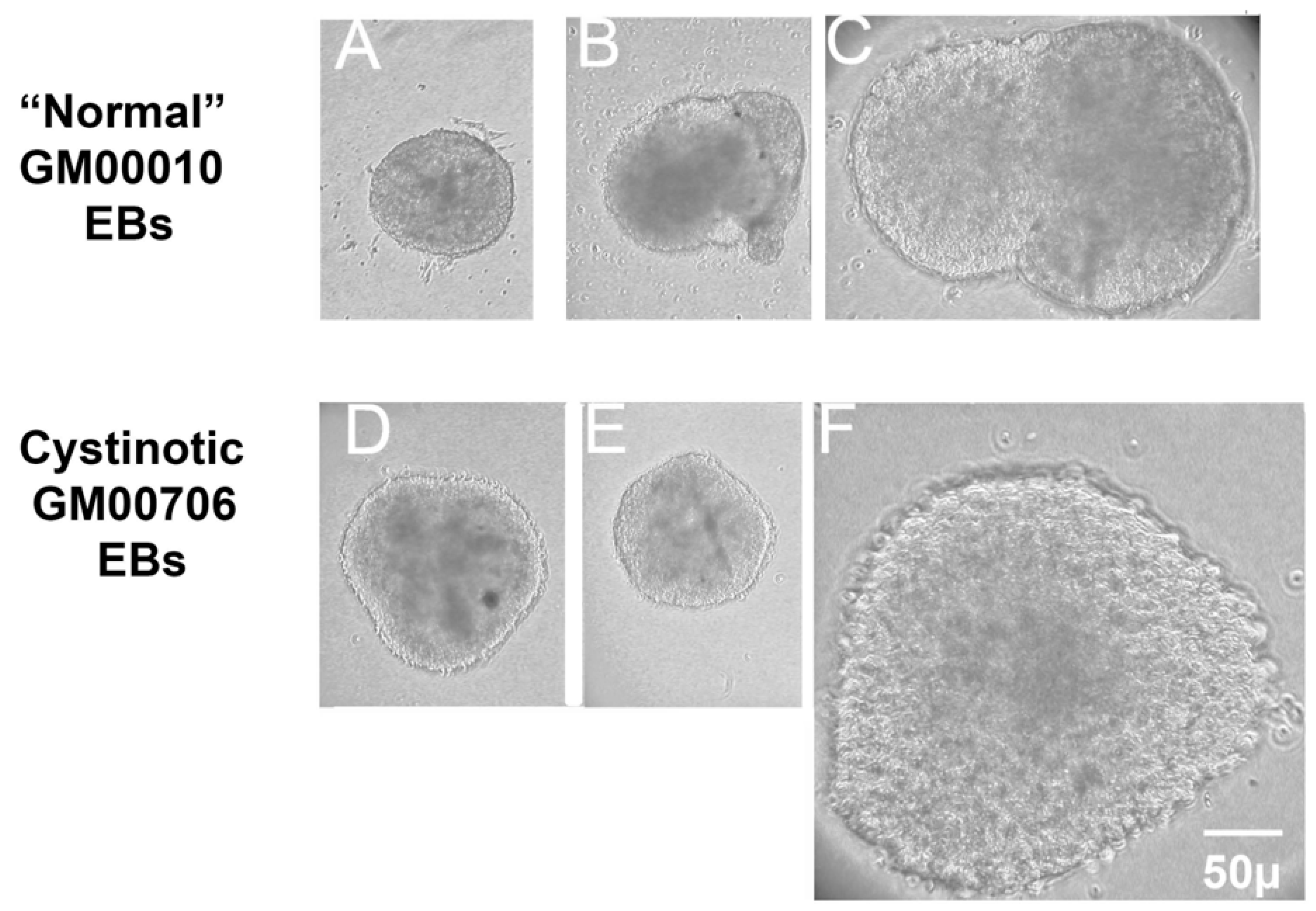
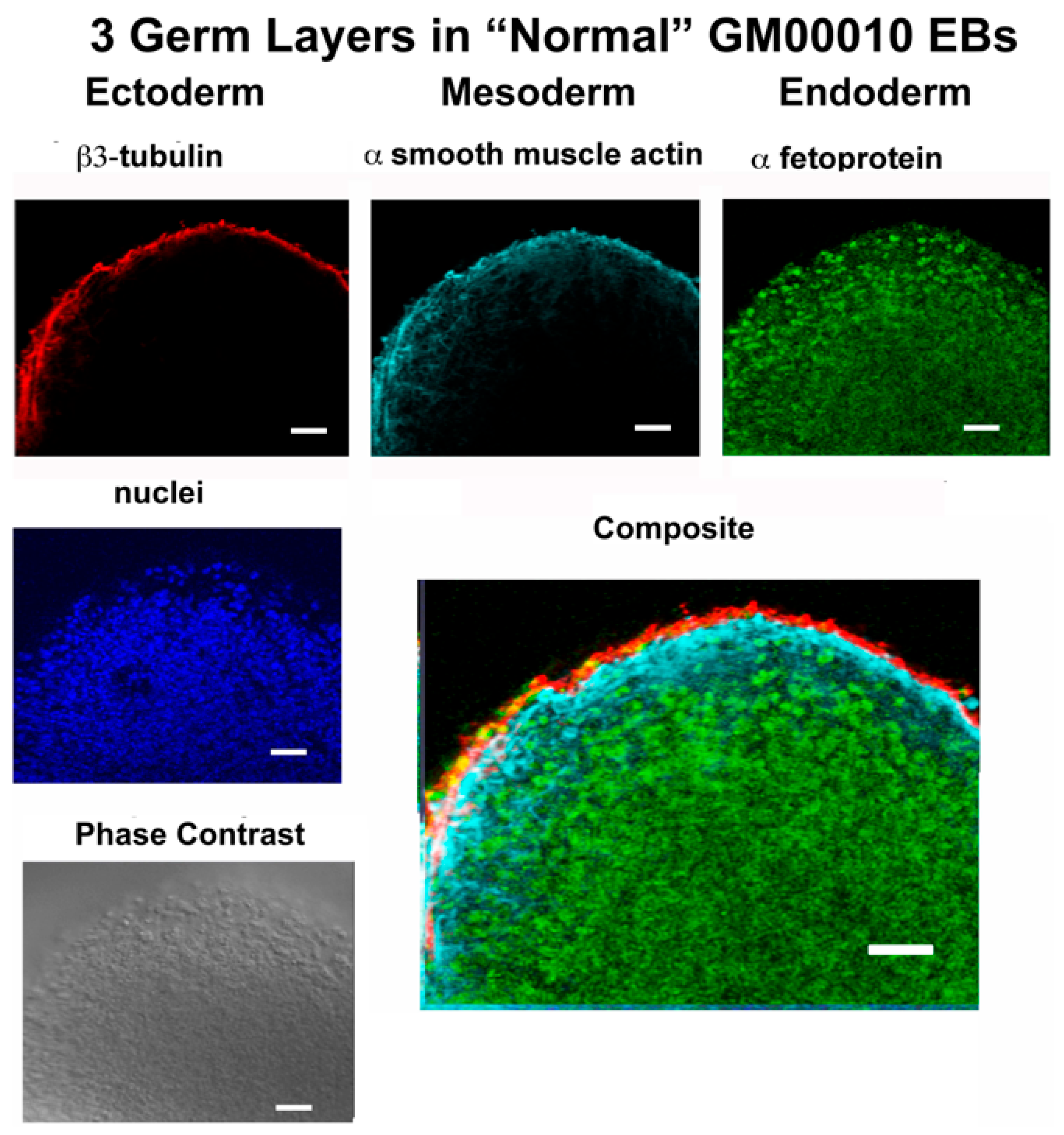
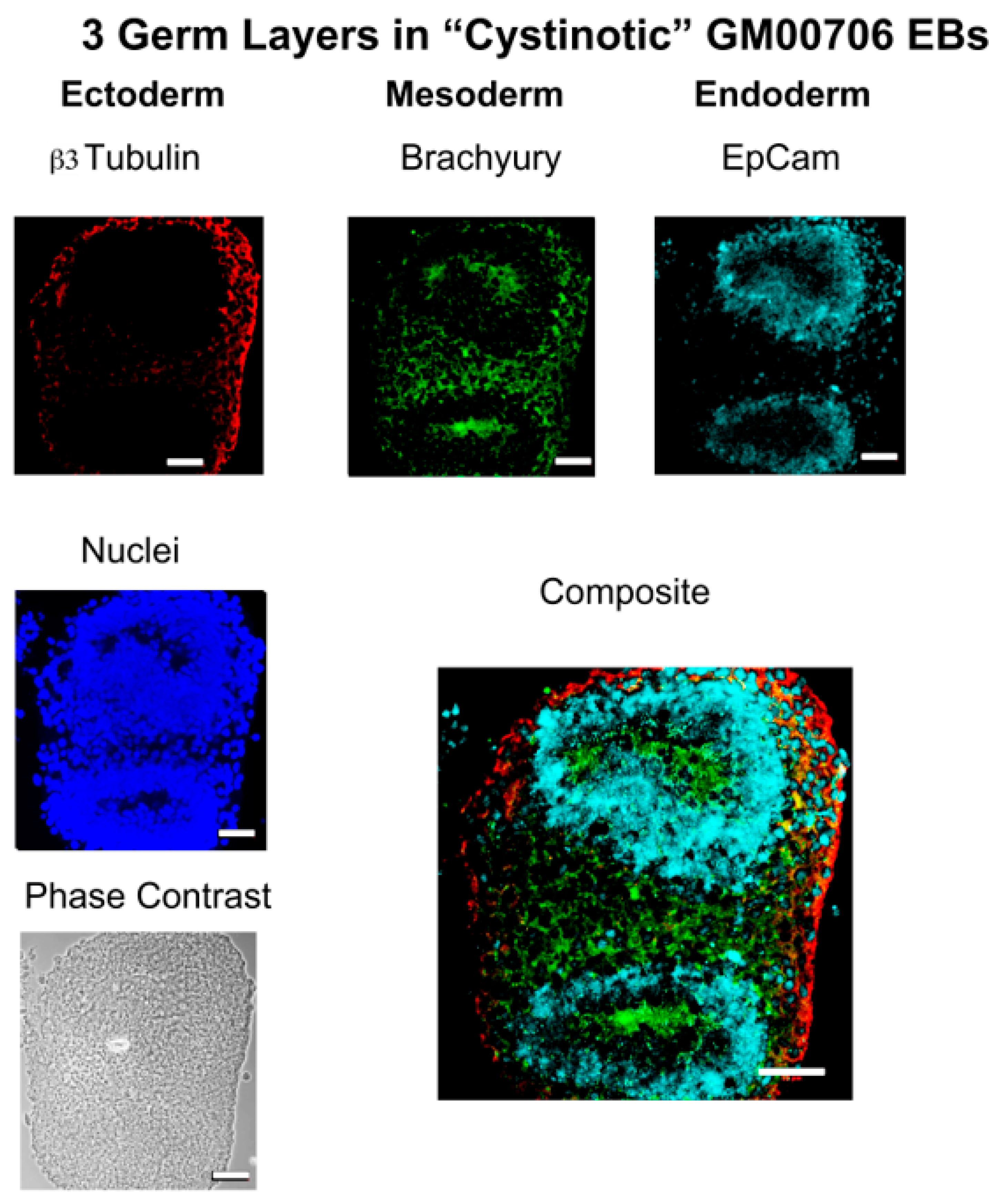
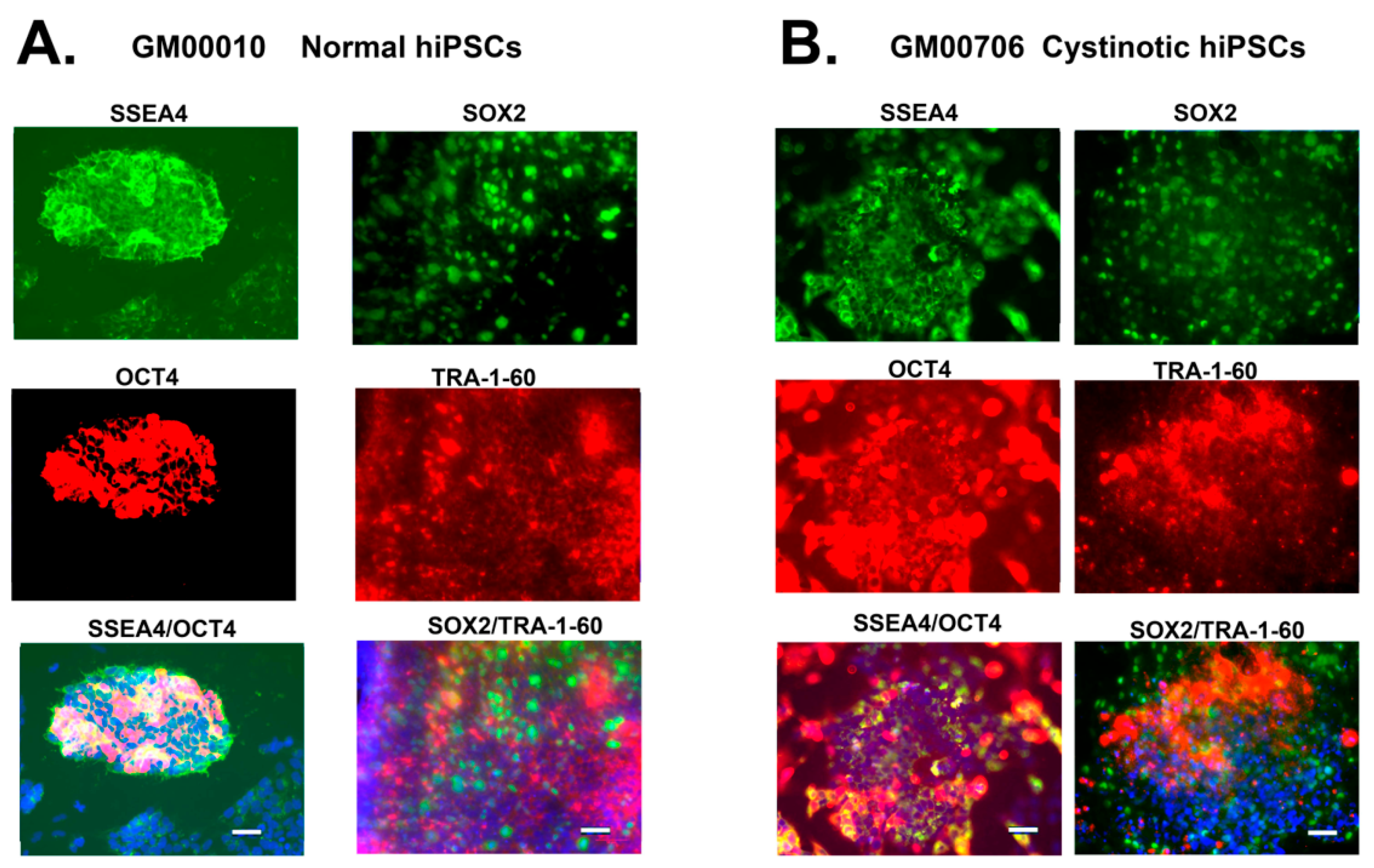
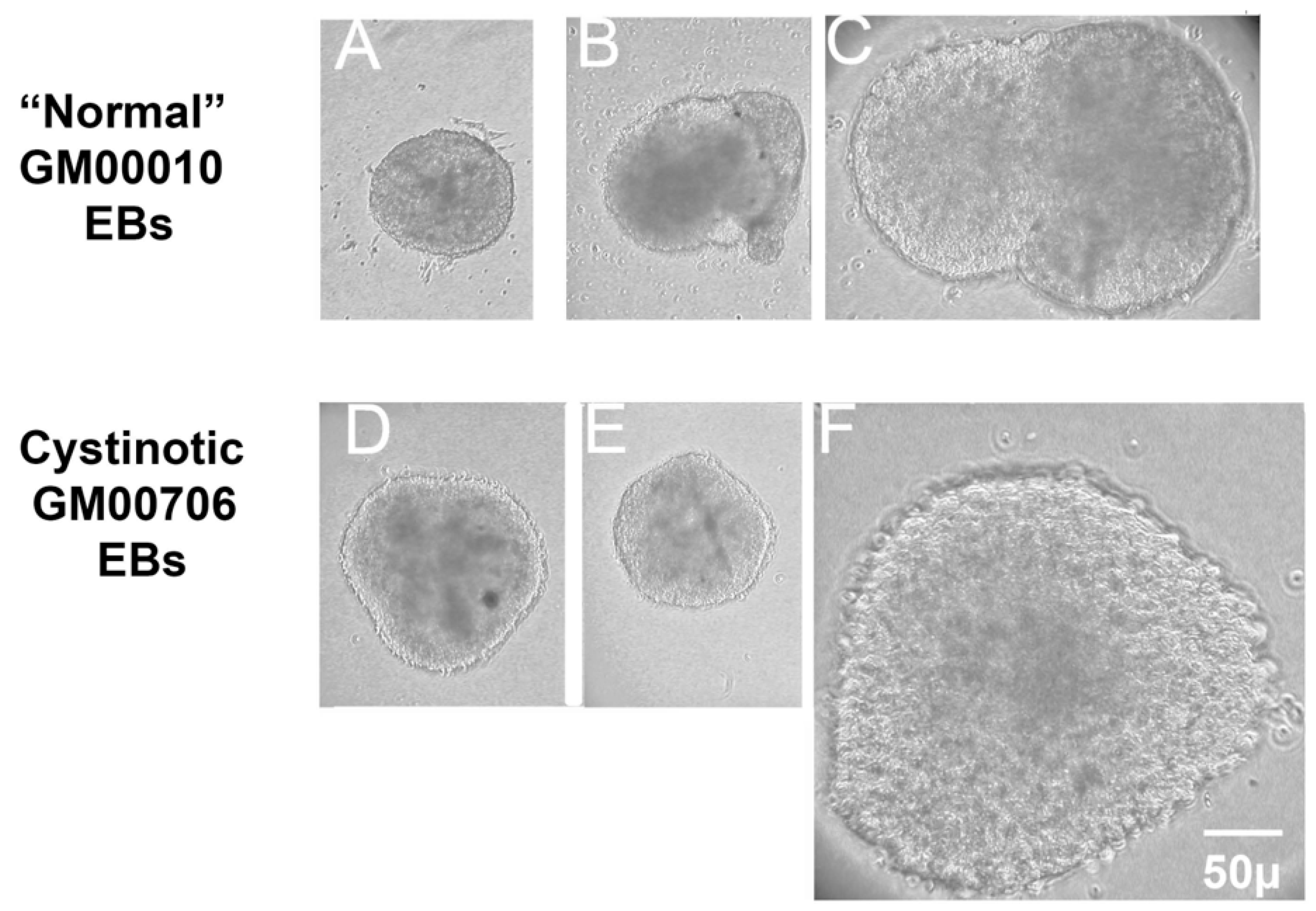
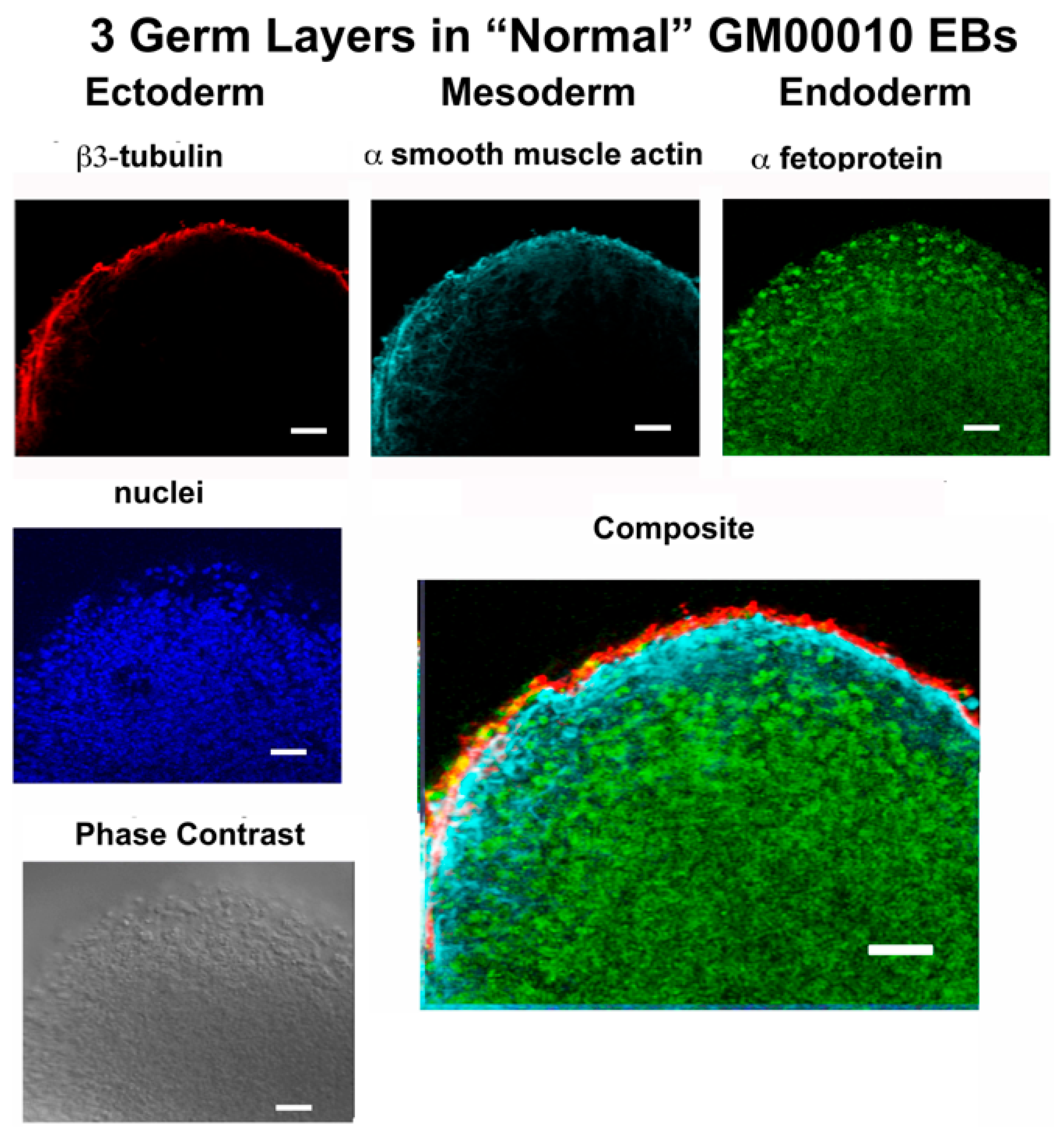
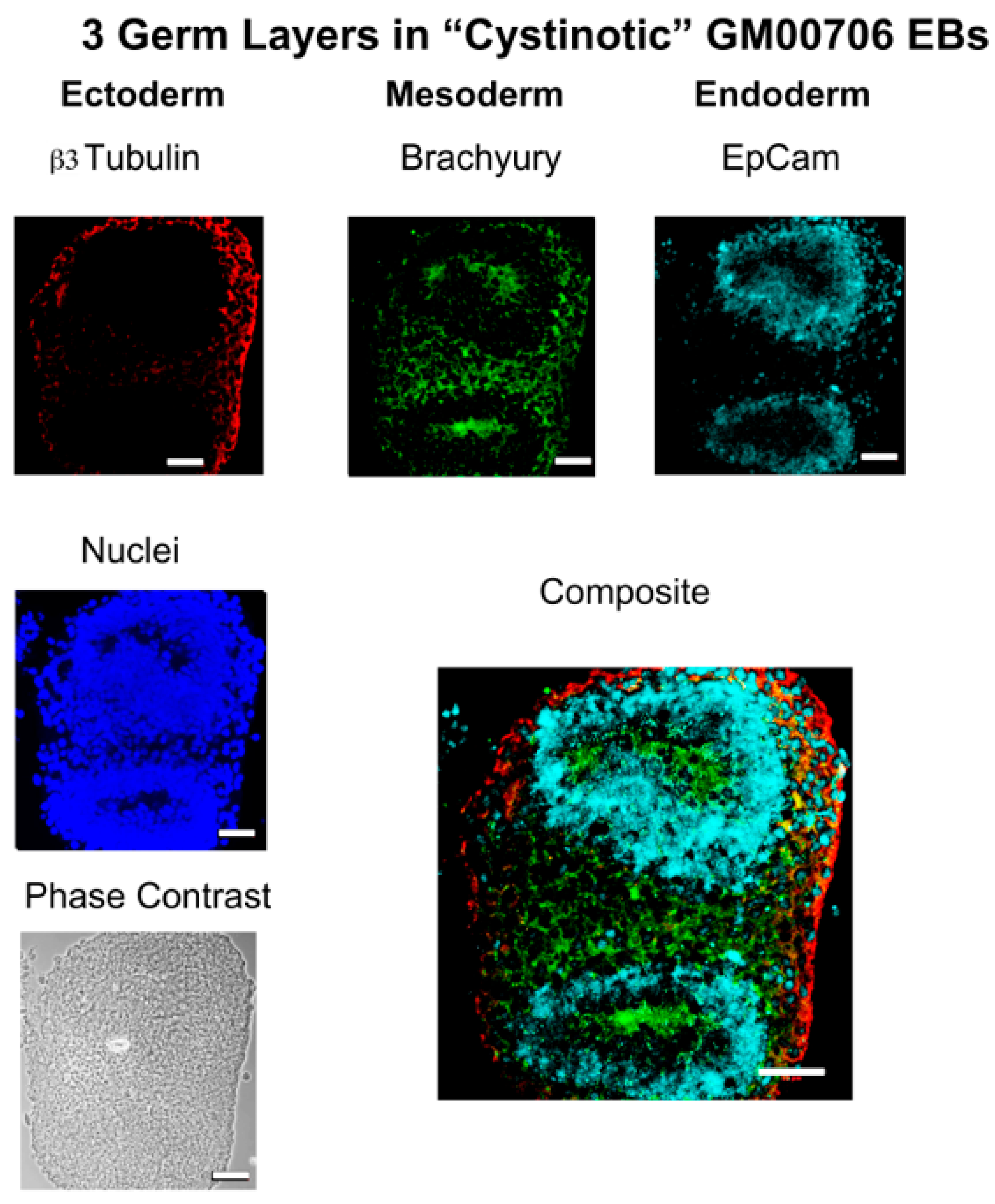
In this report, studies have been conducted with hiPSCs derived from the human dermal fibroblasts of an individual with infantile cystinosis, as well as an unaffected individual. Evidence has been presented indicating that both normal and cystinotic hiPSCs are indeed pluripotent. Not only did the normal and cystinotic hiPSCs exhibit distinctive stem cell markers, but in addition both normal and cystinotic derived hiPSCs were able to form EBs containing the three primordial germ layers, which include ectoderm, mesoderm, and endoderm. Thus, these results indicated that our hiPSCs can differentiate into derivatives of all three germ layers.
In order to examine this hypothesis further, our hiPSCs were subjected to a previously described differentiation procedure [
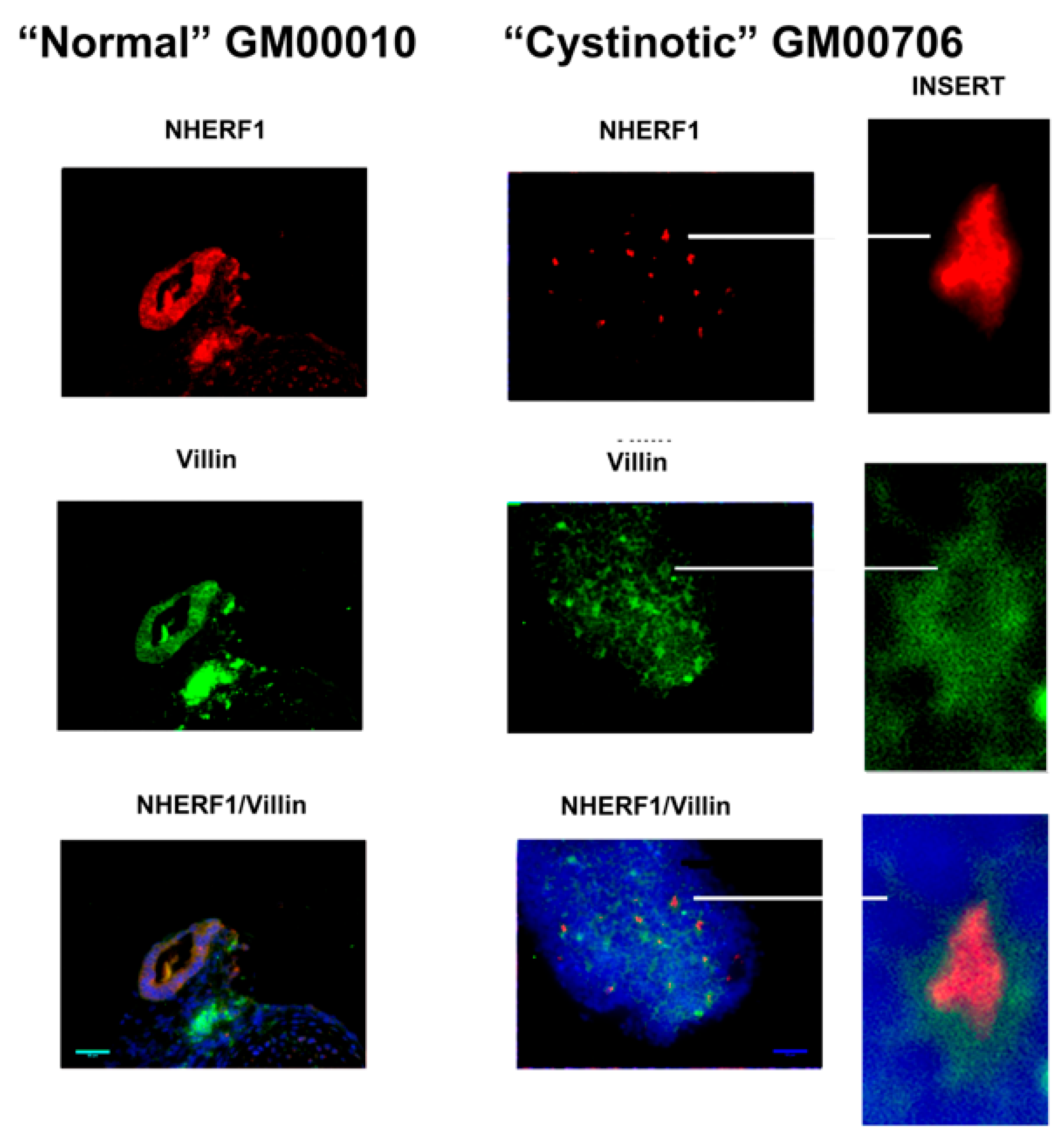
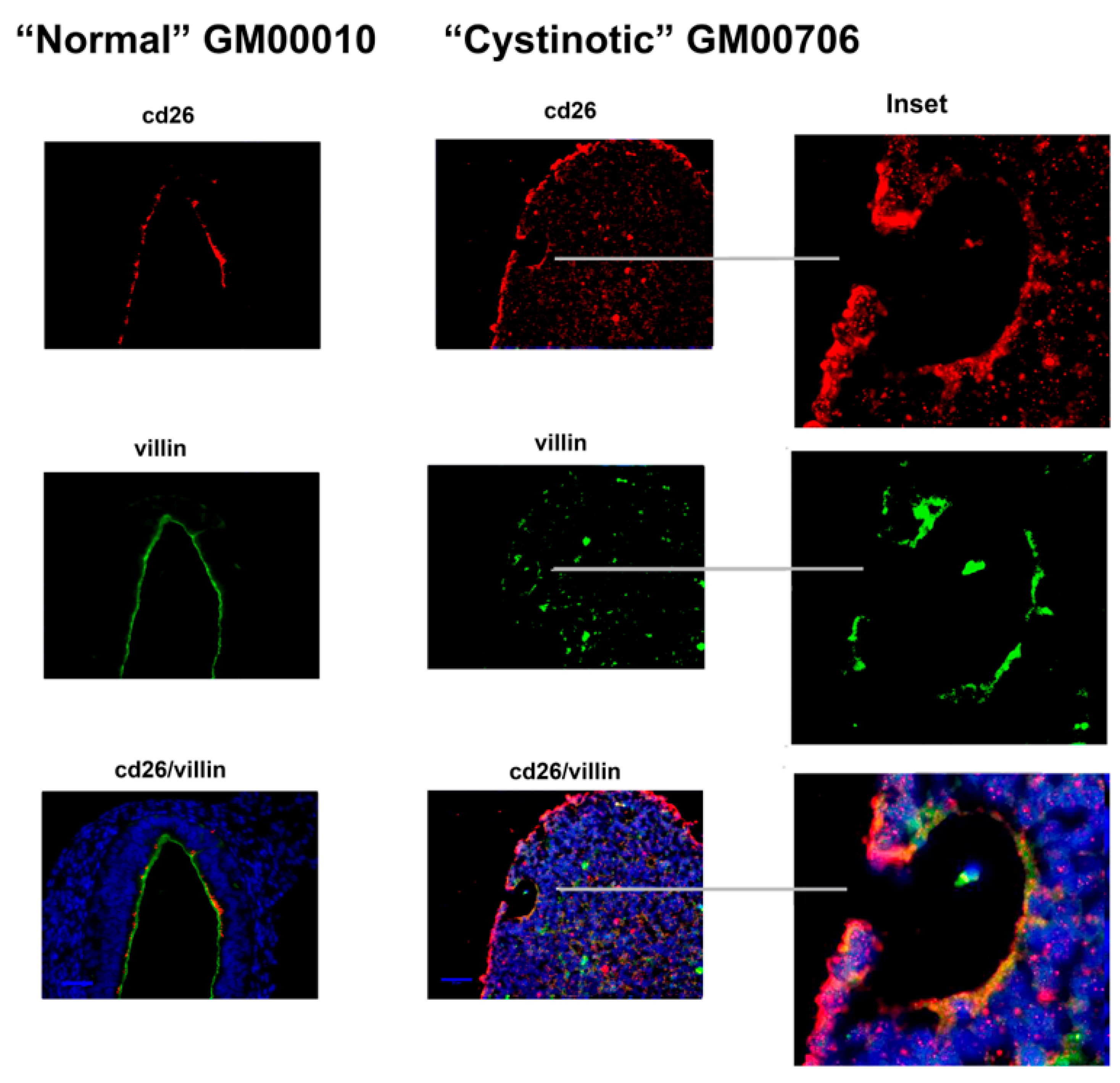
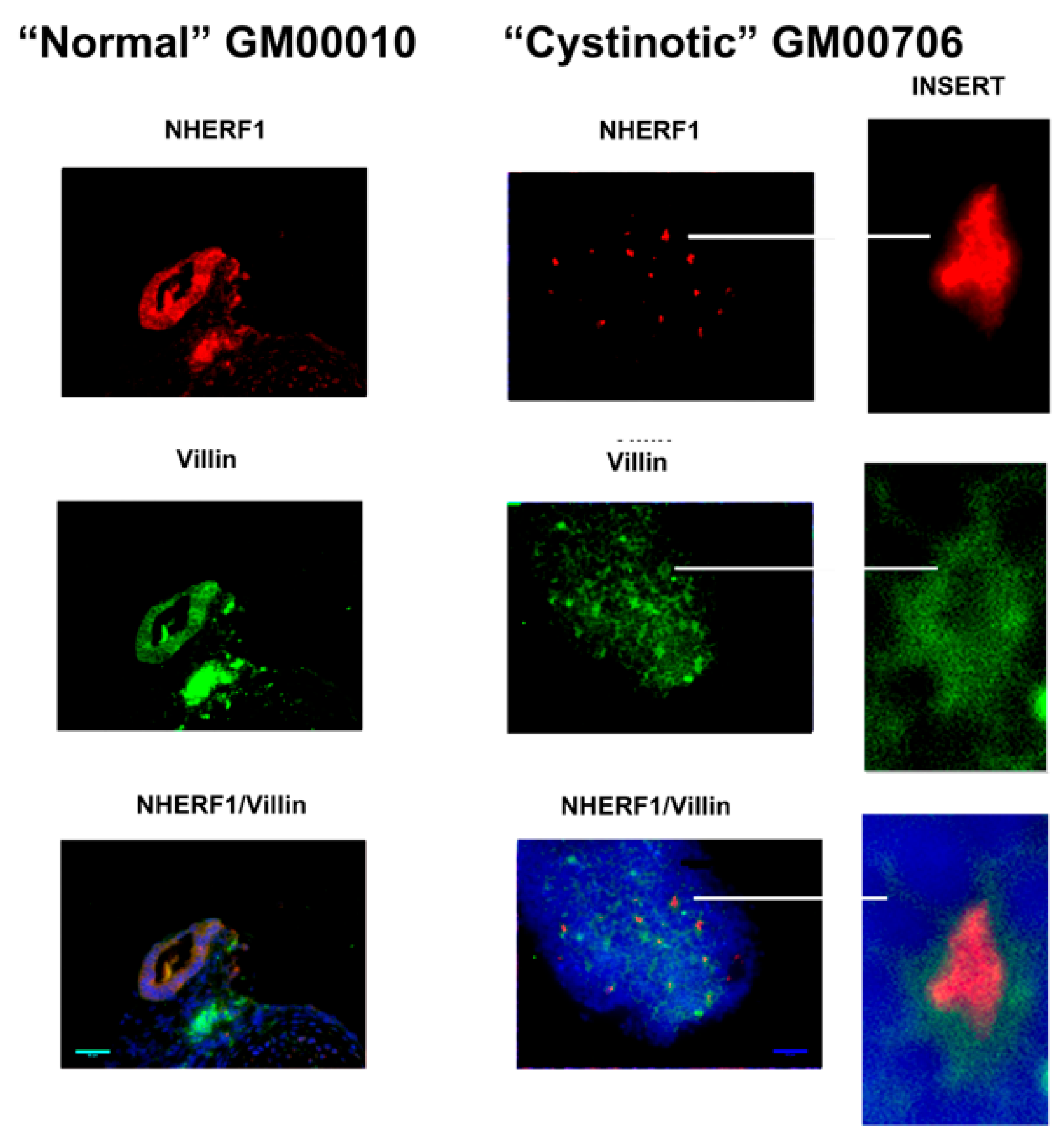
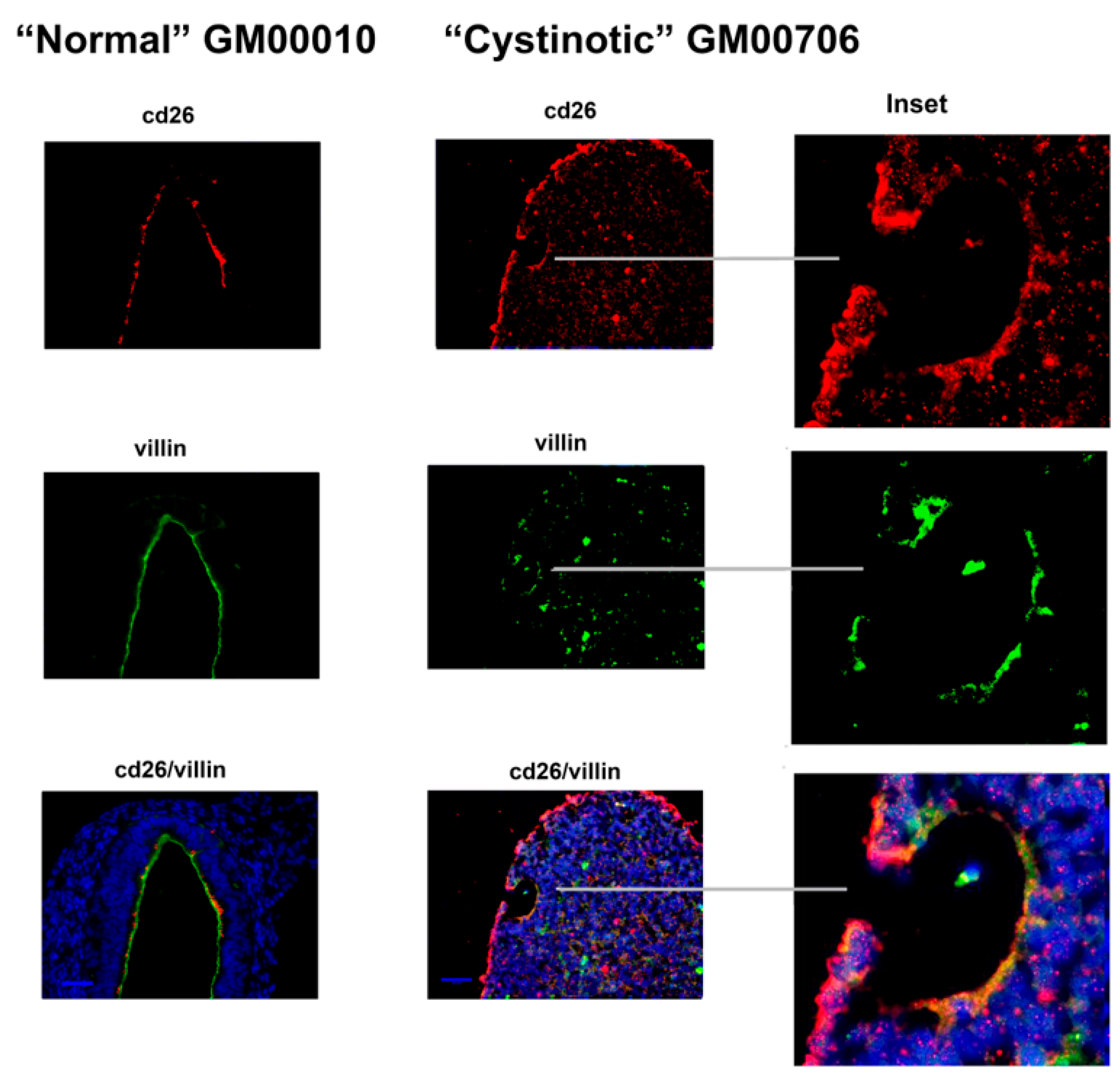
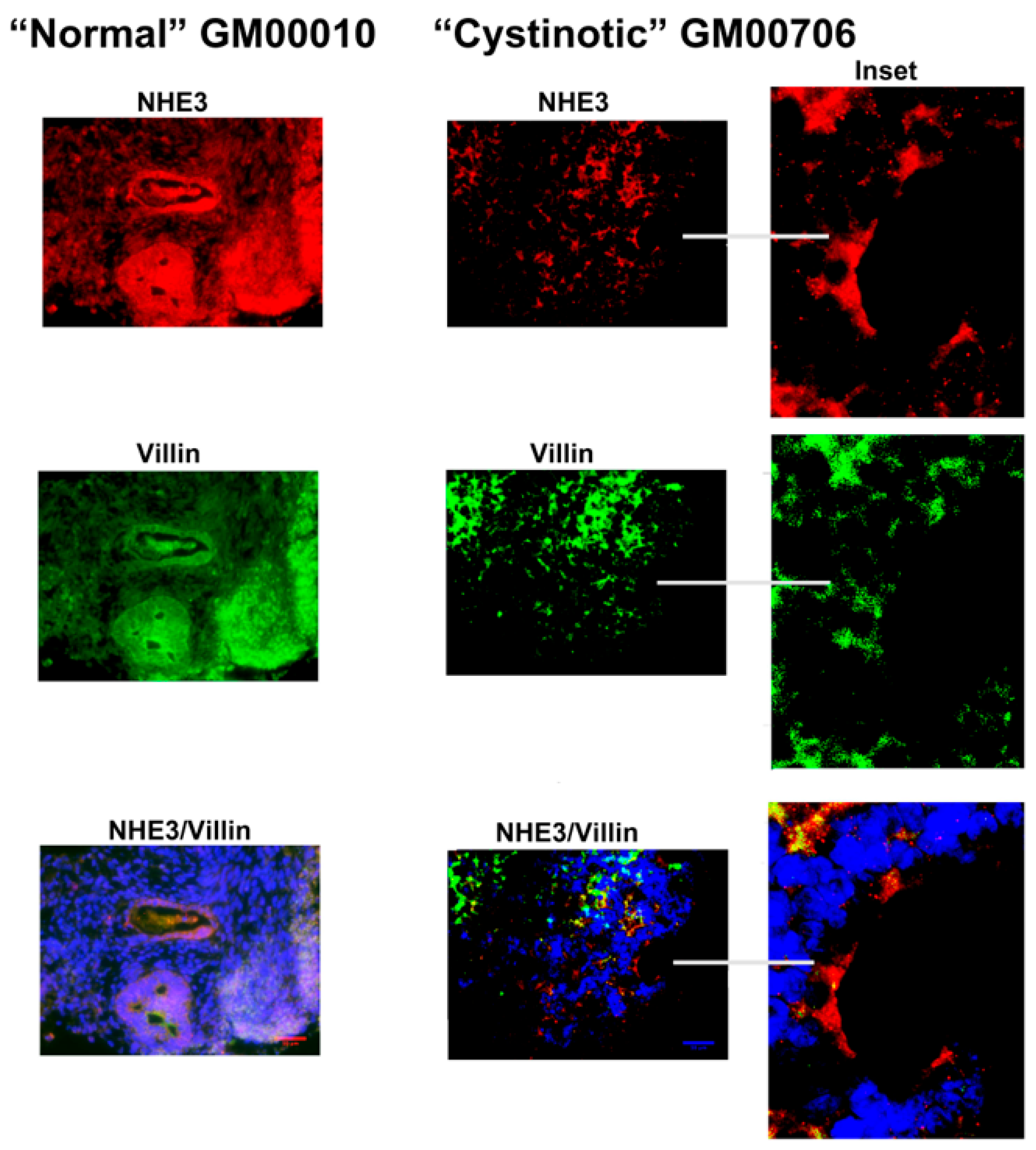
11] that has been shown to induce the formation of RPTs. After following this differentiation procedure, our cultures expressed distinctive apical markers of the RPT, including villin, cd26, NHE3, and NHERF1. In differentiated cultures derived from normal hiPSCs, structures resembling tubules with lumens emerged, and moreover, villin, cd26, NHE3, and NHERF1 were located on a surface facing a luminal space. Lumen formation was less pronounced in the “cystinotic” cultures which had undergone “differentiation”, as compared with differentiated cultures derived from normal hiPSCs. The “differentiated” cultures derived from the cystinotic hiPSCs did nevertheless express distinctive RPT markers, including villin, cd26, NHE3, and NHERF.
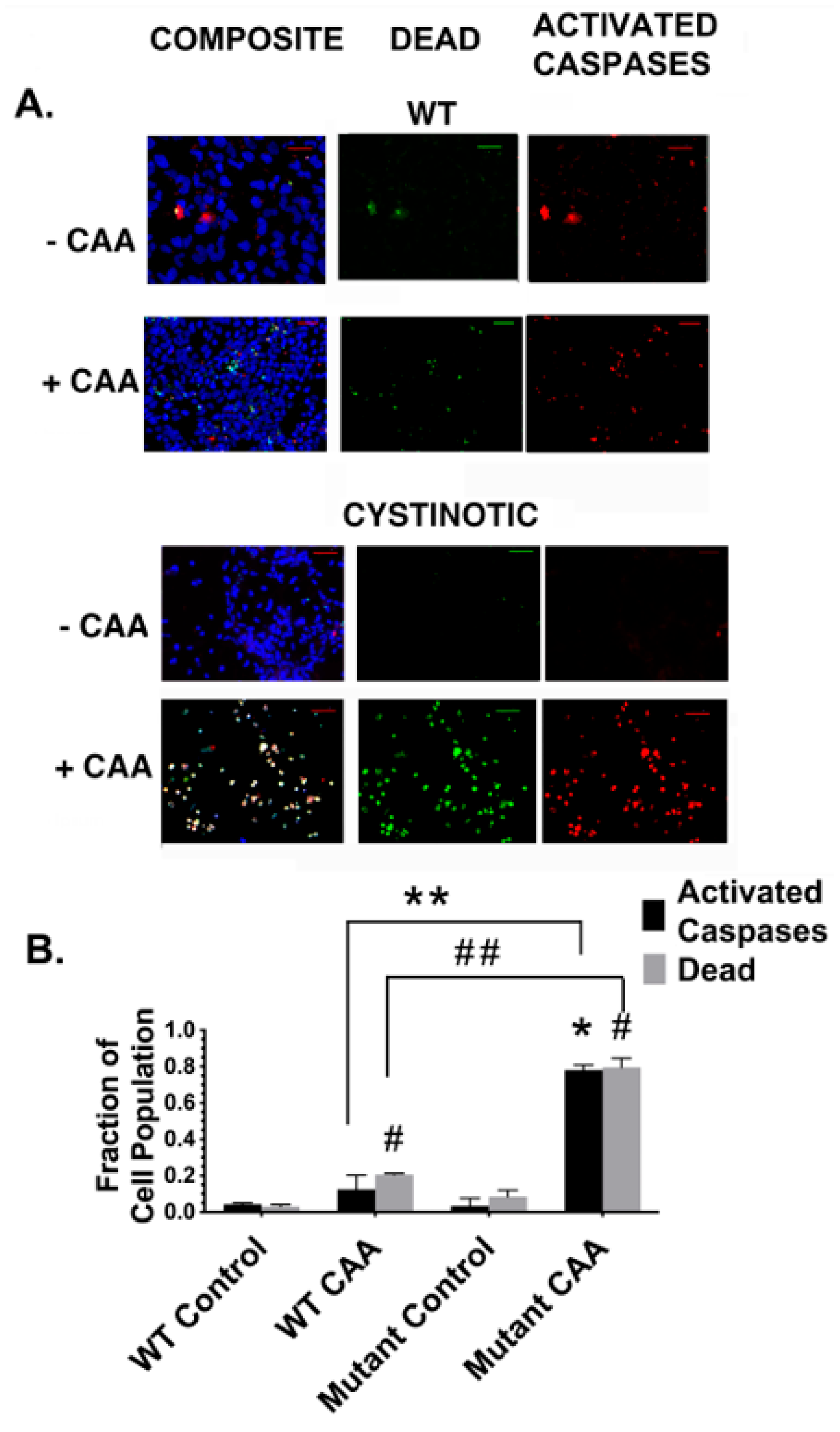
In addition, following their differentiation into putative RPTs, cultures derived from cystinotic derived cells exhibited an increased sensitivity to killing by CAA when compared to differentiated RPTs derived from normal hiPSCs. The studies with CAA are of particular importance, given that CAA is an ifosfamide metabolite that contributes to the Fanconi Syndrome that often develops in children undergoing a cancer chemotherapy regime that utilizes ifosfamide. The increased sensitivity of the differentiated cells derived from cystinotic hiPSCs to killing by CAA is consistent with previous observations regarding apoptosis in cystinotic RPTs made by Park et al. [
7], as well as by our laboratory [
16]. Previously, the cytotoxic effects of CAA on a human RPT cell line (i.e., RPTEC) were associated with an increase in intracellular Ca
2+, while Caspase-3 was not significantly activated, which suggested that necrosis, rather than apoptosis, was involved [
18]. However, an increase in intracellular Ca
2+ has been associated with pyroptosis, a mechanism of programmed cell death which involves Caspase-1 rather than Caspase-3 [
19]. Of particular interest in these regards, recently NLRP3 (Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing 3) was reported as being overexpressed in cystinotic RPT cell cultures [
20]. NLRP3 is an integral component of inflammasomes, which are capable of initiating pyroptosis [
20]. Moreover, evidence has been presented that the elevated cystine in cystinotic patients not only activates inflammasomes, but additionally activates caspase-1 [
21].
In our study, we observed a close correlation between increased caspase activation and cell death in cystinotic RPT organoids treated with CAA, as compared with with normal RPT organoids. Nevertheless, we cannot exclude the possibility that other alterations that have occurred in cystinotic RPT organoids that also contribute to the increase in cell death that occurred in cystinotic RPT organoids treated with CAA. Of particular interest in these regards is autophagy. Autophagy functions to maintain energy metabolism and viability under such conditions as starvation by delivering damaged or potentially harmful cellular components to the lysosomes for digestion and subsequent removal. However, overactivated, autophagy can potentially result in cell death [
22]. Indeed, previously, Sansanwal et al. [
23] observed an increased number of autophagic vacuoles in cultured human cystinotic RPT cells as compared to normal human RPT cells. In addition, an increased level of markers of autophagosomes was observed, including LC3-II and beclin-1. These authors suggested that increased autophagy contributed to the increased apoptosis observed in their cystinotic cultures. However, Napolitano et al. [
24] observed that chaperone-mediated autophagy was defective in their cystinotic RPT cells rather than general macroautophagic flux.
Further studies need to be conducted to resolve these issues. Renal organoids are a powerful tool for this purpose, given that (a) 3-dimensional (3D) culture systems more closely resemble cell in vivo than 2D systems, and (b) they have been derived from hiPSCs, which in turn are readily developed from dermal fibroblasts of individuals with inherited diseases.
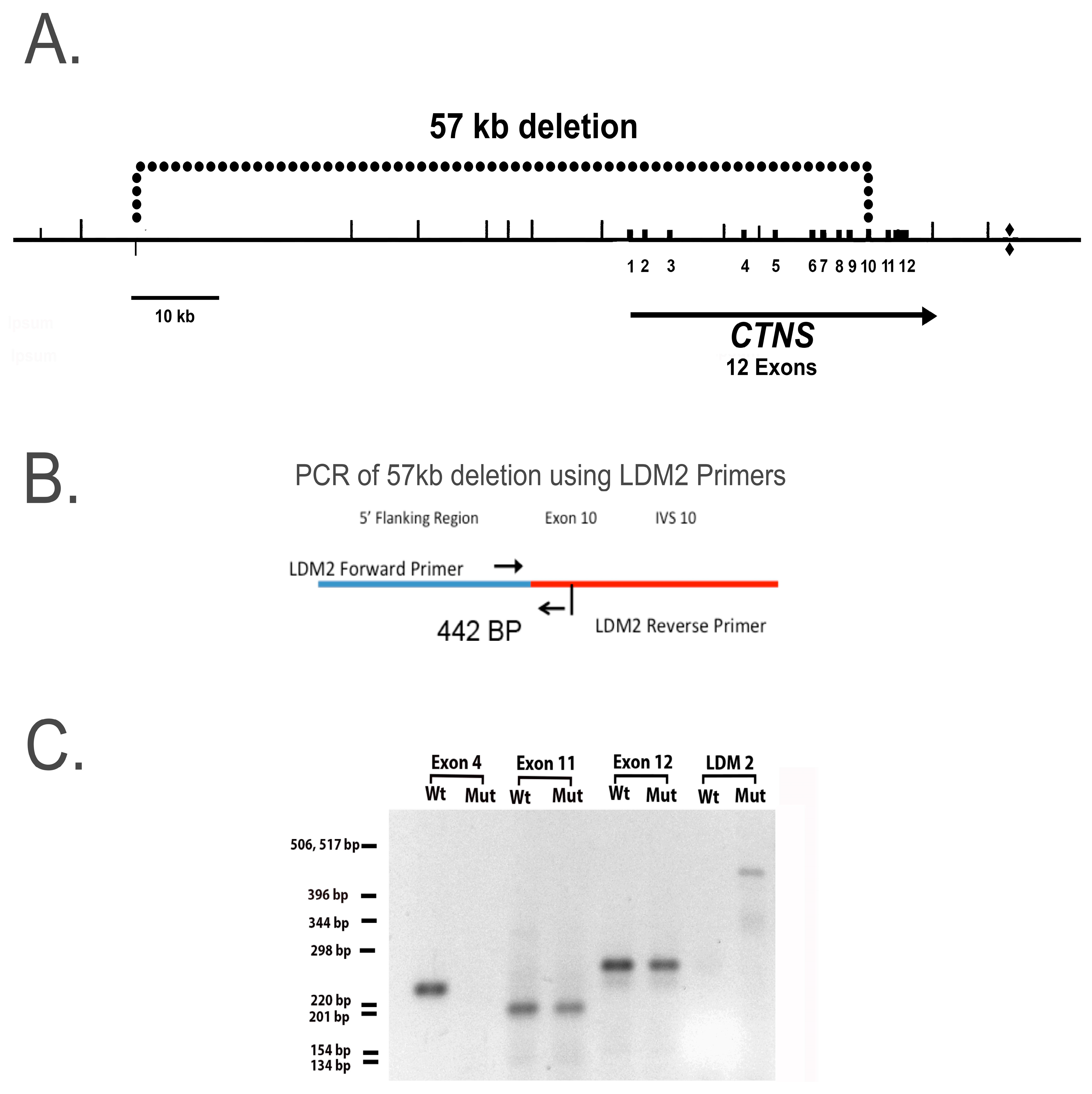
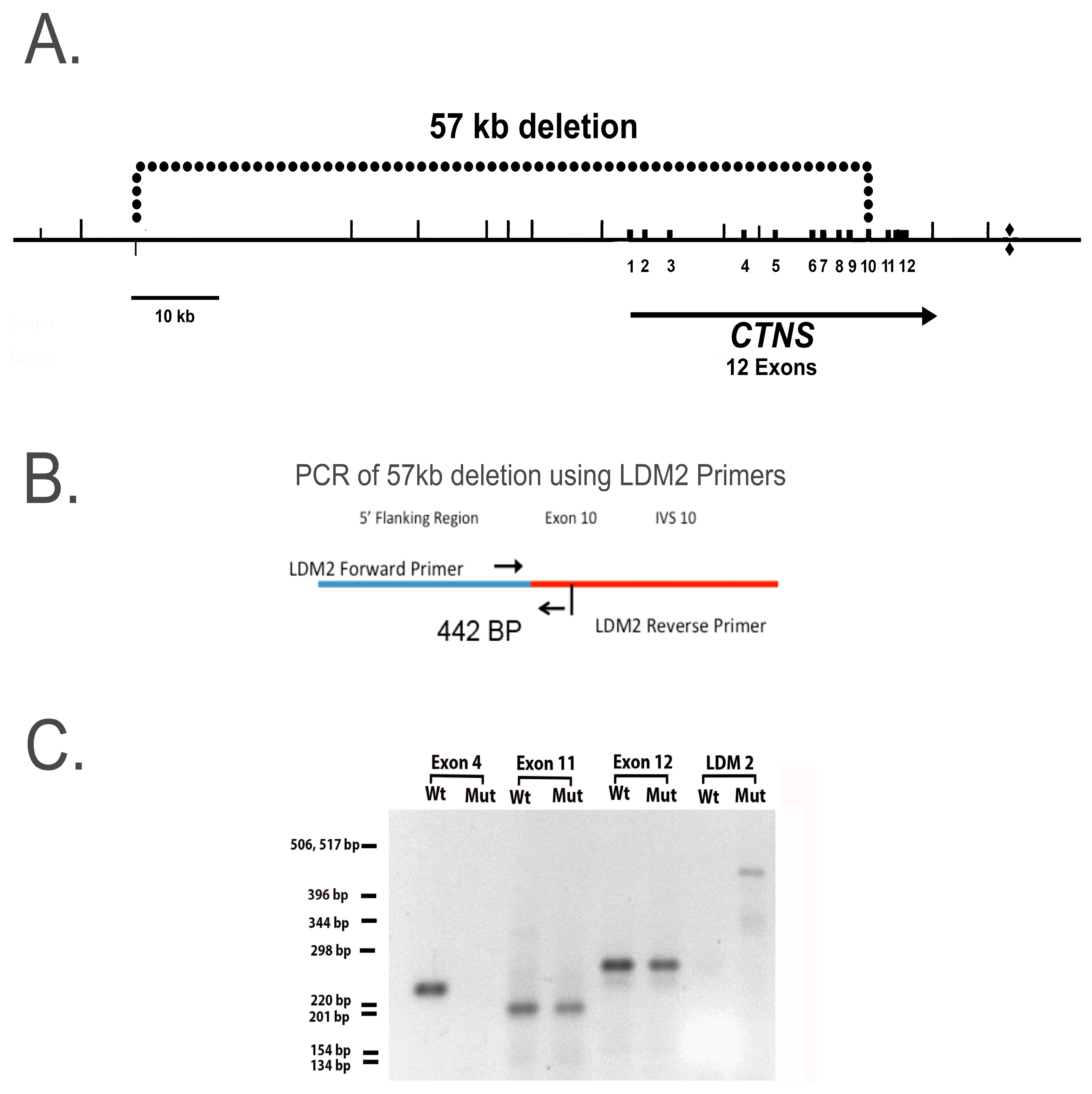
In this report, we developed hiPSCs from an individual who is homozygous for a 57-kb deletion which removes the first 9 exons in the
ctns structural gene, causing the gene product, cystinosin, to be nonfunctional. Studies of RPT cell cultures containing this deletion are particularly important because the 57-kb deletion accounts for approximately 60% of the
ctns mutations in American patients, and this mutation results in nephropathic cystinosis [
13]. The
ctns gene has been mapped to chromosome 17p13 in 1995; the region has been sequenced and over 140 pathogenic
ctns mutations have been identified, including the 57-kb deletion, whose sequence is highly conserved [
25,
26]. Indeed, the 57-kb deletion is thought to have arisen from a single individual in Northern Europe of the first millennium AD [
12,
13]. Nevertheless, in the absence of a DNA sequence analysis, we cannot exclude the possibility that other mutations have occurred in our human cystinotic RPT cells, although the results of our PCR analysis indicate that the genome of our cystinotic cell cultures is homozygous with respect to the 57-kb deletion.
Organs other than the kidney are eventually affected in infantile cystinosis after the emergence of the renal Fanconi Syndrome [
27,
28]. These organs include the eye, endocrine organs, the Gastrointestinal tract, muscles, and bones, as well as the central and peripheral nervous system. The other two forms of cystinosis also affect other organs that the kidney, including late-onset juvenile nephropathic cystinosis (where extra-renal dysfunctions observed in infantile cystinosis also develop) as well as non-nephropathic cystinosis, primarily characterized by photophobia. Despite these differences in disease presentation, the three major forms of cystinosis are all the consequence of mutations in the
ctns gene, which cause alterations in cystinosin function.
The transport functions of the
ctns gene product, cystinosin, have been studied extensively, including structural studies with three dimensional models [
28,
29]. Cystinosin, similar to other members of the LCT family, has significant homology with microbial rhodopsins (MRs). Not only do members of the LCT family, including cystinosin, have a similar size to microbial rhodopsins, but in addition these LCTs possess seven highly conservedα -helical transmembrane segments (TMSs) similar to the MRs [
2]. The helices within the TMSs undergo large conformational changes during the migration of protons through an access channel [
29]. The changes are required because cystine transport by LCTs, including cystinosin, depends upon a proton motive force (pmf) similar to microbial MRs [
2]. In the case of cystinosin, the pmf depends upon lysosomal V-ATPase activity. Previous studies indicate that cystine binding to the luminal surface of cystinosin is coupled to proton translocation. The process depends upon two TMSs (including TMSs 1-3 and TMSs 5-7), which are highly conserved regions [
2,
29], presumably being critical for normal transporter function).
A number of the mutations in the
ctns gene that result in cystinosis have been observed to alter the cystine transport capabilities of the cystinosin protein [
1]. The most prevalent mutation in Northern Europe and North America, the57 kb deletion, w encompasses the promoter and the first 10 exons of
ctns, as well as two upstream genes (
CARKL and
TRPV1) [
13]. In addition to the 57 kb deletion, 146 pathogenic
ctns mutations have been identified within exons 3 through 12, which encode for segments of the structural
ctns gene [
26]. Exons 1 and 2 in
ctns are noncoding. The mutations include 57 missense and nonsense mutations, as well as insertions and deletions. A number of the missense mutations that have occurred in
ctns alter the cytoplasmic and luminal gates for protons in the cystinosin protein, as well as the cystine binding site [
29]. Of particular interest in this regard,
ctns mutations in cystinosis often occur within TMSs (particularly in the case of infantile cystinosis). Mutations affecting inter-TMS regions and the N-terminus have been reported (primarily, but not exclusively, in juvenile and atypical cystinosis), including the 2 TMSs involved in proton translocation (i.e.,TMSs 1-3 and TMSs 5-7). Thus, in individuals with such
ctns mutations, lysosomal cystine transport is impaired, such that intracellular cystine increases in all cells in the body. Nevertheless, such
ctns mutations primarily have deleterious effects on specific organs, including the kidney. Little is understood about how these mutations cause the functional abnormalities observed in specific tissues in cystinosis. Further studies with cystinotic hiPSCs should prove to be invaluable in these regards.
Initial studies of altered cystine transport by mutated cystinosins led investigators to suggest that the
ctns mutations that cause infantile cystinosis have more deleterious effects on transport function than the transport alterations caused by
ctns mutations which result in juvenile or non-nephropathic cystinosis [
1]. However, more recent studies of 31
ctns mutations (using constructs that translocate to the plasma membrane) have indicated that some
ctns mutations that cause infantile cystinosis (including
ctns S298N and
ctns W182R) permit normal cystine transport activity to be retained, whereas several mutations causing juvenile cystinosis (including
ctns N323K and
ctns K280R) result in a complete loss of cystine transport activity [
1]. These observations are not consistent with the hypothesis that lysosomal cystine accumulation is aalswaysthe primary pathogenic cause of cystinosis.
Of particular interest in these regards are recent studies indicating that cystinosin (similar to other LCTs and MRs) has other functions in addition to transport. One such function involves the regulation of Chaperone Mediated Autophagy (CMA) due to the role played by cystinosin in trafficking the CMA receptor (i.e., LAMP2A) [
30], while another such function is the regulation of the mammalian Target of Rapamycin Complex 1 (mTORC1). Recent studies indicate that cystinosin is a component of the V-ATPase-Ragulator-Rag complex, that controls mTORC1 activation [
31,
32]. The interaction of the V-ATPase-Ragulator-Rag complex, including cystinosin, with mTORC1 promotes mTORC1 activation, an event which stimulates anabolic programs. Of particular interest in these regards is the recent report by Berquez et al. [
32]—that the lysosomal cystine storage that occurs in cystinosis stimulates the Ragulator-Rag GTPase-dependent recruitment of mTORC1, resulting in its constitutive activation. The studies conducted by Berquez et al. [
32] also suggest that the constitutive activation of mTORC1 caused by lysosomal cystine storage diverts RPT cells towards growth and proliferation so as to disrupt the expression of their differentiated functions. The recent studies conducted by Luciani and Devuyst [
33] further supports this hypothesis, and further suggests that lysosomal cystine accumulation actually causes an impairment of cell fate decisions by presumptive RPT cells in the kidney.
In the studies reported here, cystinotic hiPSCs that were differentiated into RPTs were less likely to form tubules with lumens than normal hiPSCs which had been differentiated into RPTs. This observation is consistent with the studies of Berquez et al. [
32], as well as Luciani and Devuyst [
33], whose studies indicated that lysosomal cystine storage resulting from the loss of functional cystinosin negatively affects cell fate specialization, causing a change in RPT cells so that they are no longer in a catabolic state, which promotes differentiation, but instead are in an anabolic state that promotes growth and proliferation. According to these investigators, this change is a consequence of the constitutive activation of mTORC1 signaling in
CTNS-deficient RPT cells. These investigators propose that these changes were not observed in RPT cell line derived from cystinotic patients, presumably due to the immortalization process. However, in this report, our cystinotic and normal hiPSCs were derived using episomal vectors which were only expressed temporally and thus did not alter gene expression over the long term. Further studies are needed to examine effects of the mTORC1 complex on our cystinotic hiPSCs and their differentiation into RPTs.
4. Materials and Methods
4.1. Materials
Dulbecco’s Modified Eagle’s Medium (DME), Ham’s F12 Medium (F12), fetal bovine serum, growth factor deficient matrigel, the 1KB plus DNA Ladder, and the Image-IT LIVE Red Poly Caspase Detection Kit and SlideFlasks were from ThermoFisher (Waltham, MA, USA). Human insulin, human holo-transferrin, 2-phosphoascorbate, penicillin and streptomycin, and other chemicals were from Sigma-Aldrich Chemical Corp. (St. Louis, MO, USA). Selenium was from Difco laboratories (Detroit, MI, USA). bFGF and TGFb1 were from Novoprotein (Summit, NJ, USA). Secondary antibodies were from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA). Plasmids, including pCXLE-hUL, pCXLE-hSK, and pCXLE-hOct3/4-shp53-F, were from Addgene (Watertown, MA, USA), having been deposited by Shinya Yamanaka.
4.2. Generation of hiPSCs
Integration-free human iPSCs (hiPSCs) were generated from human dermal fibroblasts homozygous for the 57 kb deletion in
ctns, which results in nephropathic cystinosis (obtained from the Coriell Institute for Medical Research (Camden, NJ, USA), Cat. # GM00706), as well as “normal” human fibroblasts (Coriell # GM00010), used as controls in the studies of GM00706 by Park and Thoene [
7]. The fibroblasts were maintained in Dulbecco’s Modified Eagle’s Medium (DME) supplemented with 20 mM sodium bicarbonate, 10% fetal bovine serum (FBS), 0.5% penicillin, and streptomycin.
Three vectors were introduced by electroporation into the cystinotic and normal human fibroblasts, including (1) pCXLE-hUL encoding for L-myc, and Lin28, (2) pCXLE-hSK encoding for SOX2 and KLF4, as well as (3) pCXLE-hOct3/4-shp53-F, encoding for Oct 3/4 and shRNA against human p53, as described by Okita et al. [
14]. The vectors also encoded for Epstein-Barr virus nuclear antigen 1 (EBNA1). Seven days later, transfected cells were replated into culture dishes coated with mouse feeder layers, as described by Takahashi et al. [
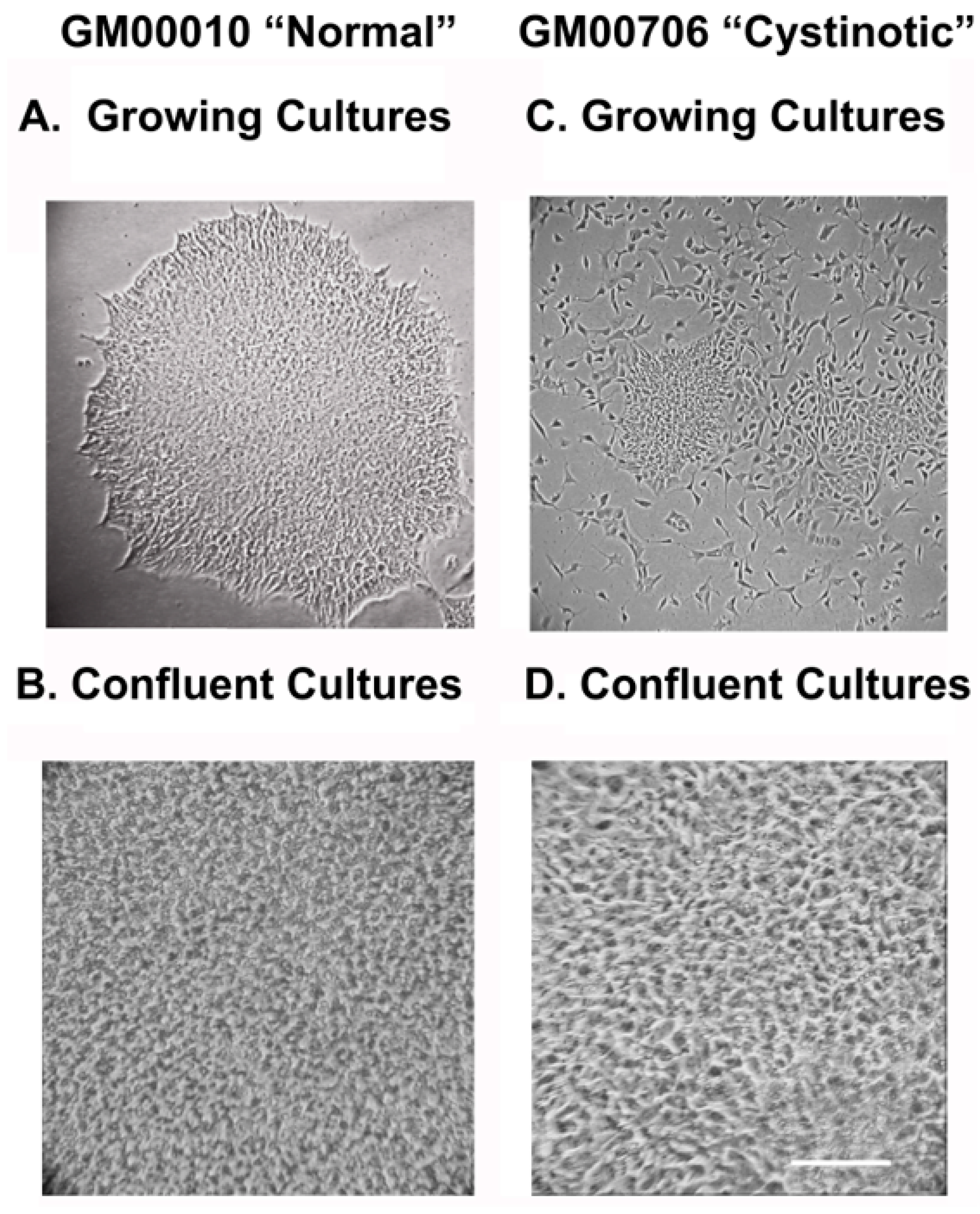
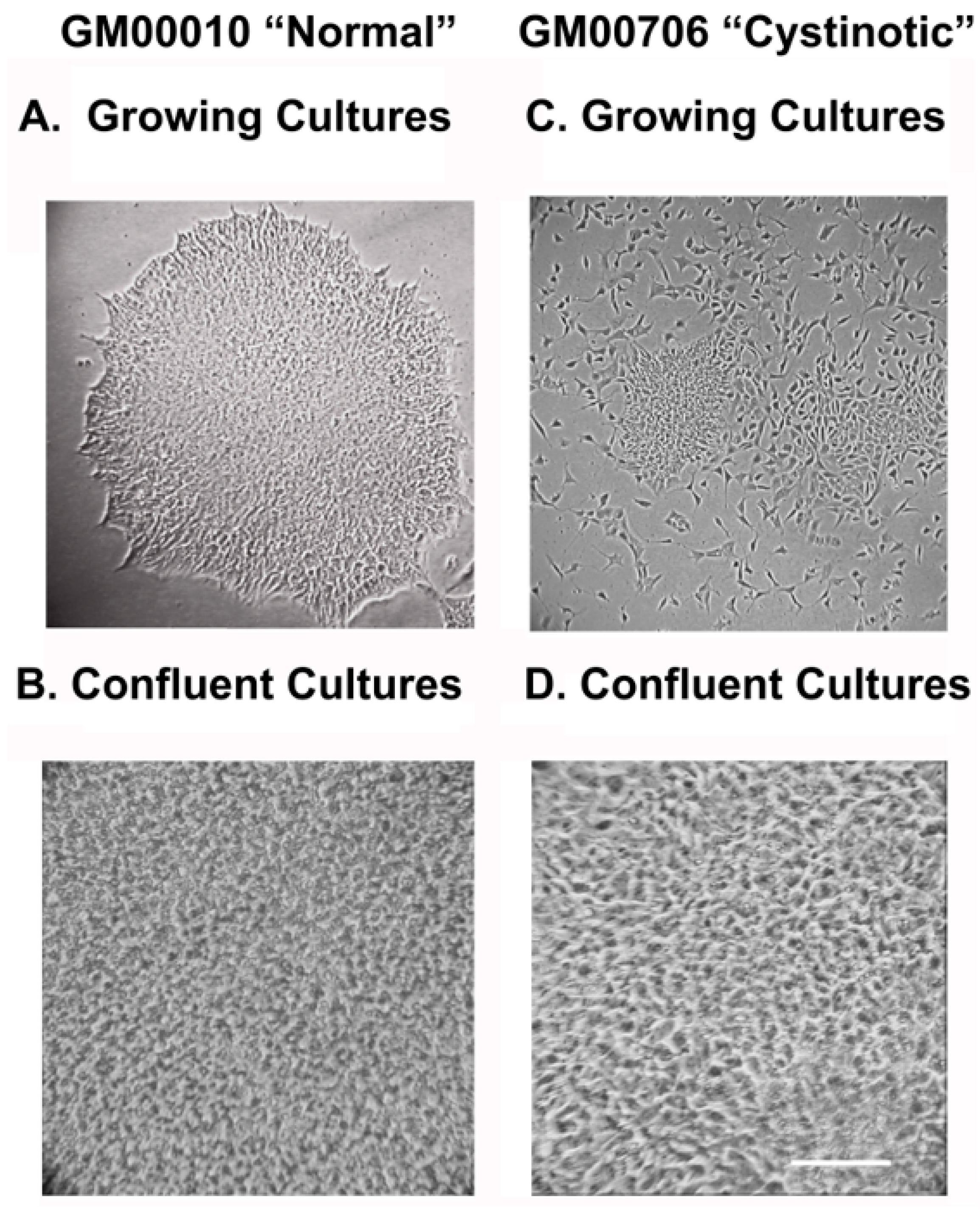
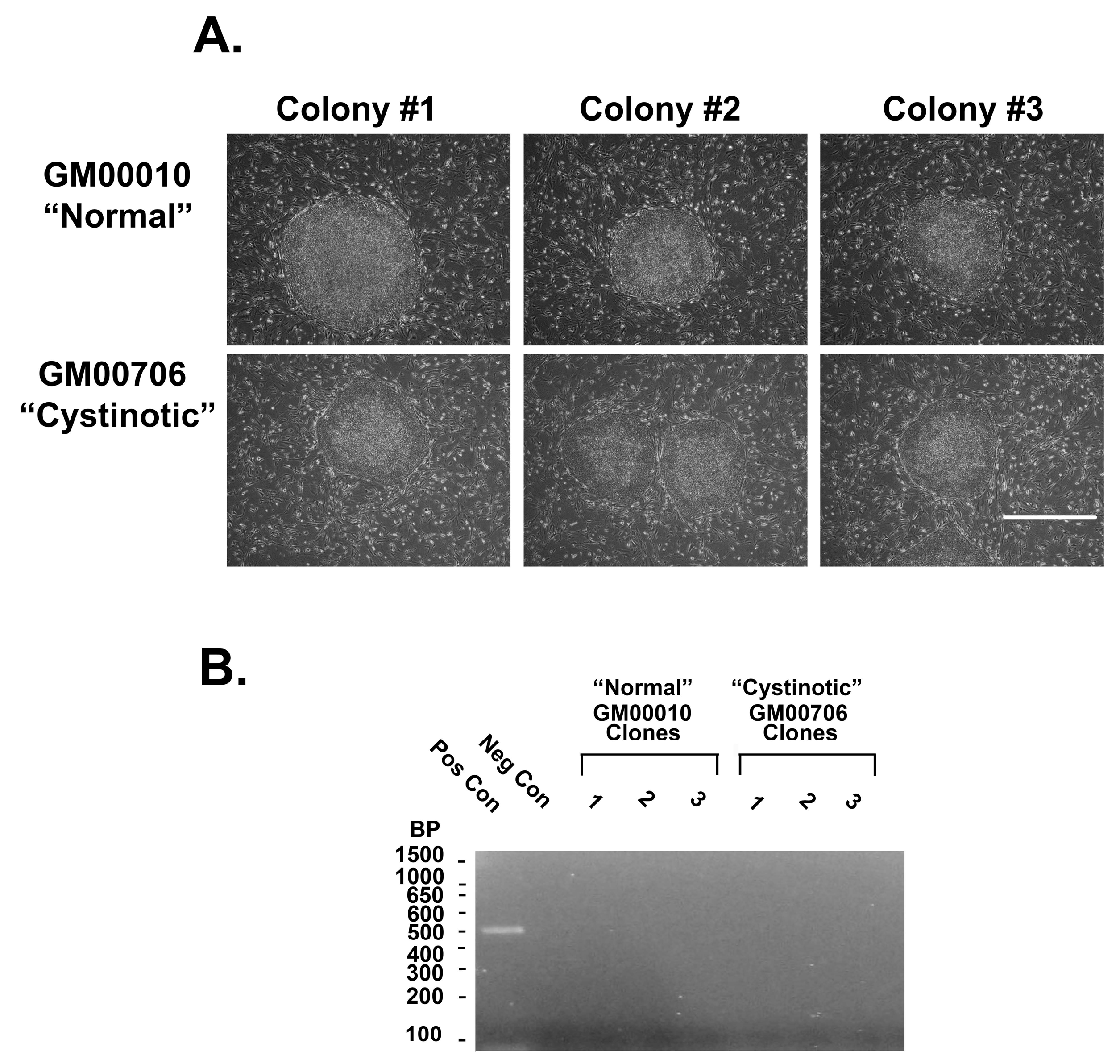
34]. Two weeks later, colonies and ESC-like morphology emerged, both from the electroporated normal (GM00010) (
Figure 12A) and cystinotic fibroblasts (GM00706) (
Figure 12A). Colonies were isolated and expanded, as described by Takahashi et al. [
34].
Previously, Yu et al. [
35] found that when using episomal vectors to obtain pluripotent hiPSCs, the vectors were spontaneously lost. Despite this, the cultures retained a stable pluripotent state. In order to determine whether the vectors were similarly lost from our hiPSCs, the presence of EBNA1 in episomal DNA was examined in individual clonal isolates of GM00010 and GM00706 hiPSCs after 3 months in cultures, as well as from positive control cultures (cultures obtained 2 weeks after transfection of dermal fibroblasts with the 3 vectors) and untransfected cultures (negative controls). A PCR analysis of EBNA1 in episomal DNA was conducted using forward and reverse primers (ATCGTCAAAGCTGCACACAG and CCCAGGAGTCCCAGTAGTCA, respectively), as described by Yu et al. [
35].
Figure 12B shows the absence of EBNA1 3 months after transfection (unlike the positive control), consistent with the loss of the original episomal vectors.
4.3. PCR Analysis of CTNS
Genomic DNA was purified from normal and cystinotic hiPSCs using a Monarch Genomic DNA Purification Kit (NEB #T3010) and PCR amplifications conducted using a Bio-Rad CFX96 Touch qPCR System (Bio-Rad Laboratories, Hercules, CA, USA.) as described by Anikster et al. [
13]. Primers employed to amplify specific regions in the
ctns gene included (a) LDM2 primers (Forward, 5′-ACCTCTCTGATGTGTCCAAG-3′, Reverse, 5′-AGCCAAAGGCATCAGGAAAG-3′), which amplify the breakpoint in the 57 kb deletion (illustrated in
Figure 3B, and described by Anikster et al. [
13]), as well as (b) primers that amplify specific exon regions in
ctns as described by Town et al. [
25], including primers for exon 4 (forward 5′-GTCATTGATTTGGGTCC-3′; reverse 5′-TAGGGCTTGTCTTACAGGTA-3′), exon 10 (forward 5′-GGCCTCTGTGTGGGTCC-3′; reverse 5′-GGCCATGTAGCTCTCACCTC-3′), and exon 11 (forward 5′-GCCCTCCGTCTGTATGTCCG-3′; reverse 5′-GCCCGATGCCCCAGC-3′). PCR products were separated on agarose gels and visualized on a Fotodyne Photo Prep UV transilluminator Model 3-3500 (Fotodyne, Inc., Hartland, WI, USA).
4.4. Culture of hiPSCs
Both normal and cystinotic hiPSCs were cultured in chemically defined E8 medium on matrigel coated dishes in a 5% CO
2/95% air humidified environment at 37 °C, as described by Chen et al. [
36]. E8 medium was prepared from a basal medium (DME/F12) which consisted of a 1:1 mixture of Dulbecco’s Modified Eagle’s Medium and Ham’s F12 containing a total of 20 mM sodium bicarbonate, 118 mM NaCl, penicillin (92 unit/mL), and streptomycin (200 µg/mL). E8 medium was obtained by further supplementing DME/F12 with 20 µg/mL human insulin, 10 µg/mL human holo-transferrin, 100 µg/mL FGF2, 2 ng/mL TGFβ, 64 µg/mL 2-phospho-L-ascorbate, and 5 × 10
−8 M selenium.
The medium was changed daily until the cultures became confluent. Confluent hiPSC cultures were passaged by first incubating the hiPSCs for 5 min at 37 °C in Phosphate Buffered Saline (PBS) supplemented with 0.5 mM EDTA (EDTA/PBS solution), followed by centrifugation (in EDTA/PBS) and resuspension of the cultures in E8 medium supplemented with Rock Inhibitor Y27632 (10 µM). Cultures were then plated into culture dishes coated with matrigel, as detailed by Lin and Chen [
37].
4.5. Pluripotency Detection: Identification of Stem Cell Markers
Immunostaining of hiPSCs was conducted using SlideFlasks, as described by Marti et al. [
38]. To summarize, the medium was removed from the cultures in the SlideFlasks prior to microscopy, and cultures were fixed with 4%
w/
v p-formaldehyde. The cultures were blocked (with TBS + 0.5%
v/
v Triton X-100 + 6%
v/
v donkey serum), followed by incubation with primary antibodies in TBS++ (i.e., TBS + 0.1%
v/
v Triton X-100 and 3% donkey serum). Antibody combinations used included (A) rabbit anti-Oct4 (A24867 ThermoFisher, Waltham, MA, USA) and mouse anti-SSEA4 (A24866 ThermoFisher), as well as (B) rat anti-SOX2 (A24759, ThermoFisher) and mouse anti-TRA-1-60 (A24868, ThermoFisher). After the incubation with primary antibodies, the cultures were washed 3 times with TBS++ and incubated 2 h at 37 °C with secondary antibodies in TBS++ (including AlexaFluor 594 donkey anti-rabbit and AlexaFluor 488 goat anti-mouse IgG3 in the case of combination A, as well as Alexa Fluor 488 donkey anti-rat and Alexa Fluor 595 goat anti-mouse IgM in the case of combination B). After washing 3 times with TBS, the slides were incubated with DAPI, mounted, and examined under a Zeiss Axio Observer Fluorescent Microscope at 100×.
4.6. Pluripotency Detection: Formation of Embryoid Bodies (EBs) with Three Germ Layers
EBs were generated from hiPSCs that were 60–80% confluent, as described by Lin and Chen [
37]. After washing the cultures twice with EDTA/PBS, the cells were incubated with EDTA/PBS for 10 min. Aggregates were disrupted by pipetting and transferred to a 15 mL tube containing an equal amount of E8 medium containing 5 mg/mL Polyvinyl Alcohol (PVA) (E8/PVA medium) and 110 µM Rock Inhibitor Y-27632. The cells were spun at 10,000 rpm 5 min, followed by resuspension in EB/PVA medium with 110 µM Y-27632 at 10
5 cells/mL (determined using a Coulter Counter Model Zf, Beckman Coulter, Chasman, MN, USA. To form EBs, 20 µL drops were hung on the lid of Petri dishes and were incubated for two days. The cultures were then transferred into Corning low attachment dishes for further culturing.
Subsequently, the EBs were collected and placed into a small drop of molten 2% Low Melting Point (LMP) Agarose (ThermoFisher, Waltham, MA, USA) in PBS and cooled, as described by Shamblott et al. [
39]. The EBs in the solidified LMP Agarose were fixed with 3% p-formaldehyde in PBS and embedded in paraffin. Sections (6 mm) were placed upon Probe on plus microscope slides (Fisher Sci., Hampton, NH, USA).
Sections to be used for immunostaining were dewaxed and rehydrated by serial incubation with xylene and decreasing gradations of ethanol. For antigen retrieval, the slides were incubated for 20 min at 95 °C in 10 mM Tris, pH 9.0, 1 mM EDTA, and 0.05% Tween 20. The slides were then incubated with TBS, followed by 100 mM glycine to minimize autofluorescence, permeabilized with 0.25% Triton X-100 in TBS, and blocked with TBS containing 0.25% Triton X-100 and 3% donkey serum.
Sections were then incubated overnight at 4 °C with primary antibody in TBS++. Antibody combinations included (A) mouse antiβ-tubulin (Clone TUJ1, R&D Systems, MAB1195; ectoderm), goat antiα-smooth muscle actin (R&D Systems, NB300-978; mesoderm), and rabbit anti-α-1-fetoprotein (Dako A0008; endoderm), as well as (B) mouse anti-β3-tubulin (Clone TUJ1, R&D Systems, MAB1195; ectoderm), rabbit anti-brachyury (R&D Systems MAP20851; mesoderm), and goat anti-EpCam (R&D Systems AF960; endoderm). Following the primary antibody incubation, slides were washed and incubated with secondary antibodies in TBS++ for 2 h at 37 °C. Included amongst the secondary antibodies utilized were Donkey anti-mouse Cy3, Donkey anti-goat Cy5, and Donkey anti-rabbit Cy2 in the case of antibody combination A, as well as Donkey anti-mouse Cy3, Donkey anti-rabbit Cy2, and Donkey anti-goat Cy5. After the incubation with secondary antibodies, slides were treated with DAPI, mounted, and sections were visualized under a Zeiss Axio Observer Fluorescent Microscope at 100×.
4.7. Differentiation of hiPSCs into Renal Organoids
The method of Lam et al. [
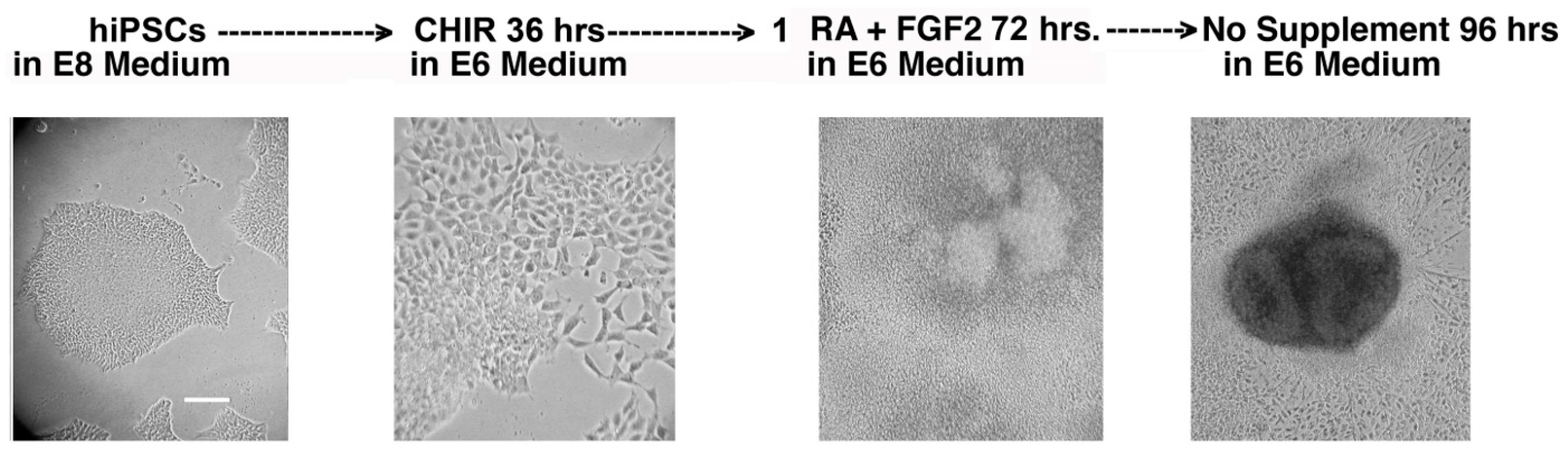
11] was employed to differentiate normal and cystinotic hiPSCs into RPTs. To summarize, (a) the hiPSCs were incubated for 36 h in E6 medium (i.e., DME/F12 supplemented 20 µg/mL human insulin and 10 µg/mL human transferrin) and were further supplemented with 5 µM CHIR99021, a glycogen synthase inhibitor (which induces the differentiation of hiPSCs into mesoderm via the Wnt pathway (5)). Subsequently, (b) the cultures were incubated for 72 h in E6 medium supplemented with 100 ng/mL FGF2 and 1 µM retinoic acid (which induces the formation of inner mesoderm (IM) from mesoderm) (5). Finally, (c) the cultures were maintained for ≥7 days in E6 medium with no further supplements, resulting in the emergence of organoids which expressed distinctive RPT characteristics, including Lotus Tetragonolobus Lectin binding and junctional N-cadherin (5).
The renal organoids that appeared after 7 days were removed from culture dishes with a rubber policeman, transferred to a 15 mL tube, and were centrifuged at 500 rpm. The culture medium was removed and replaced with PBS containing 3.7% p-formaldehyde (PFA). After an overnight incubation, the cultures were washed by centrifuged to remove the PBS + PFA, followed by 2 washes with PBS. The tube containing the hiPSCs was put into a 37 °C waterbath. After adding molten 2% low melting point agarose in PBS to the tube, the hiPSCs in the agarose were transferred to parafilm, where the agarose solidified. The hiPSCs in the agarose were embedded in paraffin, and 6 mm sections were transferred onto Probe on Plus microscope slides.
The sections were dewaxed, rehydrated, and immunostained, as described above with EBs. Antibodies combinations employed in immunostaining included (A) rabbit polyclonal anti-cd26 (sc-9153, H-270, Santa Cruz Biotech, Dallas, TA, USA) and mouse monoclonal anti-villin (sc-58897, Santa Cruz Biotech), (B) rabbit polyclonal anti-NHERF-1 (sc-134485, H-100, Santa Cruz Biotech) and mouse monoclonal anti-villin (sc-58897, Santa Cruz Biotech), as well as (C) rabbit polyclonal anti-NHE3 (sc-28757, H-170, Santa Cruz Biotech) and mouse monoclonal anti-villin (sc-58897, Santa Cruz Biotech). Subsequently, slides were stained with a Donkey anti-mouse Cy2 secondary antibody and a Donkey anti-rabbit Cy3 secondary antibody. Slides were treated with DAPI and were mounted and visualized under a Zeiss Axio Observer Fluorescent Microscope (100×).
4.8. Quantitation of Apoptosis and Cell Death
The frequency of apoptotic and dead cells was determined using an Image-IT LIVE Red Poly Caspase Detection Kit. To summarize, apoptotic cells were identified using a Fluorescent Labelled Inhibitor of Caspases (FLICA). SRVAD-FMK FLICA reagent (that recognizes activated caspase 1, as well as caspases 3-9, with red fluorescence). To detect dead cells, the cultures were co-stained with Sytox Green (a nuclear stain that permeates cells with compromised membranes, typical of dead cells). Hoechst 33342 was employed to detect nuclei (via blue fluorescence). Cultures were examined under a Zeiss Axio Observer Fluorescence Microscope (Zeiss, Oberkochen, Germany) at 100×. The number of cells stained with these reagents was quantitated using NIH ImageJ in at least 20 microscope fields in each of the three dishes per condition.
4.9. Statistical Analysis
Statistical analyses were conducted using GraphPad Prism Version 10.0.3. Statistical results were expressed as Means ± SEM. Statistical differences between groups were determined by means of a Chi-square (and Fisher’s exact) test, also using GraphPad Prism. Differences between means were considered statistically significant when p < 0.05.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}