
Inferring the Regulatory Network of miRNAs on Terpene Trilactone Biosynthesis Affected by Environmental Conditions
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
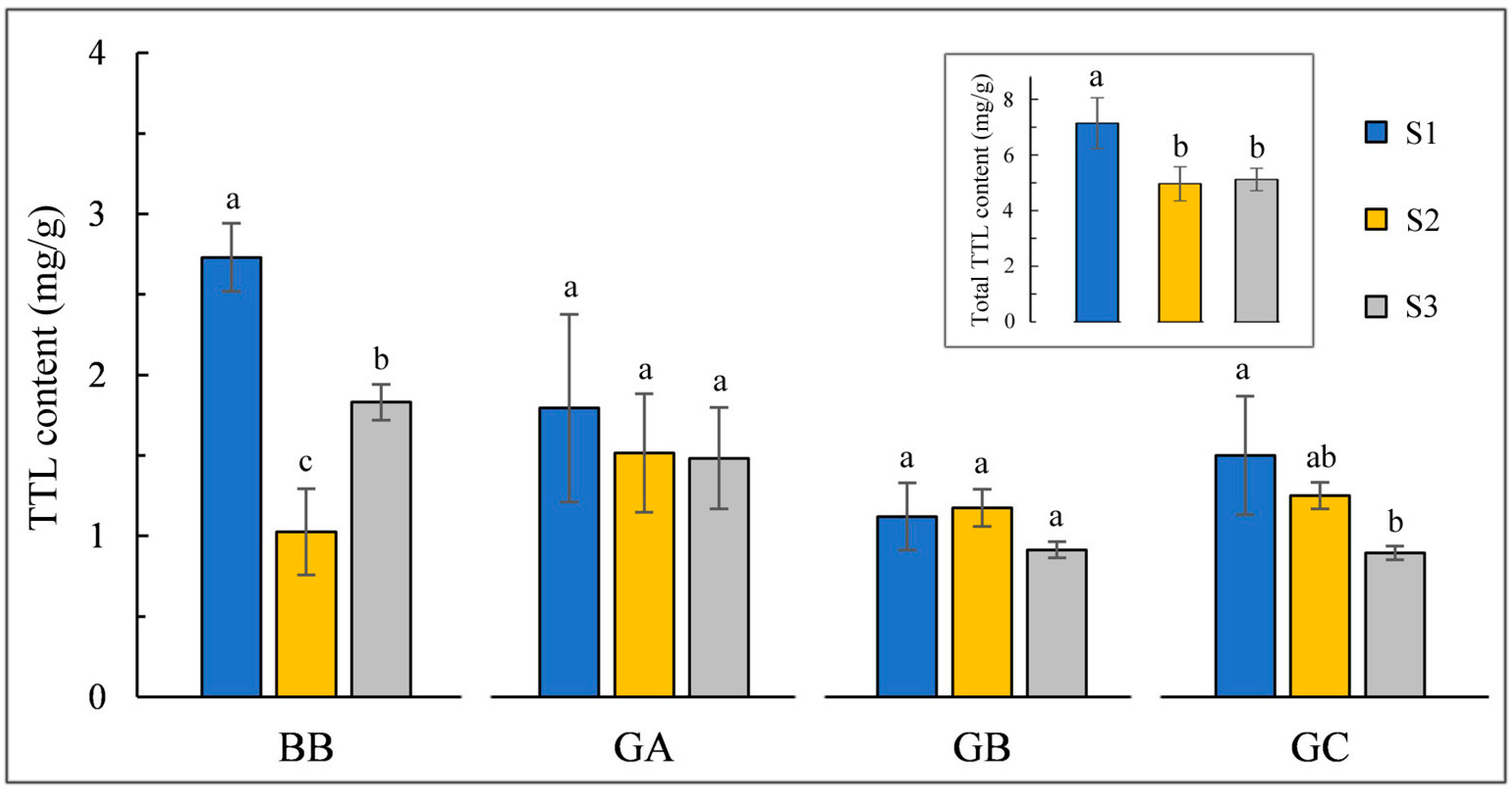
2.1. Changes in TTLs Content under Different Environments
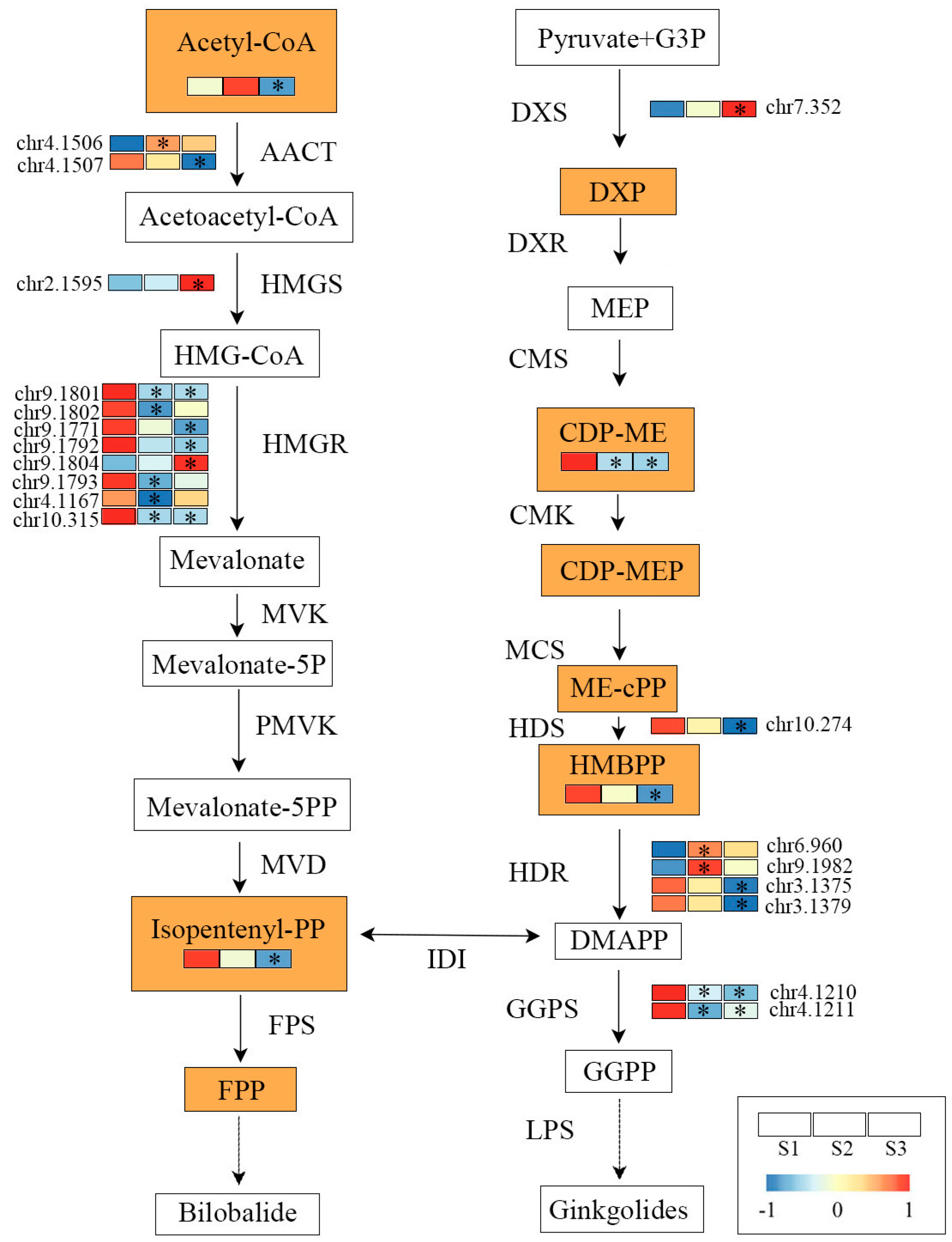
2.2. Analysis of Environment-Responsive mRNAs
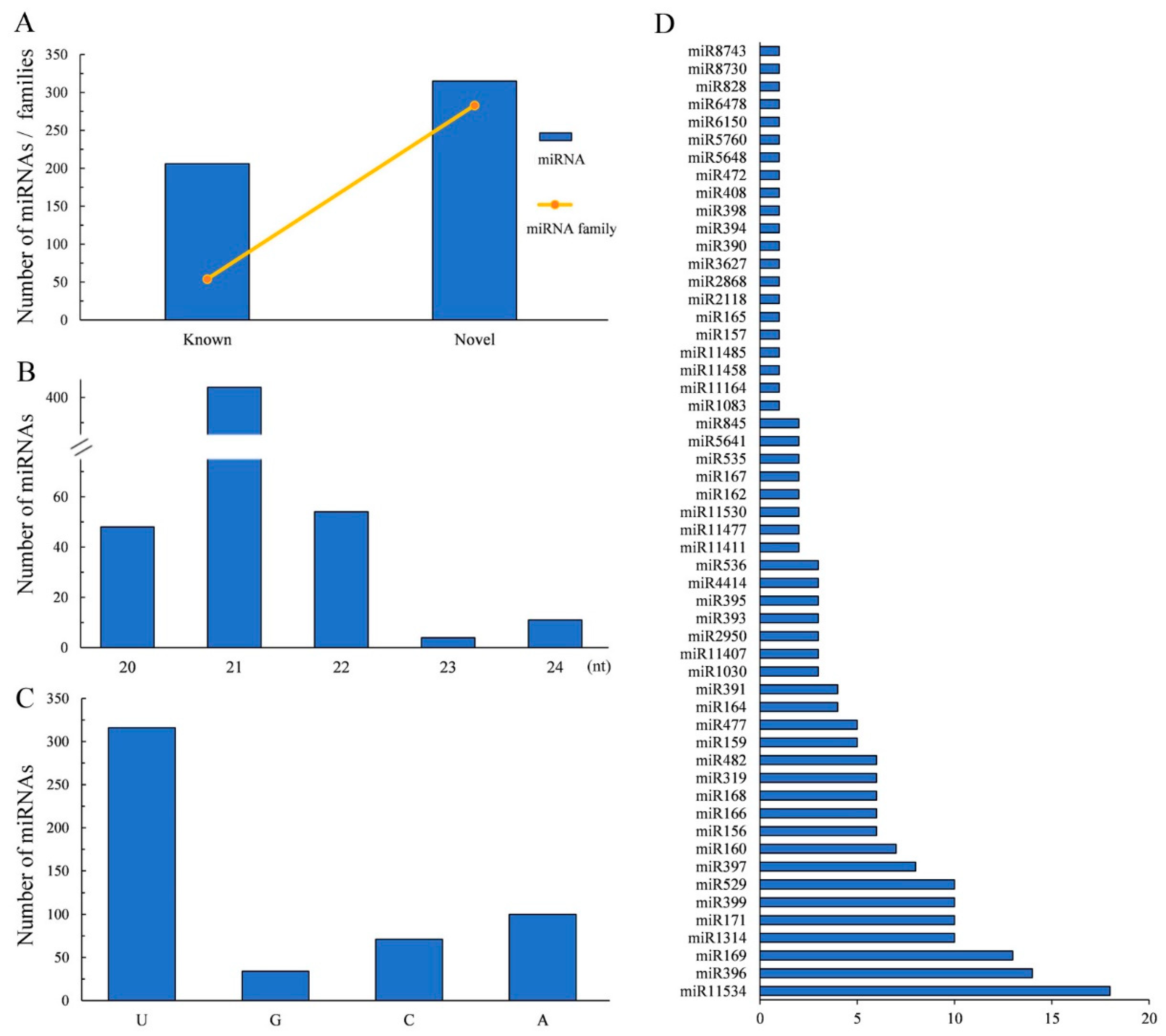
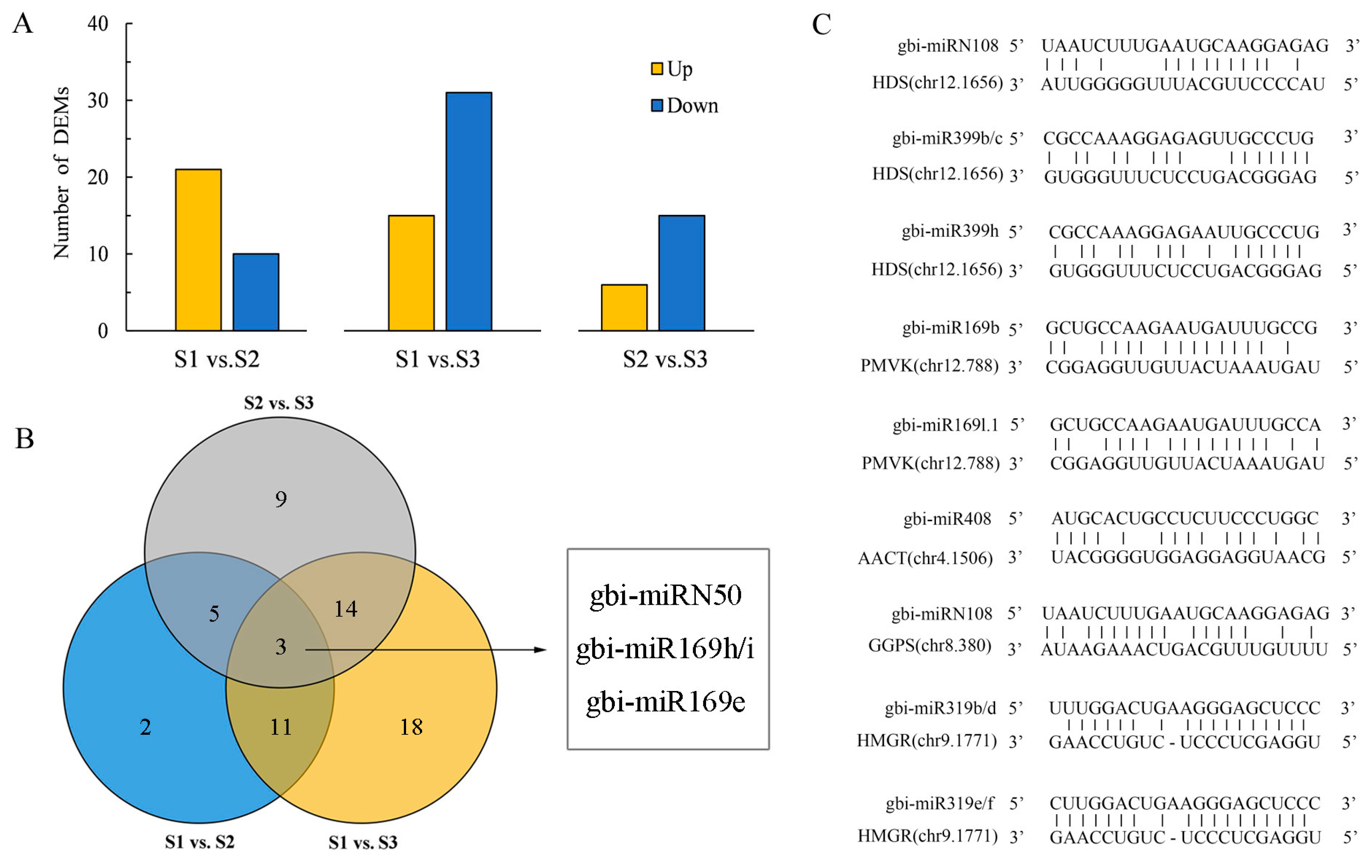
2.3. Annotation of miRNAs and Their Target Genes
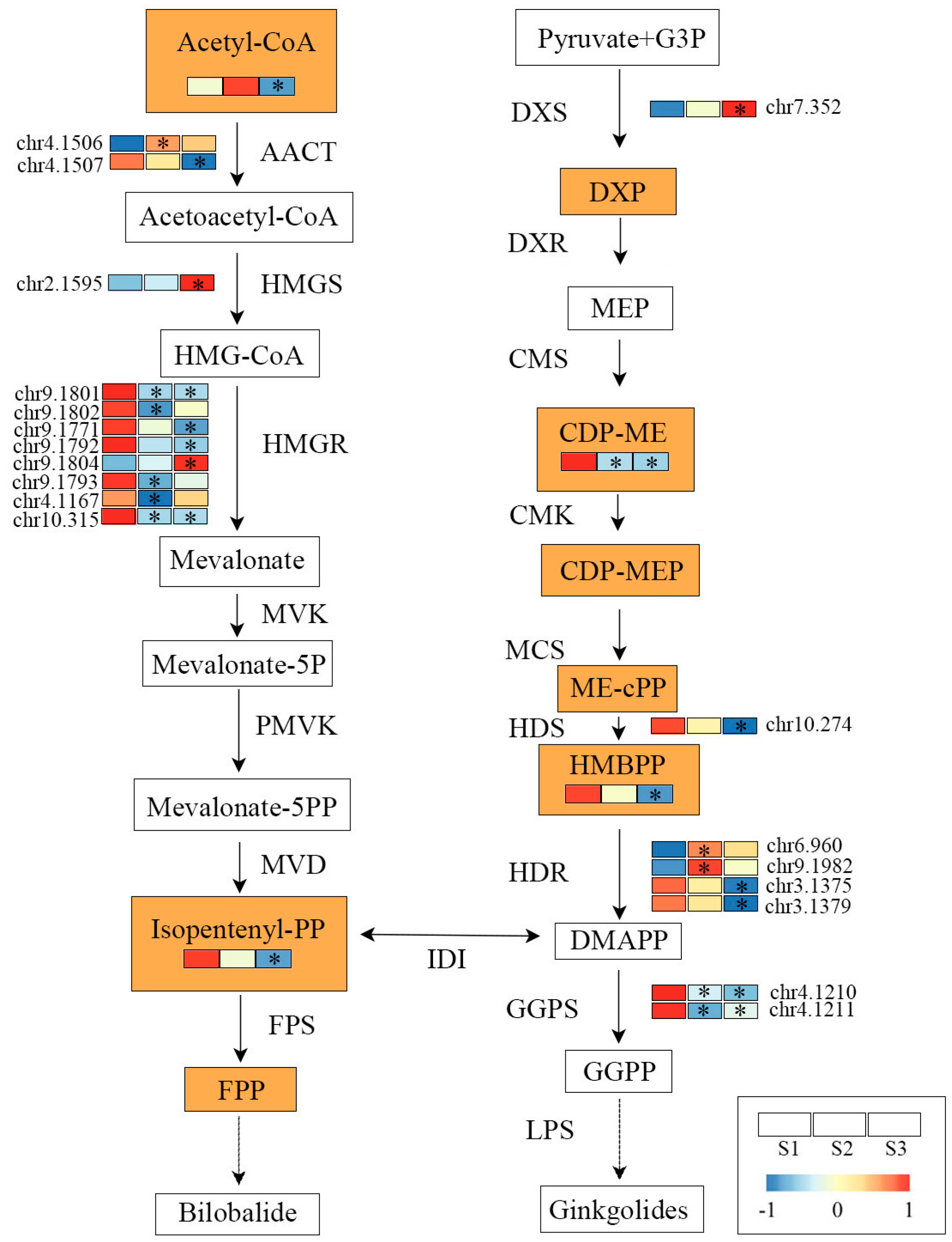
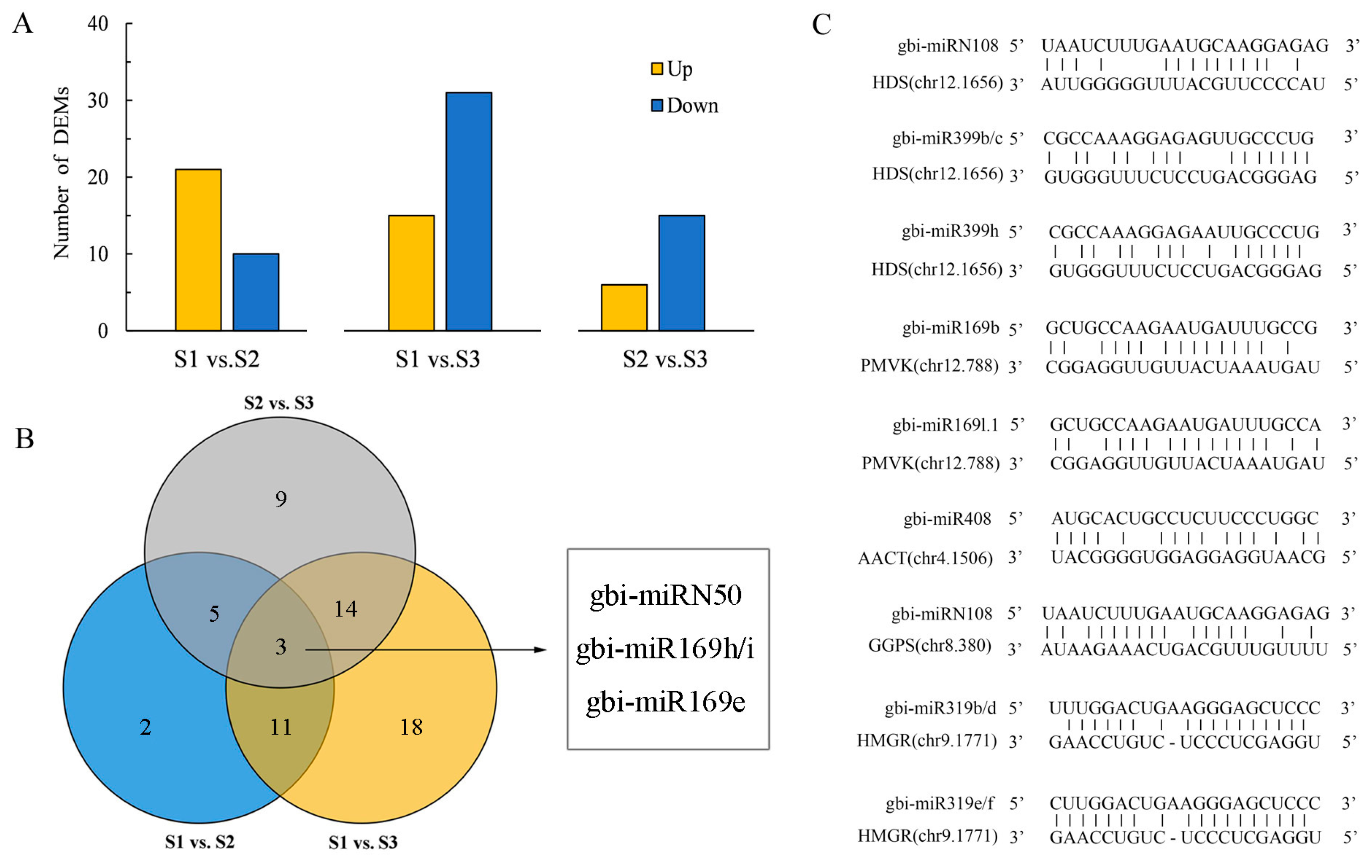
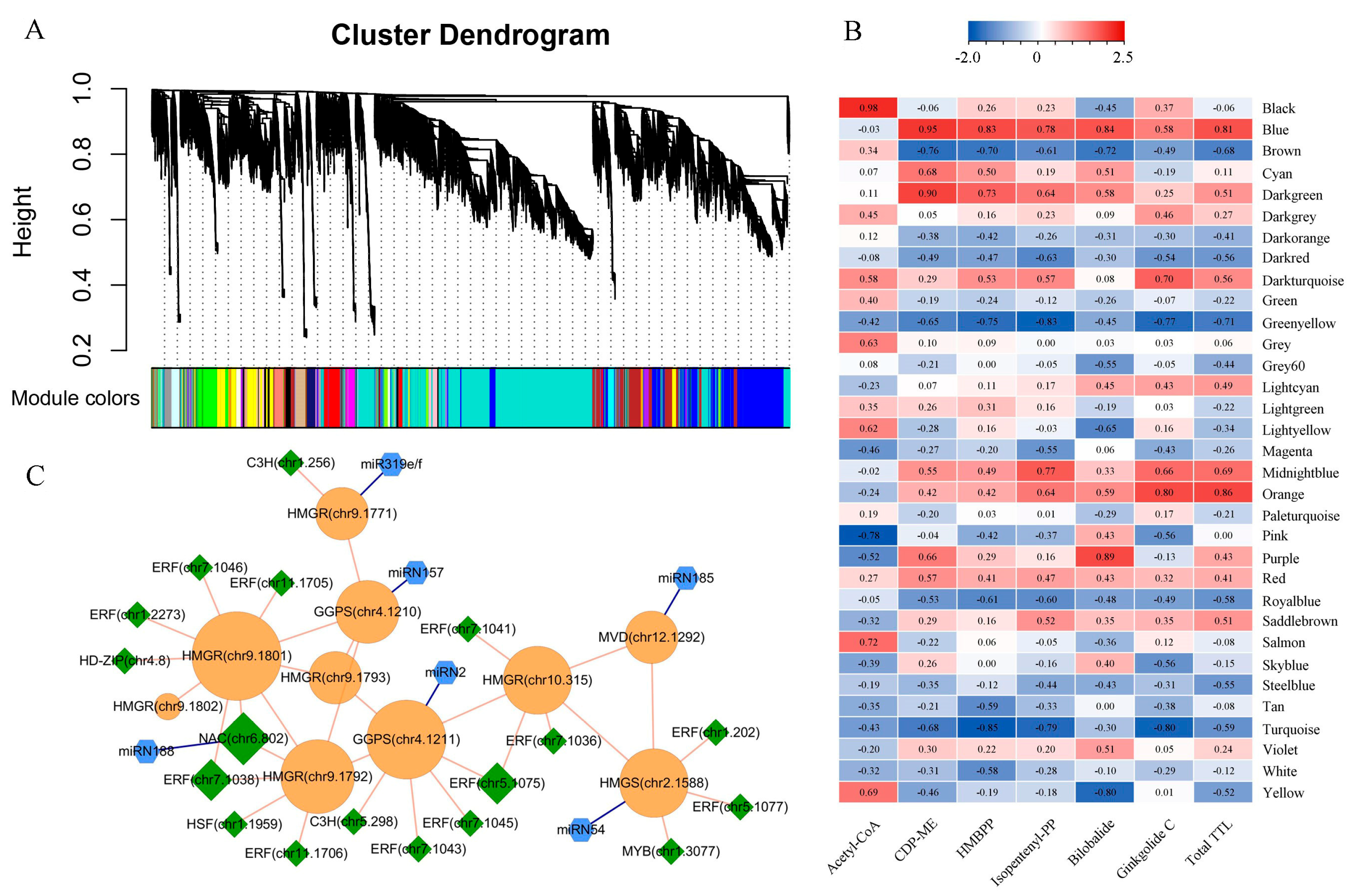
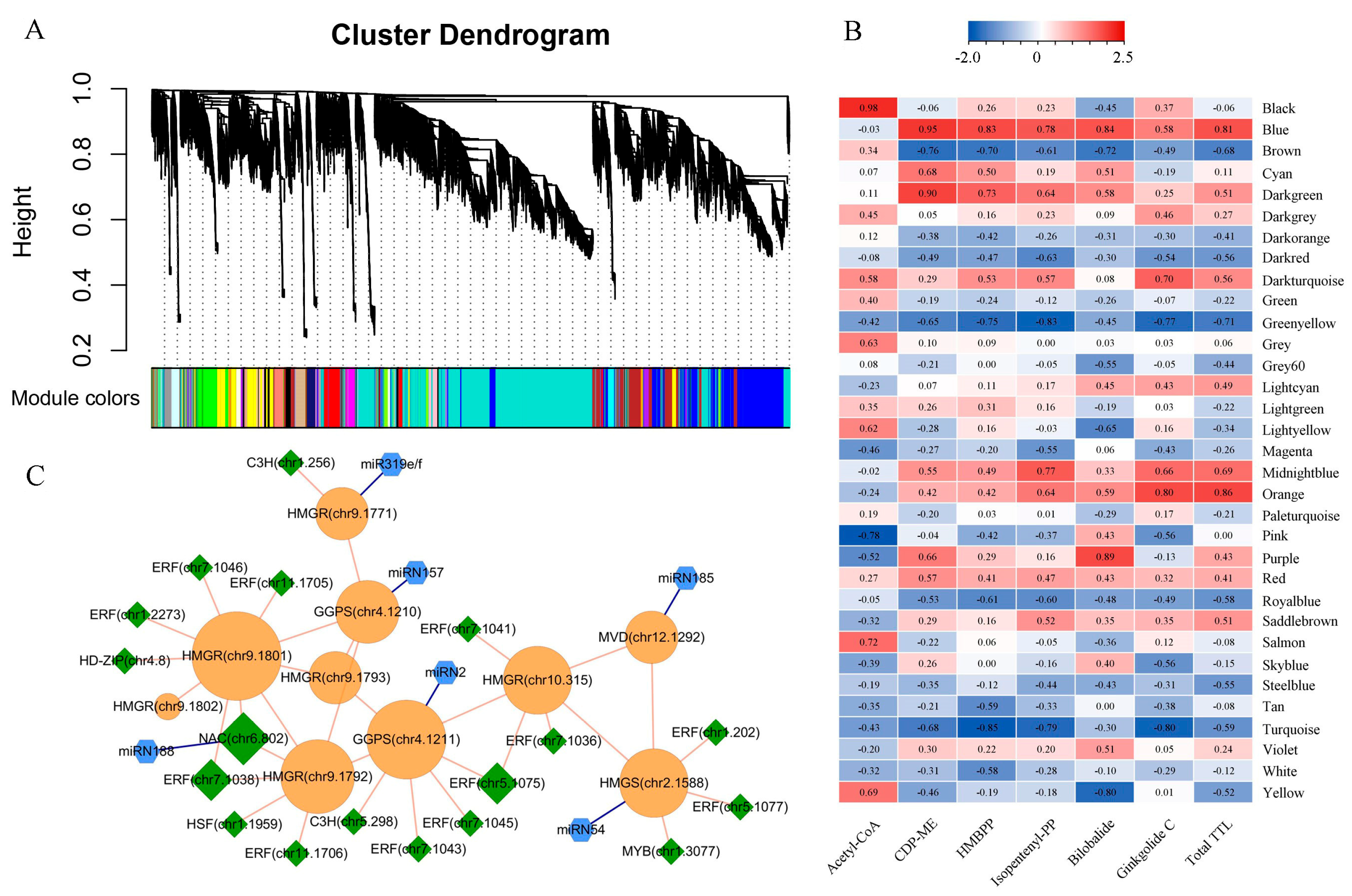
2.4. Construction of TTL-Related Gene Regulatory Network
3. Discussion
3.1. Effects of Environmental Condition on TTLs Accumulation in Ginkgo
3.2. Key Genes Regulating TTL Biosynthesis and Accumulation
3.3. MicroRNA Functions in Environmental Stress Responses
4. Materials and Methods
4.1. Plant Materials
4.2. Extraction and Determination of TTL-Related Components
4.3. RNA Sequencing (RNA-seq) and Analysis
4.4. Small RNA Sequencing (sRNA-seq) and Analysis
4.5. Constructing Co-Expression Network
4.6. Quantitative Real-Time PCR (qRT-PCR) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| TTL | terpene trefoil lactone |
| miRNA | microRNA |
| WGCNA | weighted gene co-regulatory network |
| TF | transcription factor |
| IPP | isopentenyl diphosphate |
| DMAPP | dimethyl allyl pyrophosphate |
| MVA | mevalonate |
| MEP | methylerythritol 4-phosphate |
| DXS | 1-deoxy-D-xylulose-5-phosphate synthase |
| DXR | 1-deoxy-D-xylulose-5-phosphate reductoisomerase |
| HMGR | hydroxymethylglutaryl-CoA reductase |
| GA | ginkgolide A |
| GB | ginkgolide B |
| GC | ginkgolide C |
| BB | bilobalide |
| AACT | acetyl-CoA C-acetyltransferase |
| HMGS | hydroxymethylglutaryl-CoA synthase |
| MVK | mevalonate kinase |
| PMVK | phosphomevalonate kinase |
| MVD | diphosphomevalonate decarboxylase |
| CMS | 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase |
| CMK | 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase |
| MCS | 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase |
| HDS | (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase |
| HDR | 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase |
| FPS | farnesyl diphosphate synthase |
| GGPS | geranylgeranyl diphosphate synthase |
| IDI | isopentenyl-diphosphate Delta-isomerase |
| LPS | levopimaradiene synthase |
| CYP450 | Cytochrome P450 |
| DEG | differentially expressed gene |
| DEM | differentially expressed miRNA |
| FC | fold change |
| GO | gene ontology |
| KEGG | Kyoto encyclopedia of genes and genomes |
| TPM | transcripts per million |
| sRNA | small RNA |
| ELSD | evaporative light scattering detector |
References
- Jacobs, B.P.; Browner, W.S. Ginkgo biloba: A living fossil. Am. J. Med. 2000, 108, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Boateng, I.D.; Yang, X.M.; Li, Y.; Liu, W. Effects of processing methods on quality, antioxidant capacity, and cytotoxicity of Ginkgo biloba leaf tea product. J. Sci. Food Agric. 2023, 103, 4993–5003. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhong, W.; Shu, C.; Yang, H.; Li, E. Comparative analysis of chemical constituents and bioactivities of the extracts from leaves, seed coats and embryoids of Ginkgo biloba L. Nat. Prod. Res. 2021, 35, 5498–5501. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.; Marabotti, A.; Ramteke, P.W.; Facchiano, A. Interaction of human chymase with ginkgolides, terpene trilactones of Ginkgo biloba investigated by molecular docking simulations. Biochem. Biophys. Res. Commun. 2016, 473, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.G.; Lu, X.; Gao, W.; Li, P.; Yang, H. Structure, synthesis, biosynthesis, and activity of the characteristic compounds from Ginkgo biloba L. Nat. Prod. Rep. 2022, 39, 474–511. [Google Scholar] [CrossRef] [PubMed]
- Jaracz, S.; Malik, S.; Nakanishi, K. Isolation of ginkgolides A, B, C, J and bilobalide from G. biloba extracts. Phytochemistry 2004, 65, 2897–2902. [Google Scholar] [CrossRef]
- Crimmins, M.T.; Pace, J.M.; Nantermet, P.G.; Kim-Meade, A.S.; Thomas, J.B.; Watterson, S.H.; Wagman, A.S. The total synthesis of (±)-ginkgolide B. J. Am. Chem. Soc. 2000, 122, 8453–8463. [Google Scholar] [CrossRef]
- Sabater-Jara, A.B.; Souliman-Youssef, S.; Novo-Uzal, E.; Almagro, L.; Belchí-Navarro, S.; Pedreño, M.A. Biotechnological approaches to enhance the biosynthesis of ginkgolides and bilobalide in Ginkgo biloba. Phytochem. Rev. 2013, 12, 191–205. [Google Scholar] [CrossRef]
- Zhu, C.; Cao, F.; Wang, G.; Geng, G. Effects of drought stress on annual dynamic changing pattern of the terpene lactones content in Ginkgo biloba leaves. China For. Sci. Technol. 2011, 25, 15–20. [Google Scholar]
- Lange, B.M.; Ghassemian, M. Genome organization in Arabidopsis thaliana: A survey for genes involved in isoprenoid and chlorophyll metabolism. Plant Mol. Biol. 2003, 51, 925–948. [Google Scholar] [CrossRef]
- Kang, S.M.; Min, J.Y.; Kim, Y.D.; Park, D.J.; Jung, H.N.; Karigar, C.S.; Ha, Y.L.; Kim, S.W.; Choi, M.S. Effect of supplementing terpenoid biosynthetic precursors on the accumulation of bilobalide and ginkgolides in Ginkgo biloba cell cultures. J. Biotechnol. 2006, 123, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhang, X.; Fu, M.; Zeng, H.; Ye, J.; Zhang, W.; Liao, Y.; Xu, F. Effects of different stress treatments on the total terpene trilactone content and expression levels of key genes in Ginkgo biloba Leaves. Plant Mol. Biol. Report. 2020, 38, 521–530. [Google Scholar] [CrossRef]
- Carbonell, A.; Fahlgren, N.; Garcia-Ruiz, H.; Gilbert, K.B.; Montgomery, T.A.; Nguyen, T.; Cuperus, J.T.; Carrington, J.C. Functional analysis of three Arabidopsis ARGONAUTES using slicer-defective mutants. Plant Cell 2012, 24, 3613–3629. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, X.; Tan, J.; Xu, F.; Cheng, S.; Chen, Z.; Zhang, W.; Liao, Y. Global identification of Ginkgo biloba microRNAs and insight into their role in metabolism regulatory network of terpene trilactones by high-throughput sequencing and degradome analysis. Ind. Crops Prod. 2020, 148, 112289. [Google Scholar] [CrossRef]
- Singh, N.; Srivastava, S.; Shasany, A.K.; Sharma, A. Identification of miRNAs and their targets involved in the secondary metabolic pathways of Mentha spp. Comput. Biol. Chem. 2016, 64, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Sharma, A. Turmeric (Curcuma longa): miRNAs and their regulating targets are involved in development and secondary metabolite pathways. Comptes Rendus Biol. 2017, 340, 481–491. [Google Scholar] [CrossRef]
- Aravind, J.; Rinku, S.; Pooja, B.; Shikha, M.; Kaliyugam, S.; Mallikarjuna, M.G.; Kumar, A.; Rao, A.R.; Nepolean, T. Identification, characterization, and functional validation of drought-responsive microRNAs in subtropical maize inbreds. Front. Plant Sci. 2017, 8, 941. [Google Scholar] [CrossRef]
- Yang, C.; Li, D.; Mao, D.; Liu, X.U.E.; Ji, C.; Li, X.; Zhao, X.; Cheng, Z.; Chen, C.; Zhu, L. Overexpression of microRNA319 impacts leaf morphogenesis and leads to enhanced cold tolerance in rice (Oryza sativa L.). Plant Cell Environ. 2013, 36, 2207–2218. [Google Scholar] [CrossRef]
- Wen, M.; Xie, M.; He, L.; Wang, Y.; Shi, S.; Tang, T. Expression Variations of miRNAs and mRNAs in Rice (Oryza sativa). Genome Biol. Evol. 2016, 8, 3529–3544. [Google Scholar] [CrossRef]
- Guo, Y.; Gao, C.; Wang, M.; Fu, F.-f.; El-Kassaby, Y.A.; Wang, T.; Wang, G. Metabolome and transcriptome analyses reveal flavonoids biosynthesis differences in Ginkgo biloba associated with environmental conditions. Ind. Crops Prod. 2020, 158, 112963. [Google Scholar] [CrossRef]
- Li, F.; Boateng, I.D.; Chen, S.; Yang, X.M.; Soetanto, D.A.; Liu, W. Pulsed light irradiation improves degradation of ginkgolic acids and retainment of ginkgo flavonoids and terpene trilactones in Ginkgo biloba leaves. Ind. Crops Prod. 2023, 204, 117297. [Google Scholar] [CrossRef]
- Li, L.; Stanton, J.D.; Tolson, A.H.; Luo, Y.; Wang, H. Bioactive terpenoids and flavonoids from Ginkgo biloba extract induce the expression of hepatic drug-metabolizing enzymes through pregnane X receptor, constitutive androstane receptor, and aryl hydrocarbon receptor-mediated pathways. Pharm. Res. 2009, 26, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Cai, J.; Liu, H.; Lu, Z.; Hu, J.; Lu, Y. Transcriptomic analysis reveals regulatory networks for osmotic water stress and rewatering response in the leaves of Ginkgo biloba. Forests 2021, 12, 1705. [Google Scholar] [CrossRef]
- Zhang, C.; Guo, J.; Chen, G.; Xie, H. Effects of high temperature and/or drought on growth and secondary metabolites in Ginkgo biloba leaves. J. Ecol. Rural Environ. 2005, 21, 11–15. [Google Scholar]
- Wang, H.; Xie, B.; Jiang, Y.; Wang, M. Effect of solar irradiation intensity on leaf development and flavonoid and Terpene content in Ginkgo biloba leaves. Acta Agric. Univ. Jiangxiensis 2002, 24, 617–622. [Google Scholar]
- Cardenas, P.D.; Sonawane, P.D.; Pollier, J.; Vanden Bossche, R.; Dewangan, V.; Weithorn, E.; Tal, L.; Meir, S.; Rogachev, I.; Malitsky, S.; et al. GAME9 regulates the biosynthesis of steroidal alkaloids and upstream isoprenoids in the plant mevalonate pathway. Nat. Commun. 2016, 7, 10654. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, H.; Li, F.; Chen, Z.; Li, X.; Zhu, L.; Wang, G.; Yu, J.; Huang, D.; Lang, Z. The maize transcription factor EREB58 mediates the jasmonate-induced production of sesquiterpene volatiles. Plant J. 2015, 84, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Li, J.; Yang, C.; Hu, W.; Wang, L.; Chen, X. The jasmonate-responsive AP2/ERF transcription factors AaERF1 and AaERF2 positively regulate artemisinin biosynthesis in Artemisia annua L. Mol. Plant 2012, 5, 353–365. [Google Scholar] [CrossRef]
- Wang, M.; Gao, M.; Zhao, Y.; Chen, Y.; Wu, L.; Yin, H.; Xiong, S.; Wang, S.; Wang, J.; Yang, Y.; et al. LcERF19, an AP2/ERF transcription factor from Litsea cubeba, positively regulates geranial and neral biosynthesis. Hortic. Res. 2022, 9, uhac093. [Google Scholar] [CrossRef]
- Paul, P.; Singh, S.K.; Patra, B.; Sui, X.; Pattanaik, S.; Yuan, L. A differentially regulated AP2/ERF transcription factor gene cluster acts downstream of a MAP kinase cascade to modulate terpenoid indole alkaloid biosynthesis in Catharanthus roseus. New Phytol. 2017, 213, 1107–1123. [Google Scholar] [CrossRef]
- Nieuwenhuizen, N.J.; Chen, X.; Wang, M.Y.; Matich, A.J.; Perez, R.L.; Allan, A.C.; Green, S.A.; Atkinson, R.G. Natural variation in monoterpene synthesis in kiwifruit: Transcriptional regulation of terpene synthases by NAC and ETHYLENE-INSENSITIVE3-like transcription factors. Plant Physiol. 2015, 167, 1243–1258. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, X.; Wang, G.; Cui, P.; Wu, S.; Ai, C.; Hu, N.; Li, A.; He, B.; Shao, X.; et al. The nearly complete genome of Ginkgo biloba illuminates gymnosperm evolution. Nat. Plants 2021, 7, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Jatan, R.; Lata, C. Role of microRNAs in abiotic and biotic stress resistance in plants. Proc. Indian Natl. Sci. Acad. 2019, 85, 553–567. [Google Scholar]
- Luan, M.; Xu, M.; Lu, Y.; Zhang, L.; Fan, Y.; Wang, L. Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene 2015, 555, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Moradi, K.; Khalili, F. Assessment of pattern expression of miR172 and miR169 in response to drought stress in Echinacea purpurea L. Biocatal. Agric. Biotechnol. 2018, 16, 507–512. [Google Scholar] [CrossRef]
- Wu, J.; Wang, L.; Wang, S. MicroRNAs associated with drought response in the pulse crop common bean (Phaseolus vulgaris L.). Gene 2017, 628, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Serivichyaswat, P.T.; Susila, H.; Ahn, J.H. Elongated Hypocotyl 5-Homolog (HYH) negatively regulates expression of the ambient temperature-responsive microRNA gene MIR169. Front. Plant Sci. 2017, 8, 2087. [Google Scholar] [CrossRef]
- Sun, X.; Wang, C.; Xiang, N.; Li, X.; Yang, S.; Du, J.; Yang, Y.; Yang, Y. Activation of secondary cell wall biosynthesis by miR319-targeted TCP4 transcription factor. Plant Biotechnol. J. 2017, 15, 1284–1294. [Google Scholar] [CrossRef]
- Liu, Y.; Li, D.; Yan, J.; Wang, K.; Luo, H.; Zhang, W. MiR319 mediated salt tolerance by ethylene. Plant Biotechnol. J. 2019, 17, 2370–2383. [Google Scholar] [CrossRef]
- National Meteorological Science Data Center. Available online: https://data.cma.cn/ (accessed on 2 January 2022).
- Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China; Chinese Medical Science and Technology Press: Beijing, China, 2010. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Langdon, W.B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. Biodata Min. 2015, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [PubMed]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, 1040–1045. [Google Scholar] [CrossRef]
- Qi, Y.; Xue, L.; El-Kassaby, Y.; Guo, Y. Identification and comparative analysis of conserved and species-specific microRNAs in four Populus sections. Forests 2022, 13, 873. [Google Scholar] [CrossRef]
- Axtell, M.J. ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinform. 2010, 32, 11.7.1–11.7.14. [Google Scholar] [CrossRef]
- Prufer, K.; Stenzel, U.; Dannemann, M.; Green, R.E.; Lachmann, M.; Kelso, J. PatMaN: Rapid alignment of short sequences to large databases. Bioinformatics 2008, 24, 1530–1531. [Google Scholar] [CrossRef]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2010, 27, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Qi, Y.; Feng, Y.; Yang, Y.; Xue, L.; El-Kassaby, Y.A.; Wang, G.; Fu, F. Inferring the Regulatory Network of miRNAs on Terpene Trilactone Biosynthesis Affected by Environmental Conditions. Int. J. Mol. Sci. 2023, 24, 17002. https://doi.org/10.3390/ijms242317002
Guo Y, Qi Y, Feng Y, Yang Y, Xue L, El-Kassaby YA, Wang G, Fu F. Inferring the Regulatory Network of miRNAs on Terpene Trilactone Biosynthesis Affected by Environmental Conditions. International Journal of Molecular Sciences. 2023; 24(23):17002. https://doi.org/10.3390/ijms242317002
Chicago/Turabian StyleGuo, Ying, Yongli Qi, Yangfan Feng, Yuting Yang, Liangjiao Xue, Yousry A. El-Kassaby, Guibin Wang, and Fangfang Fu. 2023. "Inferring the Regulatory Network of miRNAs on Terpene Trilactone Biosynthesis Affected by Environmental Conditions" International Journal of Molecular Sciences 24, no. 23: 17002. https://doi.org/10.3390/ijms242317002
APA StyleGuo, Y., Qi, Y., Feng, Y., Yang, Y., Xue, L., El-Kassaby, Y. A., Wang, G., & Fu, F. (2023). Inferring the Regulatory Network of miRNAs on Terpene Trilactone Biosynthesis Affected by Environmental Conditions. International Journal of Molecular Sciences, 24(23), 17002. https://doi.org/10.3390/ijms242317002