
Pharmacokinetics of Novel Dopamine Transporter Inhibitor CE-123 and Modafinil with a Focus on Central Nervous System Distribution
 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
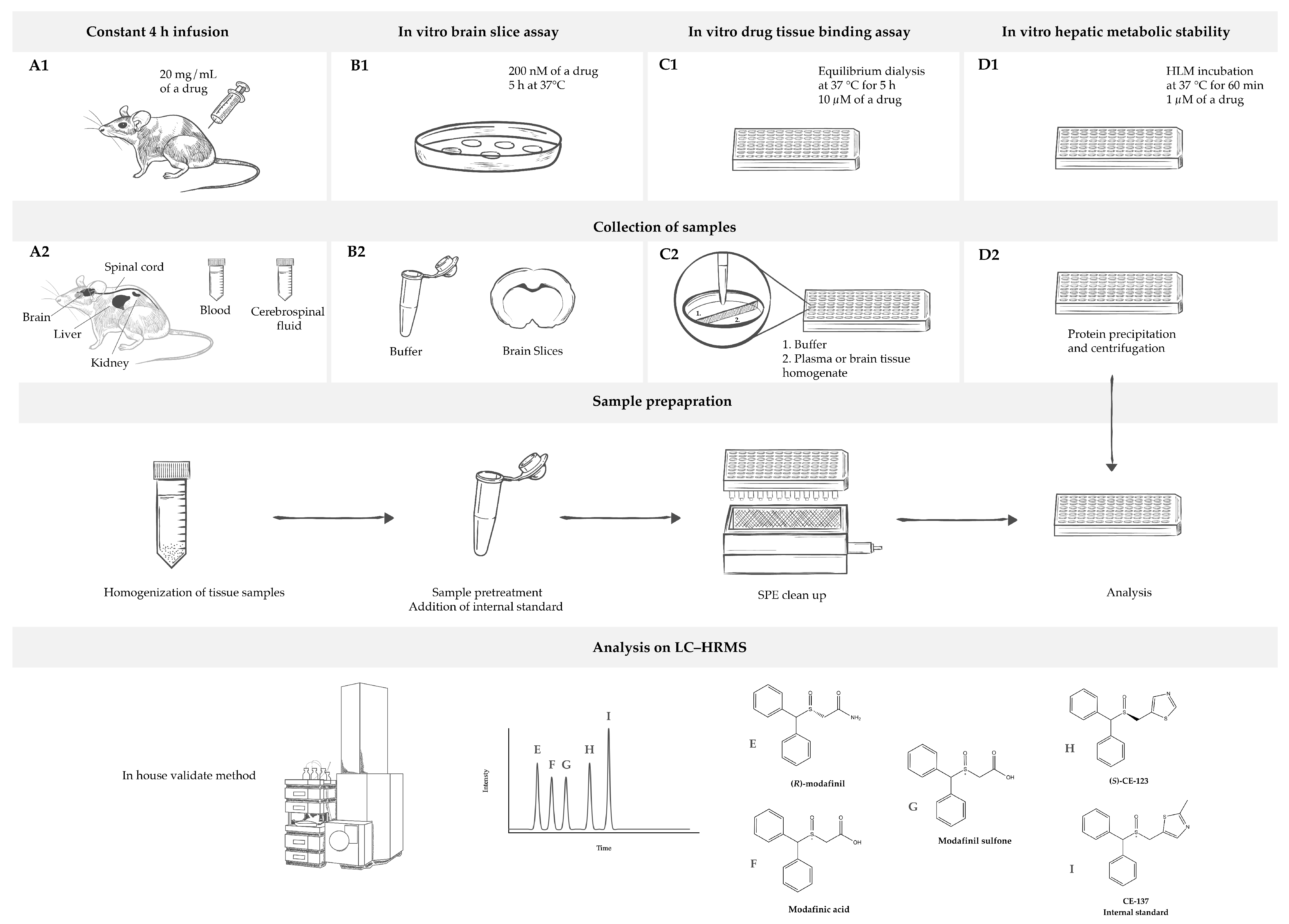
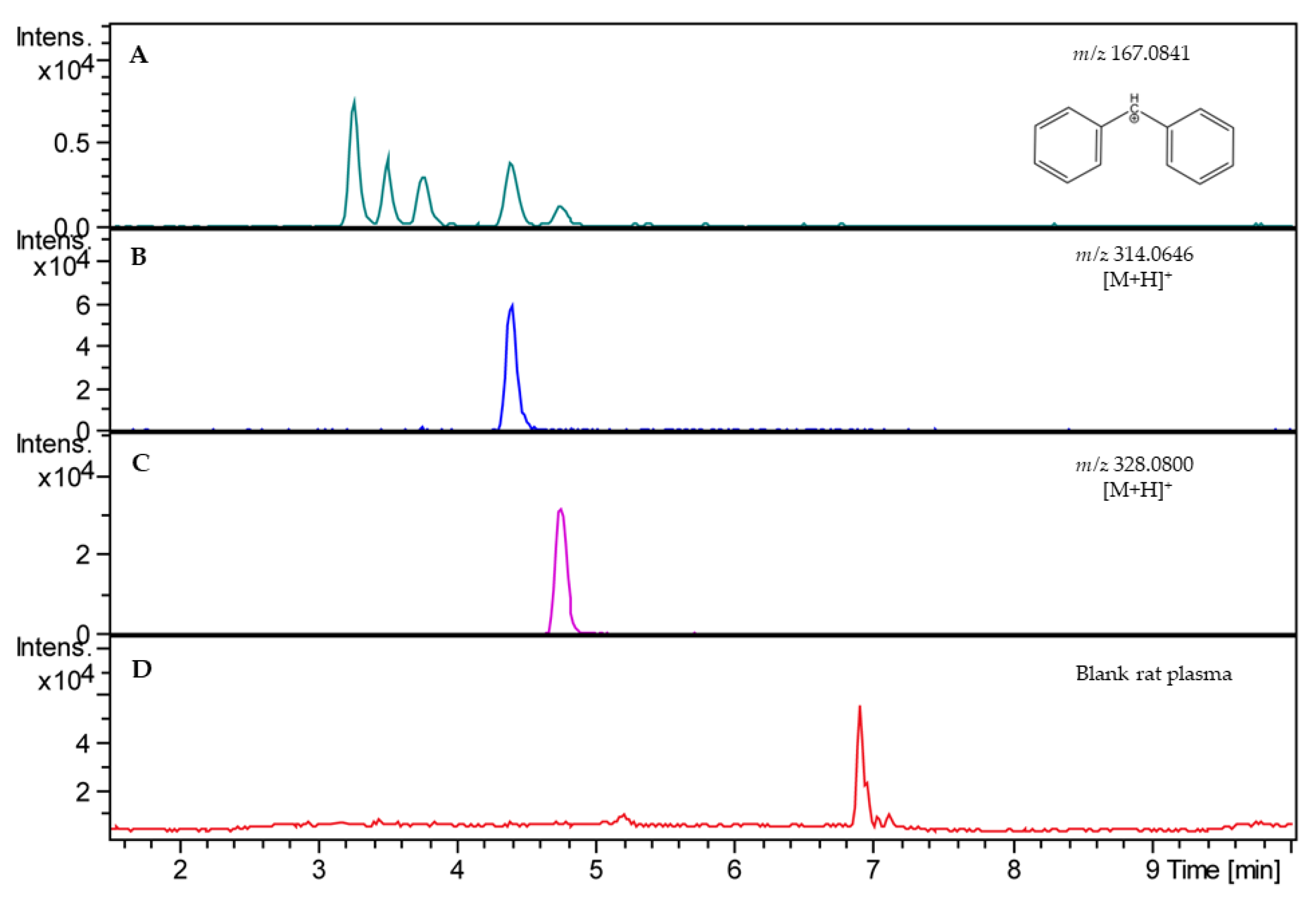
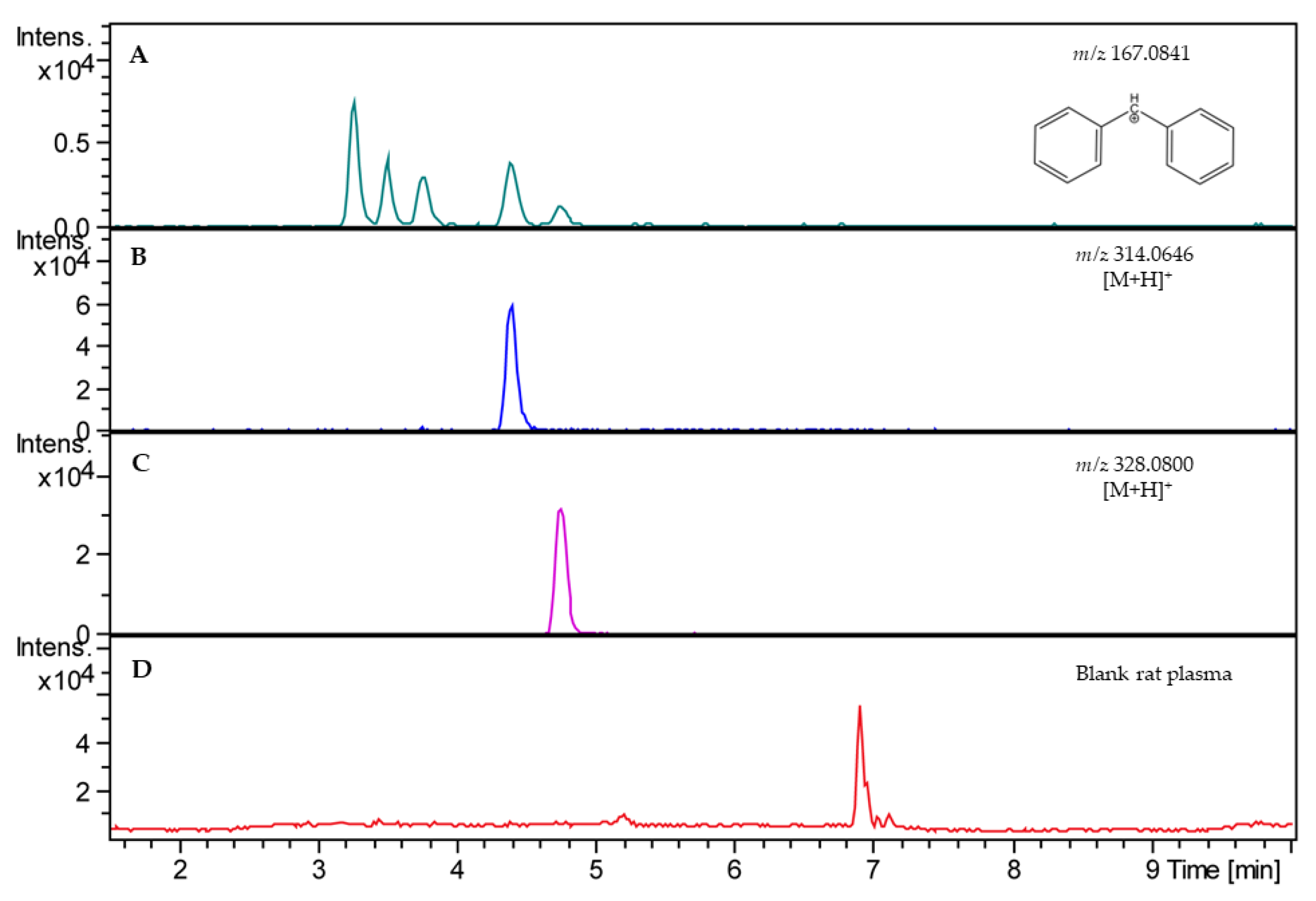
2.1. Analytical Method
2.2. Validation of Analytical Method
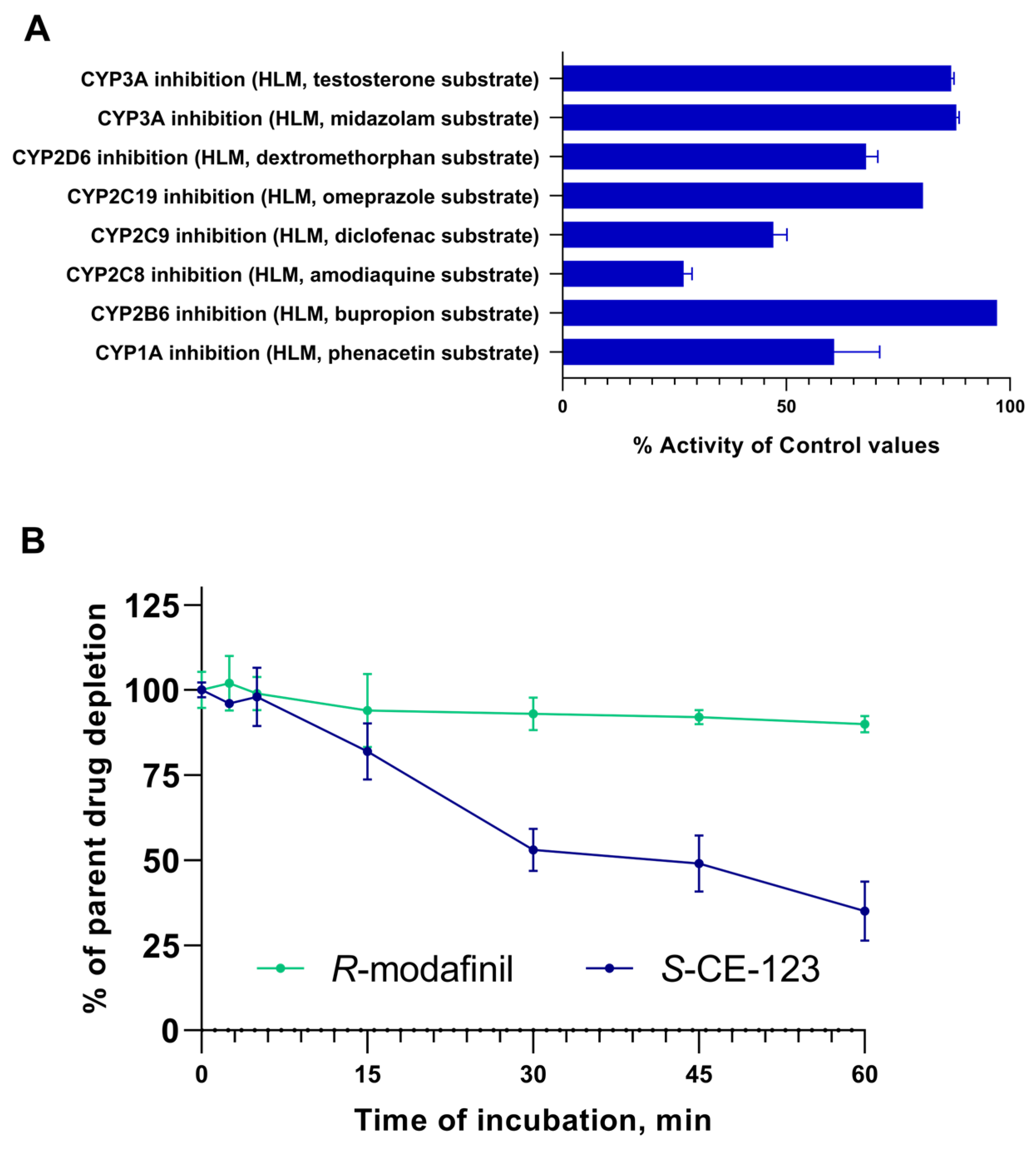
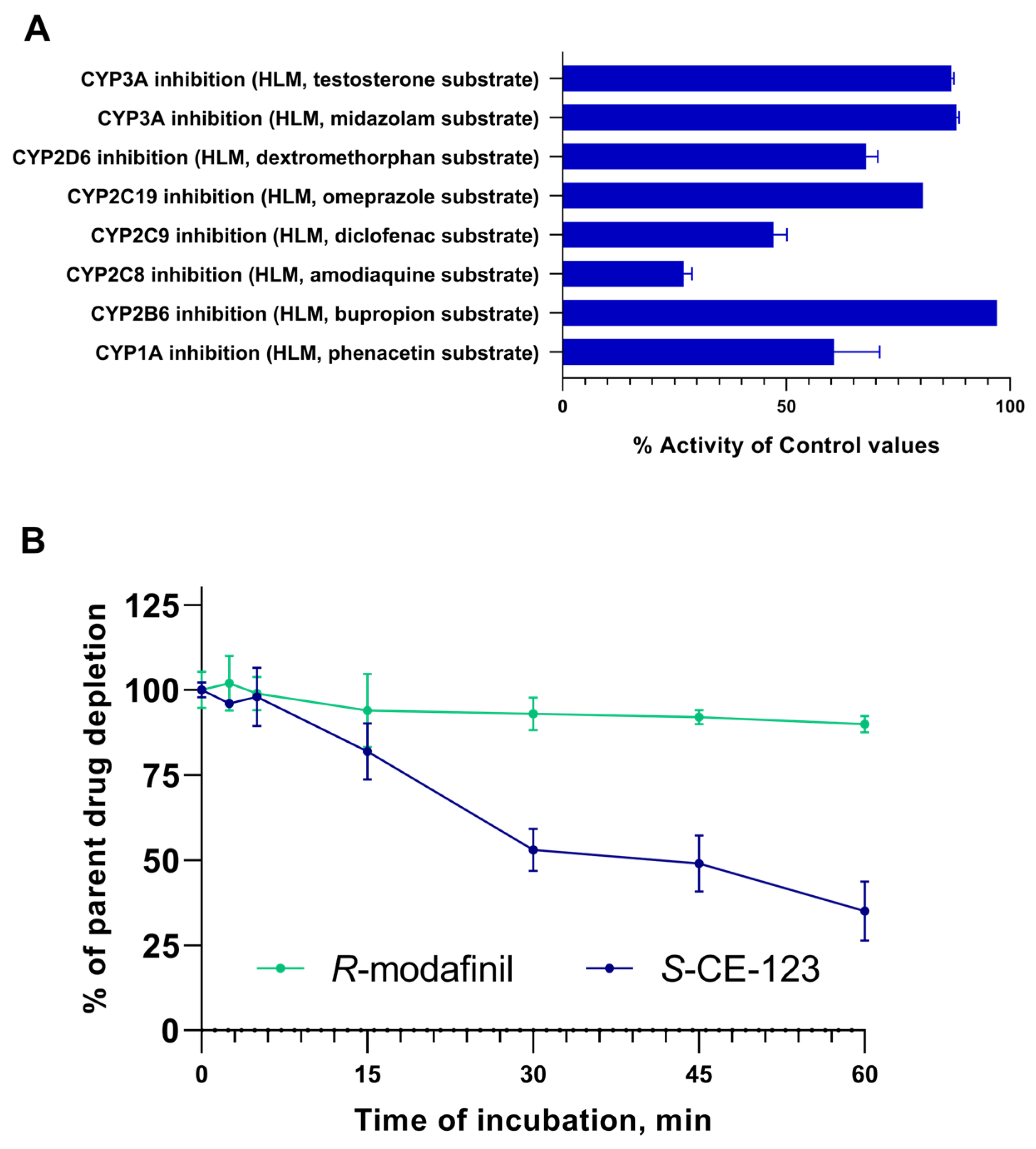
2.3. Metabolism Assays
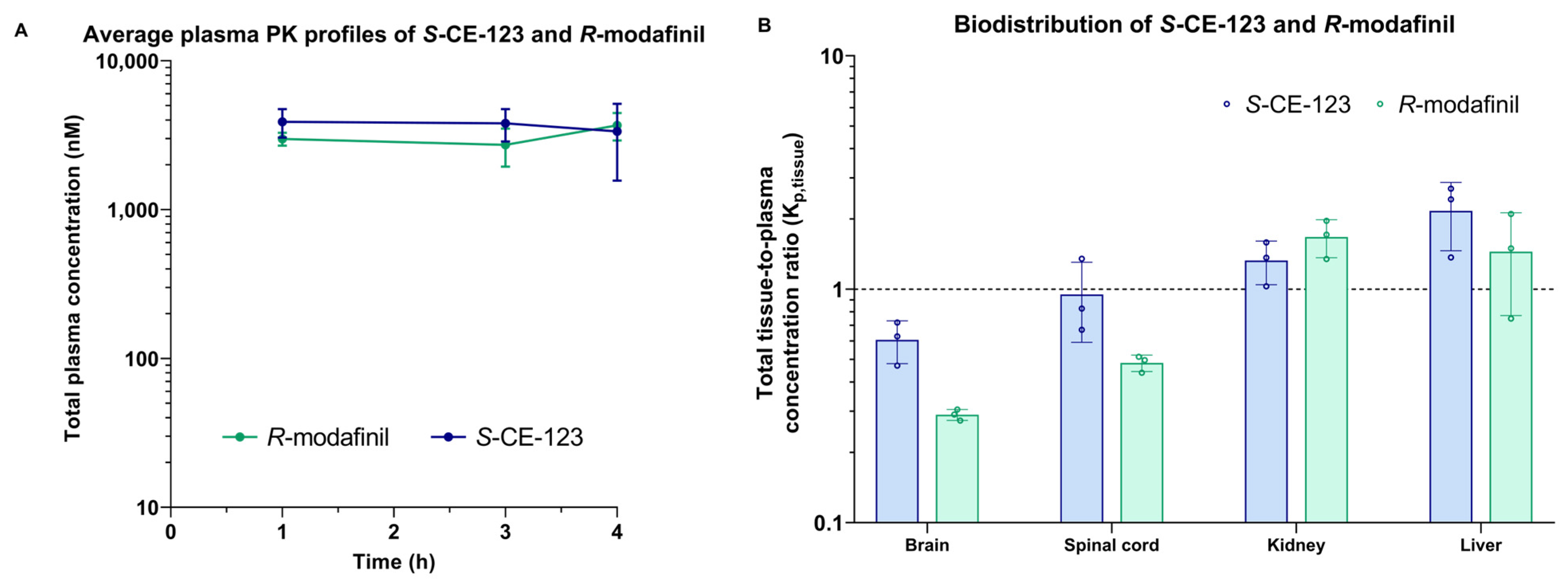
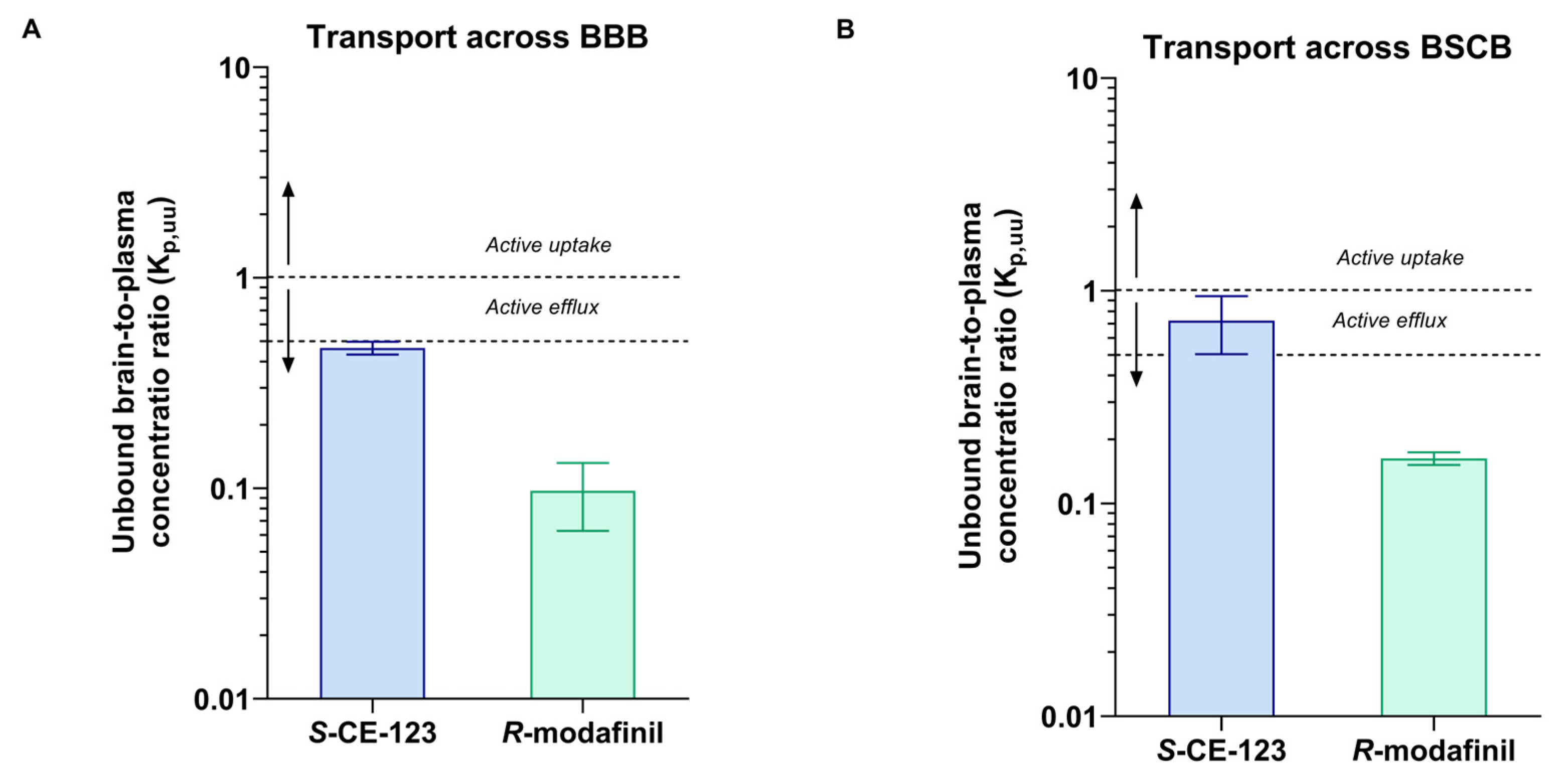
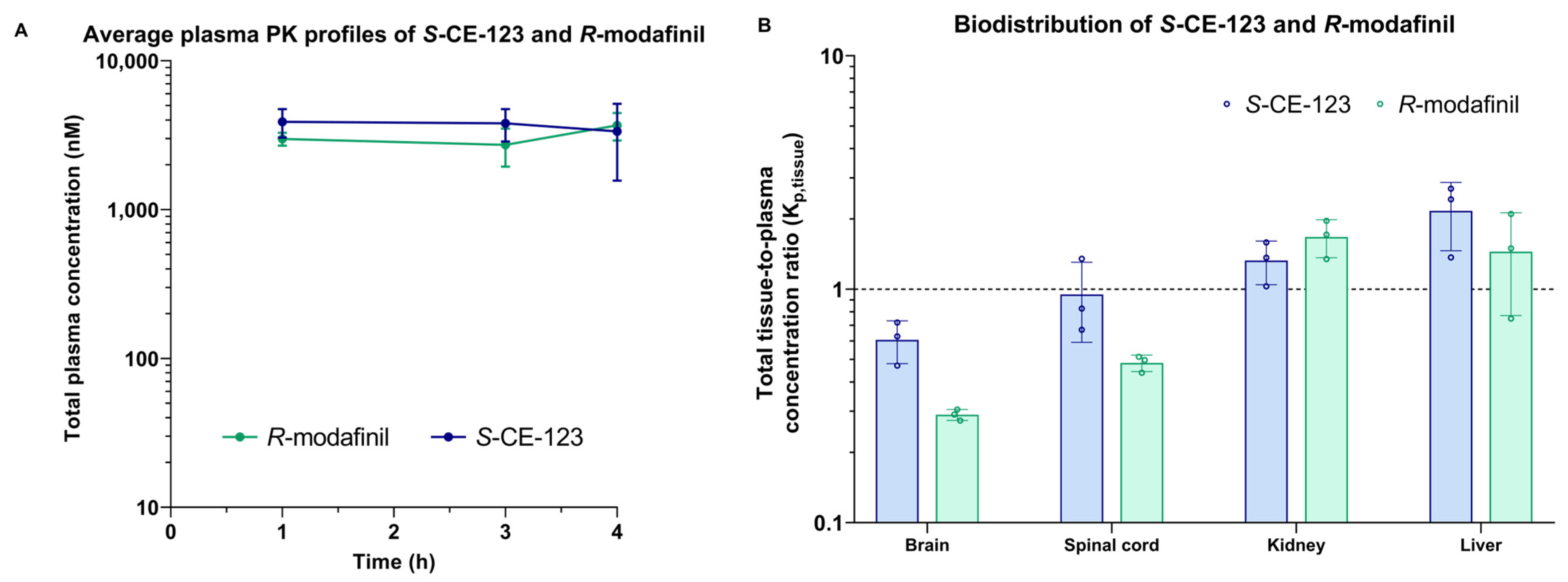
2.4. Biodistribution
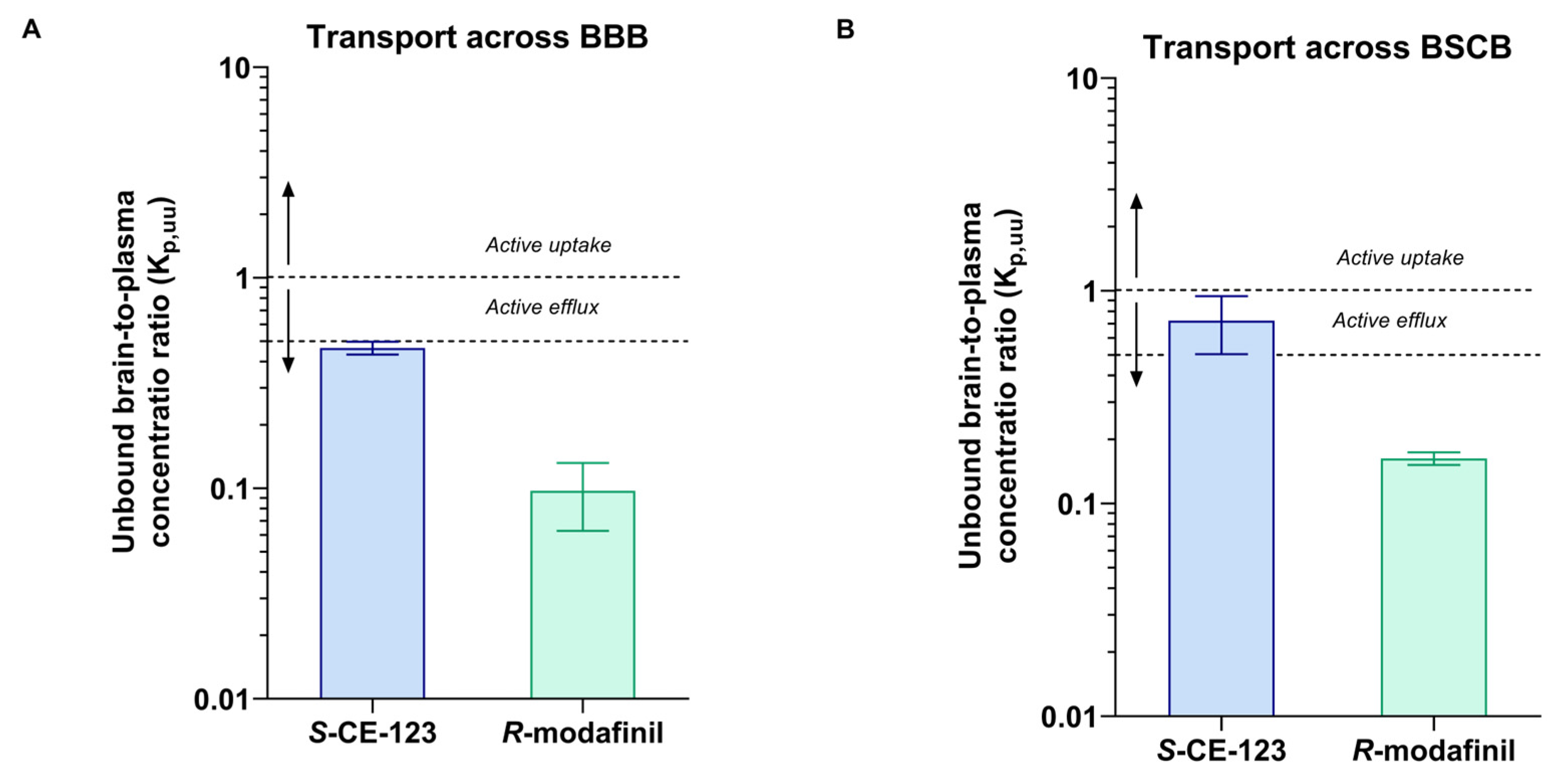
2.5. Unbound Drug PK Parameters
3. Discussion
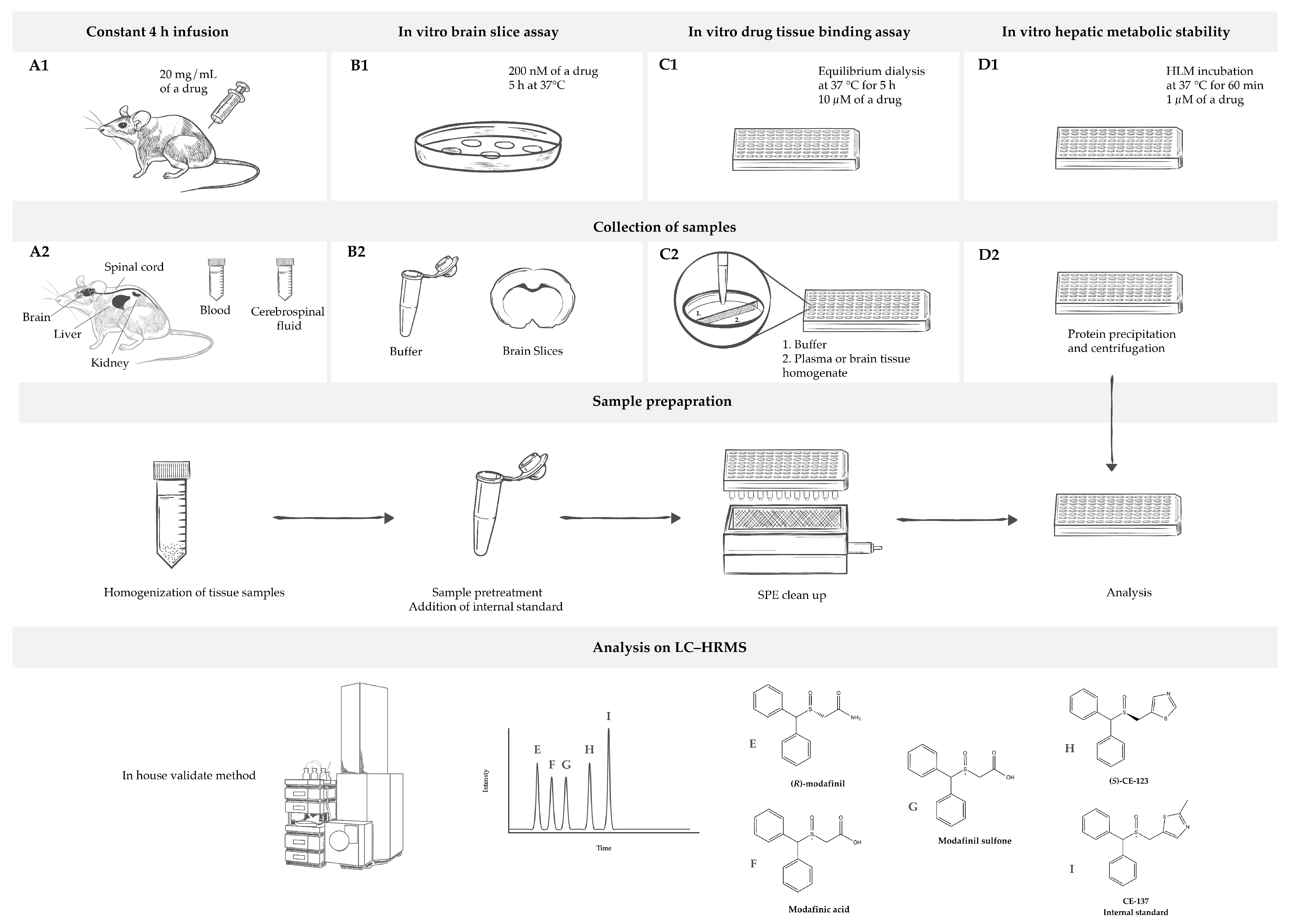
4. Materials and Methods
4.1. Materials
4.2. Preparation of Stock Solutions and Internal Standard Solutions
4.3. Animals
4.4. In Vitro Drug Tissue Binding Assay
4.5. Brain Slice Assay
4.6. Tissue Biodistribution Studies
4.7. NeuroPK Parameters
4.8. Drug Metabolism
4.9. Sample Preparation for Analysis
4.10. LC–HRMS Analysis
4.11. Validation of Analytical Method
4.12. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Y.X.; Liang, N.; Li, X.L.; Yang, S.H.; Wang, Y.P.; Shi, N.N. Diagnosis and Treatment for Mild Cognitive Impairment: A Systematic Review of Clinical Practice Guidelines and Consensus Statements. Front. Neurol. 2021, 12, 719849. [Google Scholar] [CrossRef]
- Anderson, N.D. State of the science on mild cognitive impairment (MCI). CNS Spectr. 2019, 24, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Batzu, L.; Halliday, G.M.; Geurtsen, G.J.; Ballard, C.; Ray Chaudhuri, K.; Weintraub, D. Parkinson disease-associated cognitive impairment. Nat. Rev. Dis. Prim. 2021, 7, 47. [Google Scholar] [CrossRef]
- McCutcheon, R.A.; Keefe, R.S.E.; McGuire, P.K. Cognitive impairment in schizophrenia: Aetiology, pathophysiology, and treatment. Mol. Psychiatry 2023, 28, 1902–1918. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Du, Y.; Chu, L.; Zhang, Z.; Li, F.; Lyu, D.; Li, Y.; Zhu, M.; Jiao, H.; Song, Y.; et al. Prevalence, risk factors, and management of dementia and mild cognitive impairment in adults aged 60 years or older in China: A cross-sectional study. Lancet Public Health 2020, 5, e661–e671. [Google Scholar] [CrossRef]
- Vlachos, G.S.; Kosmidis, M.H.; Yannakoulia, M.; Dardiotis, E.; Hadjigeorgiou, G.; Sakka, P.; Ntanasi, E.; Stefanis, L.; Scarmeas, N. Prevalence of Mild Cognitive Impairment in the Elderly Population in Greece: Results from the HELIAD Study. Alzheimer Dis. Assoc. Disord. 2019, 34, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Mohan, D.; Iype, T.; Varghese, S.; Usha, A.; Mohan, M. A cross-sectional study to assess prevalence and factors associated with mild cognitive impairment among older adults in an urban area of Kerala, South India. BMJ Open 2019, 9, e025473. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.C.; Wu, Y.T.; Prina, M. World Alzheimer Report 2015: The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Alzheimer’s Disease International (ADI). Dementia Statistics. Available online: https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/ (accessed on 5 July 2023).
- Speranza, L.; Di Porzio, U.; Viggiano, D.; de Donato, A.; Volpicelli, F. Dopamine: The Neuromodulator of Long-Term Synaptic Plasticity, Reward and Movement Control. Cells 2021, 10, 735. [Google Scholar] [CrossRef]
- Nieoullon, A. Dopamine and the regulation of cognition and attention. Prog. Neurobiol. 2002, 67, 53–83. [Google Scholar] [CrossRef]
- Vaughan, R.A.; Foster, J.D. Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol. Sci. 2013, 34, 489–496. [Google Scholar] [CrossRef]
- Husain, M.; Mehta, M.A. Cognitive enhancement by drugs in health and disease. Trends Cogn. Sci. 2011, 15, 28–36. [Google Scholar] [CrossRef] [PubMed]
- de Jongh, R.; Bolt, I.; Schermer, M.; Olivier, B. Botox for the brain: Enhancement of cognition, mood and pro-social behavior and blunting of unwanted memories. Neurosci. Biobehav. Rev. 2008, 32, 760–776. [Google Scholar] [CrossRef]
- Venero, C. Pharmacological Treatment of Cognitive Dysfunction in Neuropsychiatric Disorders. In Cognitive Enhancement Pharmacologic, Environmental and Genetic Factors; Knafo, S., Venero, C., Eds.; Elsevier GmbH: München, Germany, 2015; pp. 233–271. ISBN 978-0-12-417042-1. [Google Scholar]
- Minzenberg, M.J.; Carter, C.S. Modafinil: A review of neurochemical actions and effects on cognition. Neuropsychopharmacology 2008, 33, 1477–1502. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.C.; Robbins, T.W.; Clark, L.; Aron, A.R.; Dowson, J.; Sahakian, B.J. Cognitive enhancing effects of modafinil in healthy volunteers. Psychopharmacology 2003, 165, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Bobo, W.V.; Woodward, N.D.; Sim, M.Y.; Jayathilake, K.; Meltzer, H.Y. The effect of adjunctive armodafinil on cognitive performance and psychopathology in antipsychotic-treated patients with schizophrenia/schizoaffective disorder: A randomized, double-blind, placebo-controlled trial. Schizophr. Res. 2011, 130, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.C.; Clark, L.; Pomarol-Clotet, E.; McKenna, P.; Robbins, T.W.; Sahakian, B.J. Modafinil improves cognition and attentional set shifting in patients with chronic schizophrenia. Neuropsychopharmacology 2004, 29, 1363–1373. [Google Scholar] [CrossRef]
- Wang, S.M.; Han, C.; Lee, S.J.; Jun, T.Y.; Patkar, A.A.; Masand, P.S.; Pae, C.U. Modafinil for the treatment of attention-deficit/hyperactivity disorder: A meta-analysis. J. Psychiatr. Res. 2017, 84, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.C.; Clark, L.; Dowson, J.; Robbins, T.W.; Sahakian, B.J. Modafinil improves cognition and response inhibition in adult attention-deficit/hyperactivity disorder. Biol. Psychiatry 2004, 55, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- DeBattista, C.; Lembke, A.; Solvason, H.B.; Ghebremichael, R.; Poirier, J. A Prospective Trial of Modafinil as an Adjunctive Treatment of Major Depression. J. Clin. Psychopharmacol. 2004, 24, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Ketter, T.A.; Yang, R.; Frye, M.A. Adjunctive armodafinil for major depressive episodes associated with bipolar I disorder. J. Affect. Disord. 2015, 181, 87–91. [Google Scholar] [CrossRef]
- Ketter, T.A.; Amchin, J.; Frye, M.A.; Gross, N. Long-term safety and efficacy of armodafinil in bipolar depression: A 6-month open-label extension study. J. Affect. Disord. 2016, 197, 51–57. [Google Scholar] [CrossRef]
- Perugi, G.; Vannucchi, G.; Bedani, F.; Favaretto, E. Use of Stimulants in Bipolar Disorder. Curr. Psychiatry Rep. 2017, 19, 7. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.C.; Reith, M.E.A. The Atypical Stimulant and Nootropic Modafinil Interacts with the Dopamine Transporter in a Different Manner than Classical Cocaine-Like Inhibitors. PLoS ONE 2011, 6, e25790. [Google Scholar] [CrossRef] [PubMed]
- Loland, C.J.; Mereu, M.; Okunola, O.M.; Cao, J.; Prisinzano, T.E.; Mazier, S.; Kopajtic, T.; Shi, L.; Katz, J.L.; Tanda, G.; et al. R-Modafinil (Armodafinil): A Unique Dopamine Uptake Inhibitor and Potential Medication for Psychostimulant Abuse. Biol. Psychiatry 2012, 72, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Marcan Pharmaceuticals Inc. Product Monograph Pr Mar-Modafinil Modafinil Tablets, USP 100 mg Central Nervous System Stimulant; Marcan Pharmaceuticals Inc.: Ottawa, ON, Canada, 2017. [Google Scholar]
- Beryozkina, T.; Bakulev, V.; Dianova, L.; Berseneva, V.; Slepukhin, P.; Leban, J.; Kalaba, P.; Aher, N.Y.; Ilic, M.; Sitte, H.H.; et al. Design and Synthesis of N-Sulfonylamidines of Modafinic Acid. Synthesis 2016, 48, 1046–1054. [Google Scholar]
- Cao, J.; Prisinzano, T.E.; Okunola, O.M.; Kopajtic, T.; Shook, M.; Katz, J.L.; Newman, A.H. SARs at the monoamine transporters for a novel series of modafinil analogues. ACS Med. Chem. Lett. 2011, 2, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Slack, R.D.; Bakare, O.M.; Burzynski, C.; Rais, R.; Slusher, B.S.; Kopajtic, T.; Bonifazi, A.; Ellenberger, M.P.; Yano, H.; et al. Novel and High Affinity 2-[(Diphenylmethyl)sulfinyl]acetamide (Modafinil) Analogues as Atypical Dopamine Transporter Inhibitors. J. Med. Chem. 2016, 59, 10676–10691. [Google Scholar] [CrossRef]
- Jung, J.C.; Lee, Y.; Son, J.Y.; Lim, E.; Jung, M.; Oh, S. Simple Synthesis of Modafinil Derivatives and Their Anti-inflammatory Activity. Molecules 2012, 17, 10446–10458. [Google Scholar] [CrossRef]
- Kalaba, P.; Aher, N.Y.; Ilić, M.; Dragačević, V.; Wieder, M.; Miklosi, A.G.; Zehl, M.; Wackerlig, J.; Roller, A.; Beryozkina, T.; et al. Heterocyclic Analogues of Modafinil as Novel, Atypical Dopamine Transporter Inhibitors. J. Med. Chem. 2017, 60, 9330–9348. [Google Scholar] [CrossRef]
- Kalaba, P.; Ilić, M.; Aher, N.Y.; Dragačević, V.; Wieder, M.; Zehl, M.; Wackerlig, J.; Beyl, S.; Sartori, S.B.; Ebner, K.; et al. Structure-Activity Relationships of Novel Thiazole-Based Modafinil Analogues Acting at Monoamine Transporters. J. Med. Chem. 2020, 63, 391–417. [Google Scholar] [CrossRef]
- Nikiforuk, A.; Kalaba, P.; Ilic, M.; Korz, V.; Dragačević, V.; Wackerlig, J.; Langer, T.; Höger, H.; Golebiowska, J.; Popik, P.; et al. A novel dopamine transporter inhibitor CE-123 improves cognitive flexibility and maintains impulsivity in healthy male rats. Front. Behav. Neurosci. 2017, 11, 222. [Google Scholar] [CrossRef]
- Kristofova, M.; Aher, Y.D.; Ilic, M.; Radoman, B.; Kalaba, P.; Dragacevic, V.; Aher, N.Y.; Leban, J.; Korz, V.; Zanon, L.; et al. A daily single dose of a novel modafinil analogue CE-123 improves memory acquisition and memory retrieval. Behav. Brain Res. 2018, 343, 83–94. [Google Scholar] [CrossRef]
- Gibula-Tarlowska, E.; Korz, V.; Lopatynska-Mazurek, M.; Chlopas-Konowalek, A.; Grochecki, P.; Kalaba, P.; Dragacevic, V.; Kotlinski, R.; Kujawski, R.; Szulc, M.; et al. CE-123, a novel dopamine transporter inhibitor, attenuates locomotor hyperactivity and improves cognitive functions in rat model of fetal alcohol spectrum disorders. Behav. Brain Res. 2021, 410, 113326. [Google Scholar] [CrossRef] [PubMed]
- Saroja, S.R.; Aher, Y.D.; Kalaba, P.; Aher, N.Y.; Zehl, M.; Korz, V.; Subramaniyan, S.; Miklosi, A.G.; Zanon, L.; Neuhaus, W.; et al. A novel heterocyclic compound targeting the dopamine transporter improves performance in the radial arm maze and modulates dopamine receptors D1–D3. Behav. Brain Res. 2016, 312, 127–137. [Google Scholar] [CrossRef]
- Hammarlund-Udenaes, M.; Fridén, M.; Syvänen, S.; Gupta, A. On the rate and extent of drug delivery to the brain. Pharm. Res. 2008, 25, 1737–1750. [Google Scholar] [CrossRef] [PubMed]
- Loryan, I.; Reichel, A.; Feng, B.; Bundgaard, C.; Shaffer, C.; Kalvass, C.; Bednarczyk, D.; Morrison, D.; Lesuisse, D.; Hoppe, E.; et al. Unbound Brain-to-Plasma Partition Coefficient, Kp,uu,brain—A Game Changing Parameter for CNS Drug Discovery and Development. Pharm. Res. 2022, 39, 1321–1341. [Google Scholar] [CrossRef]
- Summerfield, S.G.; Yates, J.W.T.; Fairman, D.A. Free Drug Theory—No Longer Just a Hypothesis? Pharm. Res. 2022, 39, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Loryan, I.; Sinha, V.; Mackie, C.; Van Peer, A.; Drinkenburg, W.; Vermeulen, A.; Morrison, D.; Monshouwer, M.; Heald, D.; Hammarlund-Udenaes, M. Mechanistic understanding of brain drug disposition to optimize the selection of potential neurotherapeutics in drug discovery. Pharm. Res. 2014, 31, 2203–2219. [Google Scholar] [CrossRef]
- Fridén, M.; Ducrozet, F.; Middleton, B.; Antonsson, M.; Bredberg, U.; Hammarlund-Udenaes, M. Development of a high-throughput brain slice method for studying drug distribution in the central nervous system. Drug Metab. Dispos. 2009, 37, 1226–1233. [Google Scholar] [CrossRef]
- Loryan, I.; Fridén, M.; Hammarlund-Udenaes, M. The brain slice method for studying drug distribution in the CNS. Fluids Barriers CNS 2013, 10, 1226–1233. [Google Scholar] [CrossRef]
- van Liempd, S.; Morrison, D.; Sysmans, L.; Nelis, P.; Mortishire-Smith, R. Development and validation of a higher-throughput equilibrium dialysis assay for plasma protein binding. J. Lab. Autom. 2011, 16, 56–67. [Google Scholar] [CrossRef]
- Kalvass, J.C.; Maurer, T.S. Influence of nonspecific brain and plasma binding on CNS exposure: Implications for rational drug discovery. Biopharm. Drug Dispos. 2002, 23, 327–338. [Google Scholar] [CrossRef]
- Wan, H.; Rehngren, M.; Giordanetto, F.; Bergström, F.; Tunek, A. High-throughput screening of drug-brain tissue binding and in silico prediction for assessment of central nervous system drug delivery. J. Med. Chem. 2007, 50, 4606–4615. [Google Scholar] [CrossRef]
- Fridén, M.; Gupta, A.; Antonsson, M.; Bredberg, U.; Hammarlund-Udenaes, M. In Vitro Methods for Estimating Unbound Drug Concentrations in the Brain Interstitial and Intracellular Fluids. Drug Metab. Dispos. 2007, 35, 1711–1719. [Google Scholar] [CrossRef]
- Li, P.; Bartlett, M.G. A review of sample preparation methods for quantitation of small-molecule analytes in brain tissue by liquid chromatography tandem mass spectrometry (LC-MS/MS). Anal. Methods 2014, 6, 6183–6207. [Google Scholar] [CrossRef]
- Nasiri, A.; Jahani, R.; Mokhtari, S.; Yazdanpanah, H.; Daraei, B.; Faizi, M.; Kobarfard, F. Overview, consequences, and strategies for overcoming matrix effects in LC-MS analysis: A critical review. Analyst 2021, 146, 6049–6063. [Google Scholar] [CrossRef] [PubMed]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Validation of analytical procedures: Text and methodology Q2(R1). Available online: https://www.ich.org/page/quality-guidelines (accessed on 25 July 2022).
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Bioanalyitical Method Validation and Study Sample Analysis M10. Available online: https://www.ema.europa.eu/en/ich-m10-bioanalytical-method-validation-scientific-guideline (accessed on 25 July 2022).
- Gorman, S.H. Determination of modafinil, modafinil acid and modafinil sulfone in human plasma utilizing liquid-liquid extraction and high-performance liquid chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 767, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Loryan, I.; Hoppe, E.; Hansen, K.; Held, F.; Kless, A.; Linz, K.; Marossek, V.; Nolte, B.; Ratcliffe, P.; Saunders, D.; et al. Quantitative Assessment of Drug Delivery to Tissues and Association with Phospholipidosis: A Case Study with Two Structurally Related Diamines in Development. Mol. Pharm. 2017, 14, 4362–4373. [Google Scholar] [CrossRef]
- Loryan, I.; Hammarlund-Udenaes, M.; Syvänen, S. Brain Distribution of Drugs: Pharmacokinetic Considerations. Handb. Exp. Pharmacol. 2020, 273, 121–150. [Google Scholar]
- Lubec, J.; Hussein, A.M.; Kalaba, P.; Daba Feyissa, D.; Arias-Sandoval, E.; Cybulska-Klosowicz, A.; Bezu, M.; Stojanovic, T.; Korz, V.; Malikovic, J.; et al. Low-Affinity/High-Selectivity Dopamine Transport Inhibition Sufficient to Rescue Cognitive Functions in the Aging Rat. Biomolecules 2023, 13, 467. [Google Scholar] [CrossRef] [PubMed]
- Han, D.D.; Gu, H.H. Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol. 2006, 6, 6. [Google Scholar] [CrossRef]
- Robertson, P.; Hellriegel, E.T. Clinical pharmacokinetic profile of modafinil. Clin. Pharmacokinet. 2003, 42, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.H.; Guo, T.; Deng, C.H.; Guan, Z.; Li, L.; Zhou, T.Y.; Lu, W. Population pharmacokinetics of modafinil acid and estimation of the metabolic conversion of modafinil into modafinil acid in 5 major ethnic groups of China. Acta Pharmacol. Sin. 2012, 33, 1401–1408. [Google Scholar] [CrossRef]
- Hale, J.J.; Mills, S.G.; MacCoss, M.; Dorn, C.P.; Finke, P.E.; Budhu, R.J.; Reamer, R.A.; Huskey, S.E.W.; Luffer-Atlas, D.; Dean, B.J.; et al. Phosphorylated morpholine acetal human neurokinin-1 receptor antagonists as water-soluble prodrugs. J. Med. Chem. 2000, 43, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.J.; Wang, J.S.; Donovan, J.L.; Jiang, Y.; Gibson, B.B.; DeVane, C.L.; Markowitz, J.S. Interactions of attention-deficit/hyperactivity disorder therapeutic agents with the efflux transporter P-glycoprotein. Eur. J. Pharmacol. 2008, 578, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Loryan, I.; Melander, E.; Svensson, M.; Payan, M.; König, F.; Jansson, B.; Hammarlund-Udenaes, M. In-depth neuropharmacokinetic analysis of antipsychotics based on a novel approach to estimate unbound target-site concentration in CNS regions: Link to spatial receptor occupancy. Mol. Psychiatry 2016, 21, 1527–1536. [Google Scholar] [CrossRef]
- Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Lu, A.Y.H. Role of Pharmacokinetics and Metabolism in Drug Discovery and Development. Pharmacol. Rev. 1997, 49, 403–449. [Google Scholar]
- Guengerich, F.P. Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Flockhart, D.A.; Desta, Z. Pharmacogenetics of Drug Metabolism. In Clinical and Translational Science: Principles of Human Research, 1st ed.; Robertson, D., Williams, G.H., Eds.; Academic Press: Cambridge, MA, USA, 2009; pp. 301–317. [Google Scholar]
- Backman, J.T.; Filppula, A.M.; Niemi, M.; Neuvonen, P.J. Role of Cytochrome P450 2C8 in Drug Metabolism and Interactions. Pharmacol. Rev. 2016, 68, 168–241. [Google Scholar] [CrossRef] [PubMed]
- Dutheil, F.; Dauchy, S.; Diry, M.; Sazdovitch, V.; Cloarec, O.; Mellottée, L.; Bièche, I.; Ingelman-Sundberg, M.; Flinois, J.P.; de Waziers, I.; et al. Xenobiotic-Metabolizing Enzymes and Transporters in the Normal Human Brain: Regional and Cellular Mapping as a Basis for Putative Roles in Cerebral Function. Drug Metab. Dispos. 2009, 37, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhen, Y.; Miksys, S.; Beyoǧlu, D.; Krausz, K.W.; Tyndale, R.F.; Yu, A.; Idle, J.R.; Gonzalez, F.J. Potential role of CYP2D6 in the central nervous system. Xenobiotica 2013, 43, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.M.; Idle, J.R.; Gonzalez, F.J. Polymorphic Cytochrome P450 2D6: Humanized Mouse Model and Endogenous Substrates. Drug Metab. Rev. 2004, 36, 243–277. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.M.; Idle, J.R.; Byrd, L.G.; Krausz, K.W.; Küpfer, A.; Gonzalez, F.J. Regeneration of serotonin from 5-methoxytryptamine by polymorphic human CYP2D6. Pharmacogenetics 2003, 13, 173–181. [Google Scholar] [CrossRef]
- Robertson, P.; DeCory, H.H.; Madan, A.; Parkinson, A. In vitro inhibition and induction of human liver cytochrome P450 enzymes by milnacipran. Drug Metab. Dispos. 2000, 28, 664–671. [Google Scholar] [PubMed]
- Wong, Y.N.; King, S.P.; Sim, D.; Gor, S. Open-Label, Single-Dose Pharmacokinetic Study of Modafinil Tablets: Influence of Age and Gender in Normal Subjects. J. Clin. Pharmacol. 1999, 39, 281–288. [Google Scholar] [CrossRef]
- Wong, Y.N.; Simcoe, D.; Hartman, L.N.; Laughton, W.B.; King, S.P.; McCormick, G.C.; Grebow, P.E. A double-blind, placebo-controlled, ascending-dose evaluation of the pharmacokinetics and tolerability of modafinil tablets in healthy male volunteers. J. Clin. Pharmacol. 1999, 39, 30–40. [Google Scholar] [CrossRef]
- Wong, Y.N.; King, S.P.; Laughton, W.B.; McCormick, G.C.; Grebow, P.E. Single-dose pharmacokinetics of modafinil and methylphenidate given alone or in combination in healthy male volunteers. J. Clin. Pharmacol. 1998, 38, 276–282. [Google Scholar] [CrossRef]
- Chatterjie, N.; Stables, J.P.; Wang, H.; Alexander, G.J. Anti-narcoleptic agent modafinil and its sulfone: A novel facile synthesis and potential anti-epileptic activity. Neurochem. Res. 2004, 29, 1481–1486. [Google Scholar] [CrossRef]
- Zolkowska, D.; Andres-Mach, M.; Prisinzano, T.E.; Baumann, M.H.; Luszczki, J.J. Modafinil and its metabolites enhance the anticonvulsant action of classical antiepileptic drugs in the mouse maximal electroshock-induced seizure model. Psychopharmacology 2015, 232, 2463–2479. [Google Scholar] [CrossRef]
- Nageswara Rao, R.; Shinde, D.D.; Kumar Talluri, M.V.N. Development and validation of a reversed phase liquid chromatographic method for separation and determination of related-substances of modafinil in bulk drugs. Talanta 2007, 73, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, B.; Aher, Y.D.; Aradska, J.; Ilic, M.; Feyissa, D.D.; Kalaba, P.; Aher, N.Y.; Dragacevic, V.; Saber Marouf, B.; Langer, T.; et al. R-Modafinil exerts weak effects on spatial memory acquisition and dentate gyrus synaptic plasticity. PLoS ONE 2017, 12, e0179675. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, P.; Summerfield, S.G. Challenges for blood–brain barrier (BBB) screening. Xenobiotica 2008, 37, 1135–1151. [Google Scholar] [CrossRef]
- Schmid, R.; Heuckeroth, S.; Korf, A.; Smirnov, A.; Myers, O.; Dyrlund, T.S.; Bushuiev, R.; Murray, K.J.; Hoffmann, N.; Lu, M.; et al. Integrative analysis of multimodal mass spectrometry data in MZmine 3. Nat. Biotechnol. 2023, 41, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; MacLean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Korf, A.; Smirnov, A.; Schmid, R.; Fallon, T.R.; Du, X.; Weng, J.K. Metabolomics Data Analysis Using MZmine. New Dev. Mass. Spectrom. 2020, 2020, 232–254. [Google Scholar]
- Du, X.; Smirnov, A.; Pluskal, T.; Jia, W.; Sumner, S. Metabolomics Data Preprocessing Using ADAP and MZmine 2. Methods Mol. Biol. 2020, 2104, 25–48. [Google Scholar]
- Chang, H.Y.; Colby, S.M.; Du, X.; Gomez, J.D.; Helf, M.J.; Kechris, K.; Kirkpatrick, C.R.; Li, S.; Patti, G.J.; Renslow, R.S.; et al. A Practical Guide to Metabolomics Software Development. Anal. Chem. 2021, 93, 1912–1923. [Google Scholar] [CrossRef]
- Myers, O.D.; Sumner, S.J.; Li, S.; Barnes, S.; Du, X. One Step Forward for Reducing False Positive and False Negative Compound Identifications from Mass Spectrometry Metabolomics Data: New Algorithms for Constructing Extracted Ion Chromatograms and Detecting Chromatographic Peaks. Anal. Chem. 2017, 89, 8696–8703. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Matrix | Range | R2 | Level | RE% | RSDr | RSDip | LOD | LOQ | RE | ME |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ng/mL | ng/mL | % | % | % | ng/mL | ng/mL | % | % | |||
| R-MO | Plasma | 20–2000 | 0.999 | 50 | −2.1 | 10.2 | 12.9 | 10 | 20 | 112 ± 18 | 113 ± 40 |
| 500 | 1.2 | 7.5 | 11.4 | 94 ± 11 | 73 ± 4 | ||||||
| 1000 | 1.4 | 5.7 | 5.5 | 95 ± 10 | 74 ± 7 | ||||||
| Brain * | 20–2000 | 0.998 | 50 | 2.0 | 7.0 | 2.9 | 98 ± 28 | 107 ± 5 | |||
| 500 | −6.2 | 8.6 | 12.5 | 96 ± 5 | 93 ± 11 | ||||||
| 1000 | 6.7 | 3.4 | 8.2 | 109 ± 6 | 71 ± 7 | ||||||
| S-CE-123 | Plasma | 20–2000 | 0.995 | 50 | −0.3 | 3.9 | 8.9 | 10 | 20 | 102 ± 16 | 103 ± 16 |
| 500 | −6.6 | 7.6 | 11.1 | 97 ± 7 | 74 ± 7 | ||||||
| 1000 | −1.4 | 6.3 | 9.5 | 91 ± 14 | 78 ± 11 | ||||||
| Brain * | 20–2000 | 0.999 | 50 | 14.2 | 5.5 | 11.3 | 99 ± 12 | 97 ± 13 | |||
| 500 | −9.1 | 2.6 | 3.8 | 98 ± 15 | 88 ± 24 | ||||||
| 1000 | −0.5 | 2.4 | 3.5 | 102 ± 2 | 70 ± 5 |
| Parameter | Unit | S-CE-123 | R-modafinil |
|---|---|---|---|
| Total brain-to-plasma concentration ratio, Kp,brain | unitless | 0.61 ± 0.10 | 0.29 ± 0.013 |
| Unbound brain-to-plasma concentration ratio, Kp,uu,brain | unitless | 0.46 ± 0.032 § | 0.097 ± 0.034 § |
| Total steady-state plasma concentration, Cplasma,ss | nM | 4149 ± 2202 | 5574 ± 649 |
| Fraction unbound in plasma, fu,plasma | unitless | 0.252 ± 0.023 | 0.797 ± 0.15 |
| Unbound volume of distribution in brain, Vu,brain | mL/g brain | 5.21 ± 1.12 | 3.73 ± 1.38 |
| Fraction unbound in brain homogenate, fu,brain | unitless | 0.035 ± 0.012 | 0.23 ± 0.10 |
| Unbound intracellular-to-extracellular (interstitial) concentration ratio, Kp,uu,cell | unitless | 0.18 ± 0.09 § | 0.85 ± 0.6 § |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spreitzer, I.; Keife, J.; Strasser, T.; Kalaba, P.; Lubec, J.; Neuhaus, W.; Lubec, G.; Langer, T.; Wackerlig, J.; Loryan, I. Pharmacokinetics of Novel Dopamine Transporter Inhibitor CE-123 and Modafinil with a Focus on Central Nervous System Distribution. Int. J. Mol. Sci. 2023, 24, 16956. https://doi.org/10.3390/ijms242316956
Spreitzer I, Keife J, Strasser T, Kalaba P, Lubec J, Neuhaus W, Lubec G, Langer T, Wackerlig J, Loryan I. Pharmacokinetics of Novel Dopamine Transporter Inhibitor CE-123 and Modafinil with a Focus on Central Nervous System Distribution. International Journal of Molecular Sciences. 2023; 24(23):16956. https://doi.org/10.3390/ijms242316956
Chicago/Turabian StyleSpreitzer, Iva, Josefin Keife, Tobias Strasser, Predrag Kalaba, Jana Lubec, Winfried Neuhaus, Gert Lubec, Thierry Langer, Judith Wackerlig, and Irena Loryan. 2023. "Pharmacokinetics of Novel Dopamine Transporter Inhibitor CE-123 and Modafinil with a Focus on Central Nervous System Distribution" International Journal of Molecular Sciences 24, no. 23: 16956. https://doi.org/10.3390/ijms242316956
APA StyleSpreitzer, I., Keife, J., Strasser, T., Kalaba, P., Lubec, J., Neuhaus, W., Lubec, G., Langer, T., Wackerlig, J., & Loryan, I. (2023). Pharmacokinetics of Novel Dopamine Transporter Inhibitor CE-123 and Modafinil with a Focus on Central Nervous System Distribution. International Journal of Molecular Sciences, 24(23), 16956. https://doi.org/10.3390/ijms242316956