
Two High-Quality Cygnus Genome Assemblies Reveal Genomic Variations Associated with Plumage Color


Abstract
:1. Introduction
2. Results
2.1. Genome Sequencing, Assembly and Annotation
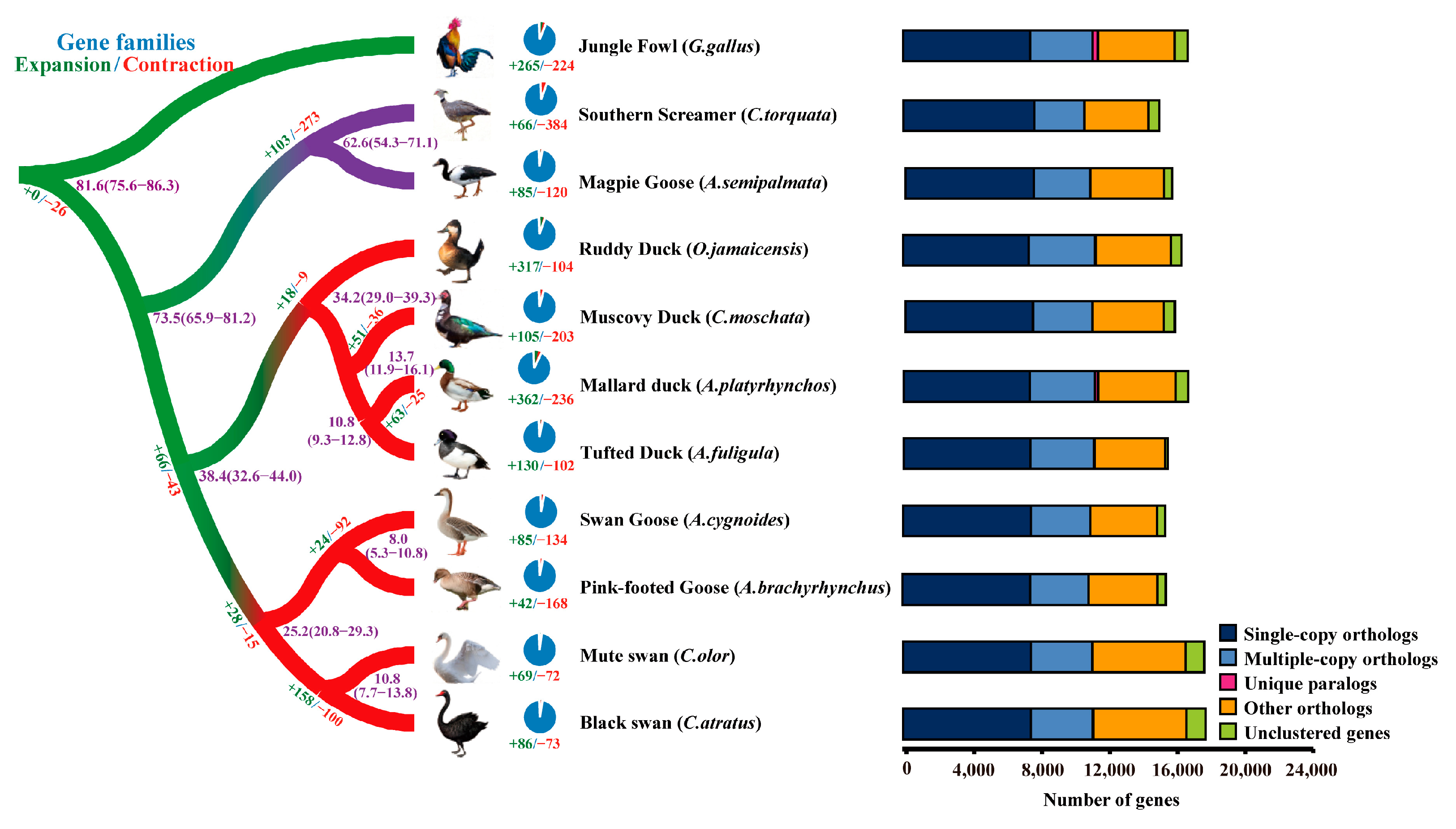
2.2. Phylogenetic Analysis
2.3. Unique and Expanded Orthologue Groups
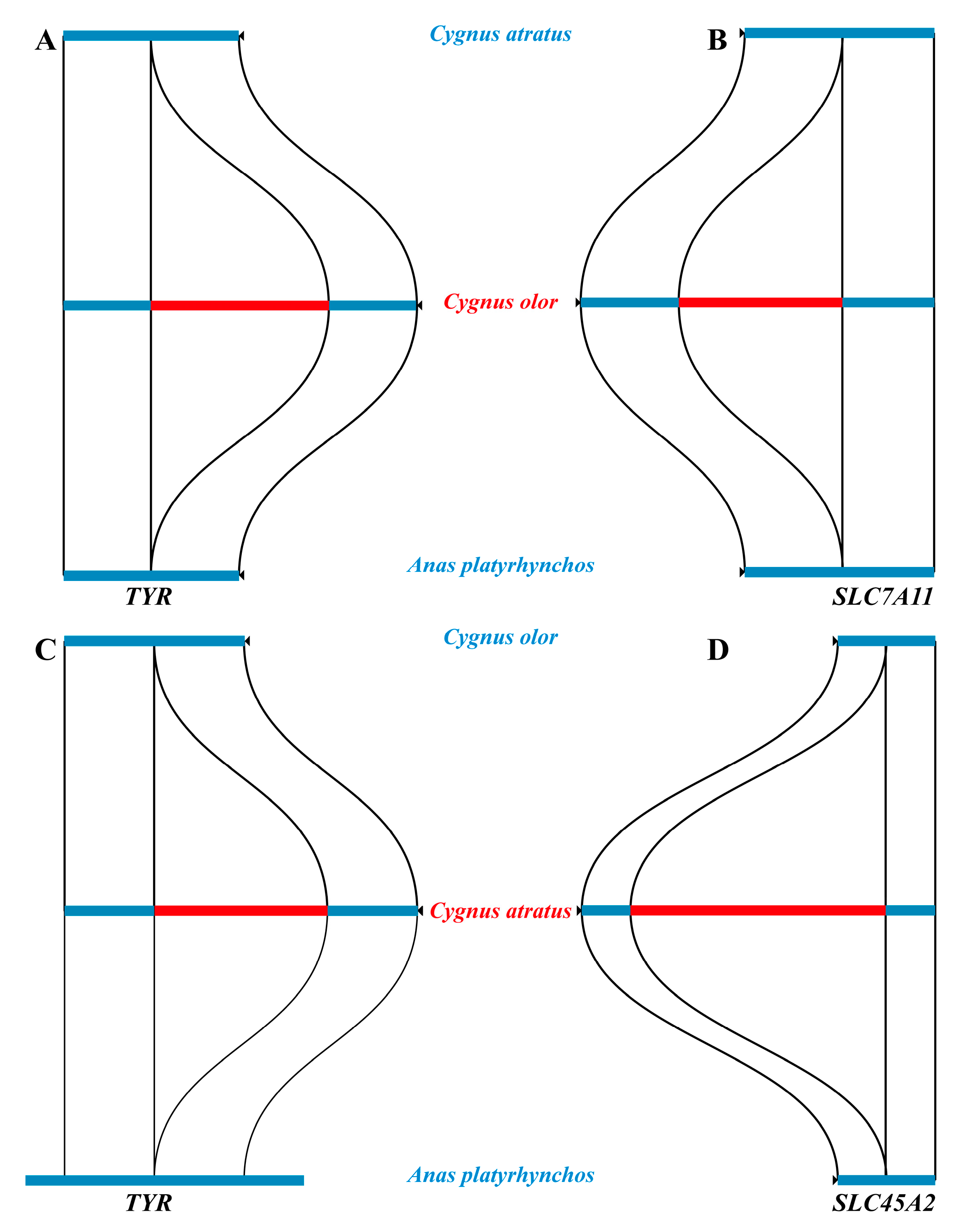
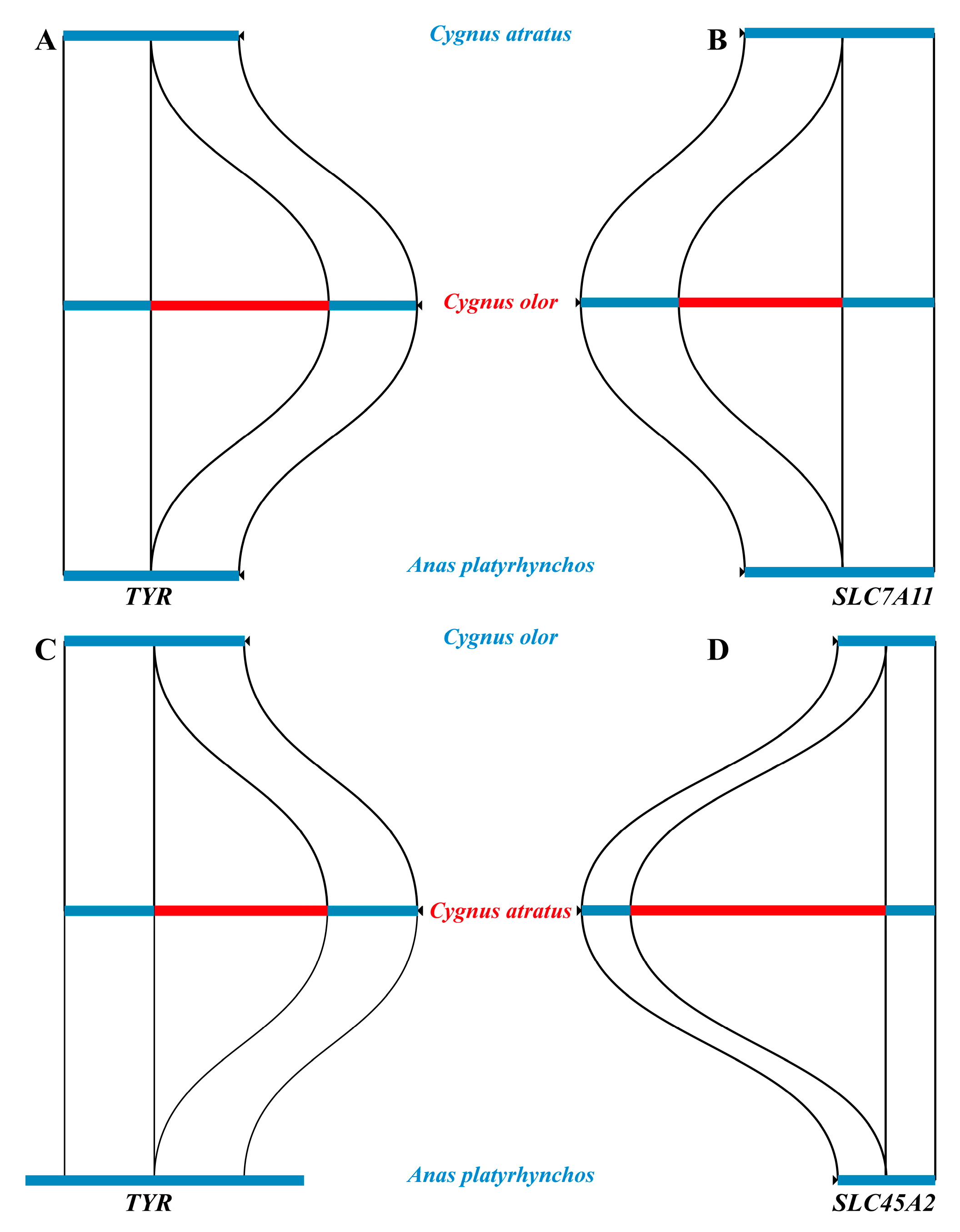
2.4. Structural Variants
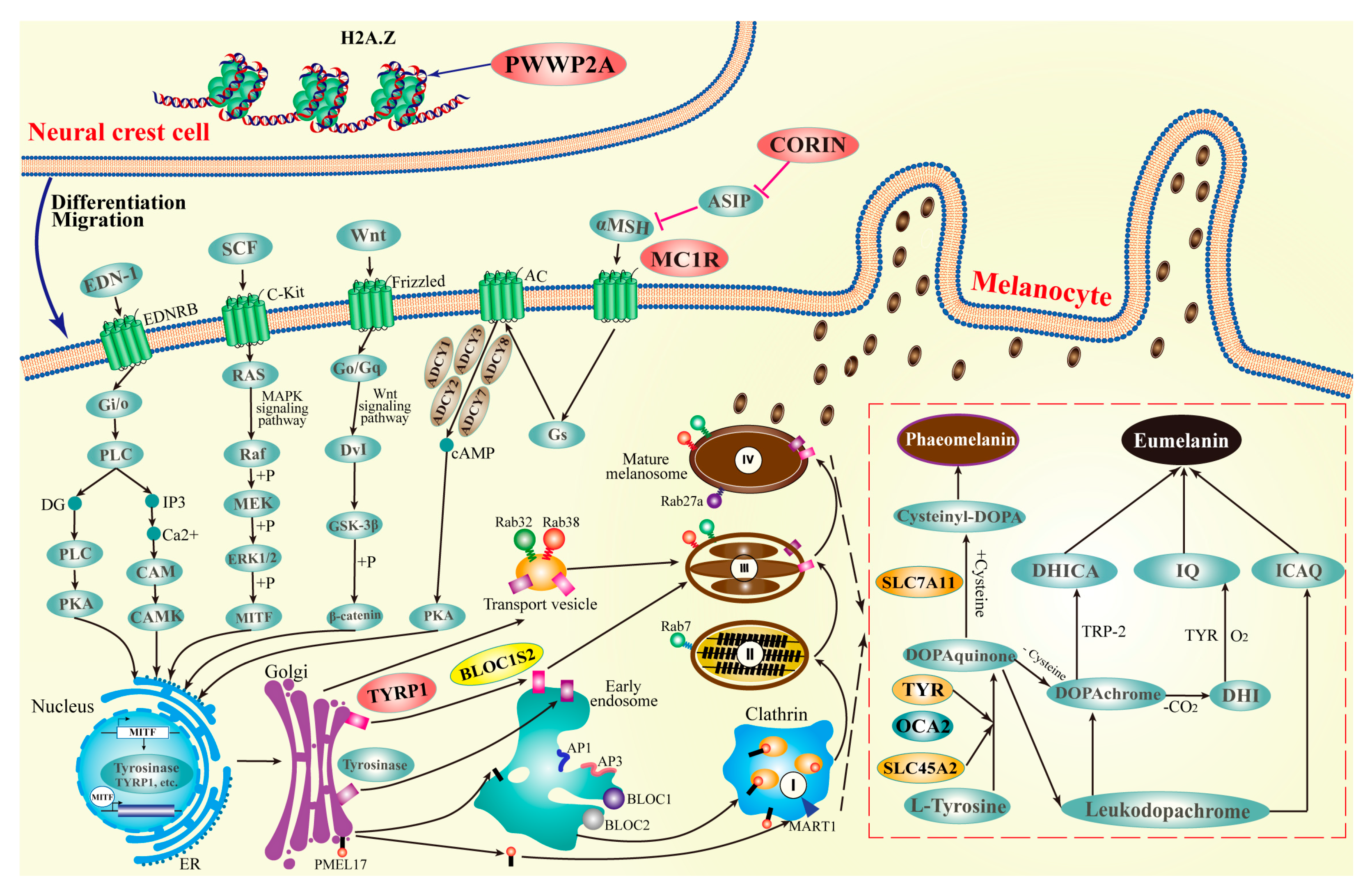
2.5. Positive Selection
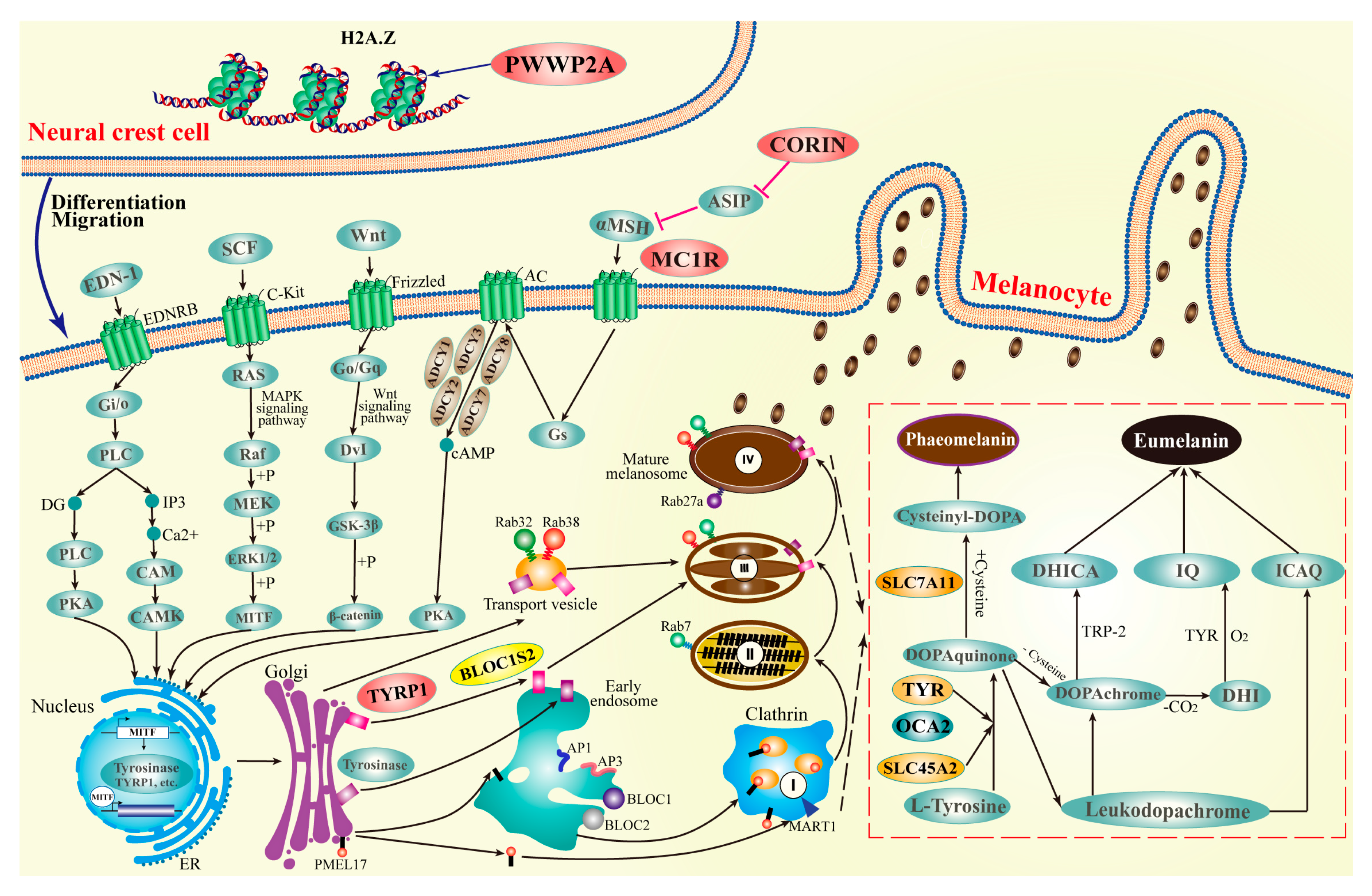
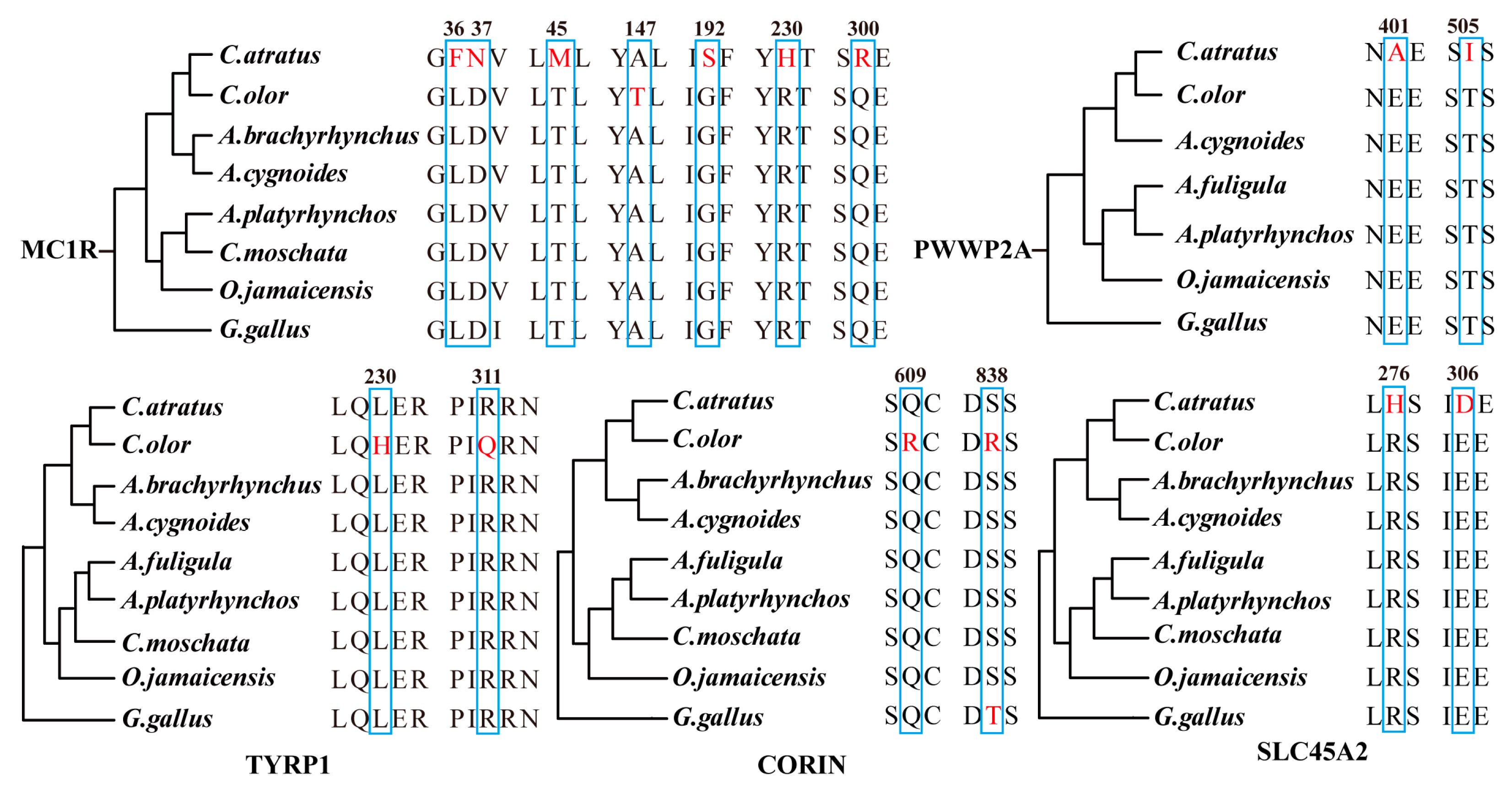
2.6. Genes with Amino Acid Replacements
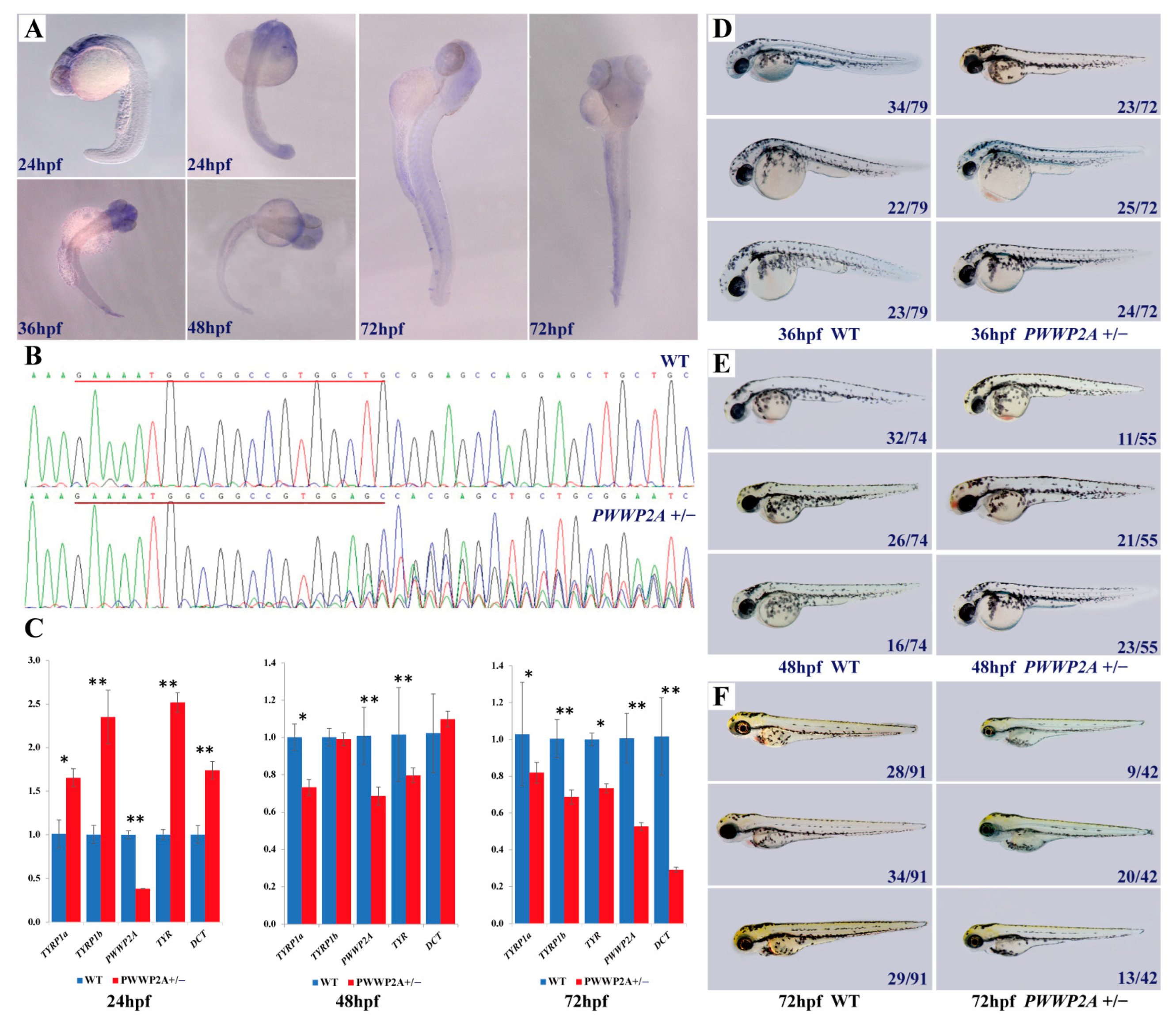
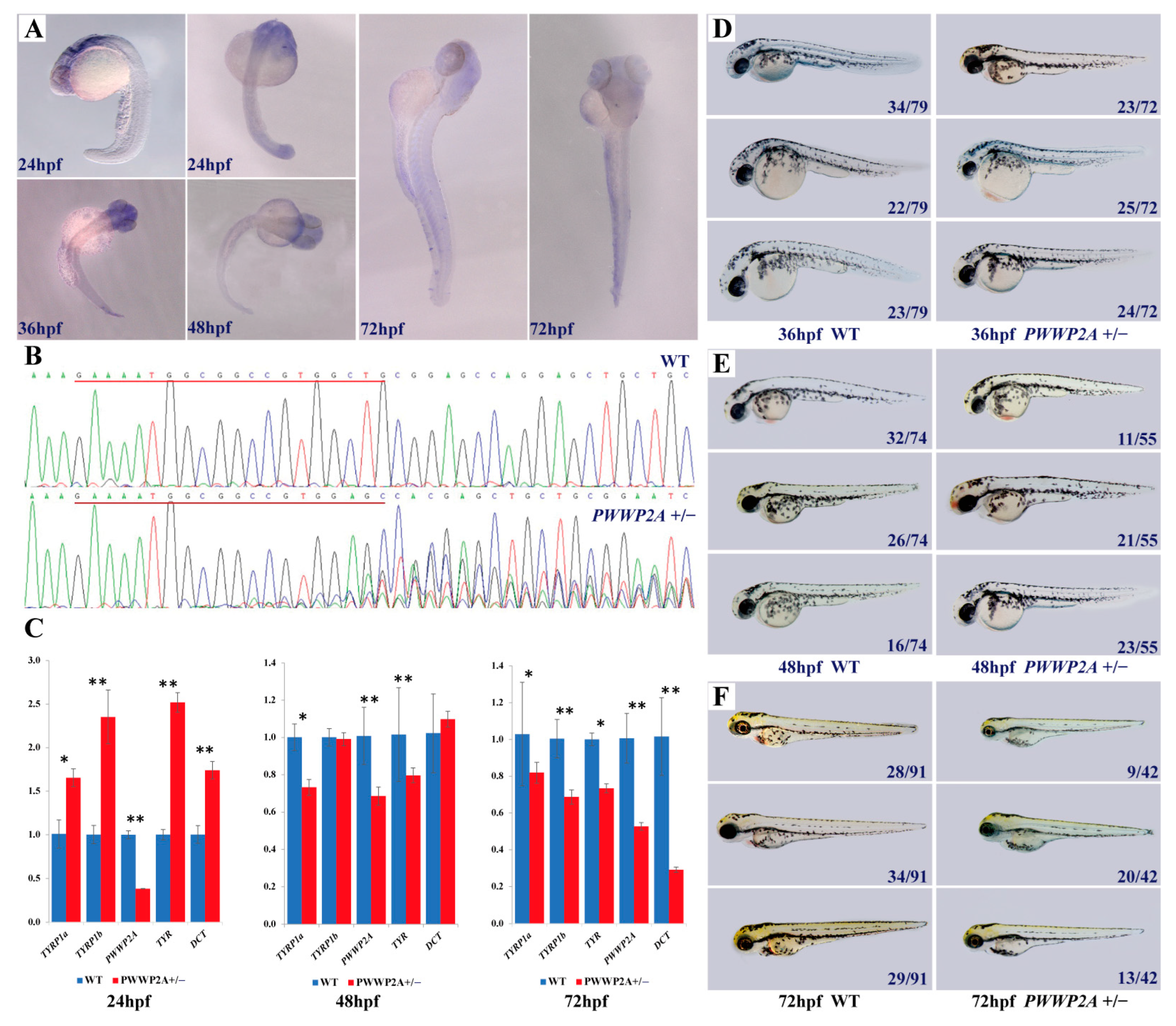
2.7. Relationship between PWWP2A Mutation and Melanin Deposition in Zebrafish
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Genomic Sequencing
4.2. Genome Size Estimation
4.3. Genomic Assembly and Evaluation
4.4. Genome Annotation
4.5. Comparative Phylogenomics
4.6. Whole-Mount In Situ Hybridization (WISH)
4.7. Generation of Mutant Fish
4.8. RT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prum, R.O. Bird Coloration: Mechanisms and Measurements; Harvard University Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Gao, G.; Xu, M.; Bai, C.; Yang, Y.; Li, G.; Xu, J.; Wei, Z.; Min, J.; Su, G.; Zhou, X. Comparative genomics and transcriptomics of Chrysolophus provide insights into the evolution of complex plumage coloration. GigaScience 2018, 7, giy113. [Google Scholar]
- Pavan, W.J.; Sturm, R.A. The Genetics of Human Skin and Hair Pigmentation. Annu. Rev. Genomics Hum. Genet. 2019, 20, 41–72. [Google Scholar] [CrossRef]
- McConnell, A.M.; Zon, L.I. Dissecting melanocytes to predict melanoma. Nat. Cell Biol. 2021, 23, 930–931. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Shi, H.; Ma, P.; Zhao, S.; Kong, Q.; Bian, T.; Gong, C.; Zhao, Q.; Liu, Y.; Qi, X.; et al. Darwinian Positive Selection on the Pleiotropic Effects of KITLG Explain Skin Pigmentation and Winter Temperature Adaptation in Eurasians. Mol. Biol. Evol. 2018, 35, 2272–2283. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Khan, B.; Simoes-Costa, M. Neural crest metabolism: At the crossroads of development and disease. Dev. Biol. 2021, 475 (Suppl. S1), 245–255. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, E.K. Melanocyte stem cells: A melanocyte reservoir in hair follicles for hair and skin pigmentation. Pigment Cell Melanoma Res. 2011, 24, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Aisa, H.A. Upregulation of Melanogenesis and Tyrosinase Activity: Potential Agents for Vitiligo. Molecules 2017, 22, 1303. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Chuong, C.M.; Lei, M. Regulation of melanocyte stem cells in the pigmentation of skin and its appendages: Biological patterning and therapeutic potentials. Exp. Dermatol. 2019, 28, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Roulin, A.; Ducrest, A.L. Genetics of colouration in birds. Semin. Cell Dev. Biol. 2013, 24, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Bychkova, E.O.; Golubeva, N.A.; Filippova, E.A.; Sangina, L.O.; Markov, A.V. A New Mutation in the MC1R Gene Leads to Unique Carnelian Color in Kurilian Bobtails. Russ. J. Genet. 2020, 56, 108–111. [Google Scholar] [CrossRef]
- Deraredj Nadim, W.; Hassanaly, S.; Benedetti, H.; Kieda, C.; Grillon, C.; Morisset-Lopez, S. The GTPase-activating protein-related domain of neurofibromin interacts with MC1R and regulates pigmentation-mediated signaling in human melanocytes. Biochem. Biophys Res. Commun. 2021, 534, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.K.; Collazo-Roman, M.; Vadaparampil, S.T.; Valavanis, S.; Del Rio, J.; Soto, B.; Flores, I.; Dutil, J.; Kanetsky, P.A. MC1R variants and associations with pigmentation characteristics and genetic ancestry in a Hispanic, predominately Puerto Rican, population. Sci. Rep. 2020, 10, 7303. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ferrer, A.; Rodriguez-Lopez, J.N.; Garcia-Canovas, F.; Garcia-Carmona, F. Tyrosinase: A comprehensive review of its mechanism. Biochim. Biophys Acta. 1995, 1247, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K.; Ozeki, H. Chemical analysis of melanins and its application to the study of the regulation of melanogenesis. Pigm. Cell Res. 2000, 13, 103–109. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, M.M. The relationship between melanin production and lipofuscin formation in Tyrosinase gene knockout melanocytes using CRISPR/Cas9 system. Life Sci. 2021, 284, 119915. [Google Scholar] [CrossRef]
- Yang, Z.; Zhong, H.; Chen, J.; Zhang, X.; Zhang, H.; Luo, X.; Xu, S.; Chen, H.; Lu, D.; Han, Y.; et al. A Genetic Mechanism for Convergent Skin Lightening during Recent Human Evolution. Mol. Biol. Evol. 2016, 33, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.D.; Han, X.M.; Ma, Y.F.; Irwin, D.M.; Gao, Y.; Deng, J.K.; Adeola, A.C.; Xie, H.B.; Zhang, Y.P. Genetic variations associated with six-white-point coat pigmentation in Diannan small-ear pigs. Sci. Rep. 2016, 6, 27534. [Google Scholar] [CrossRef]
- Lopes, R.J.; Johnson, J.D.; Toomey, M.B.; Ferreira, M.S.; Araujo, P.M.; Melo-Ferreira, J.; Andersson, L.; Hill, G.E.; Corbo, J.C.; Carneiro, M. Genetic Basis for Red Coloration in Birds. Curr. Biol. 2016, 26, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Suying, B.; Qijun, Z.; Xiuhua, T.; Xue, P.; Yue, Y. Phylogenetic Relationships of the Swans Bases on COI Barcodes. Chin. J. Wildl. 2015, 36, 315–319. [Google Scholar]
- Pointer, M.A.; Mundy, N.I. Testing whether macroevolution follows microevolution: Are colour differences among swans (Cygnus) attributable to variation at the MC1R locus? BMC Evol. Biol. 2008, 8, 249. [Google Scholar] [CrossRef]
- Hillier, L.; Miller, W.; Irney, E.B.; Warren, W.; Hardison, R.C.; Ponting, C.P.; Bork, P.; Burt, D.W.; Groenen, M.; Delany, M.E. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar]
- Chen, T.; Zhang, B.; Ziegenhals, T.; Prusty, A.B.; Chen, W. A missense mutation in SNRPE linked to non-syndromal microcephaly interferes with U snRNP assembly and pre-mRNA splicing. PLoS Genet. 2019, 15, e1008460. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.M.; Hofmann, H.A. Seeing is believing: Dynamic evolution of gene families. Proc. Natl. Acad. Sci. USA 2015, 112, 1252. [Google Scholar] [CrossRef]
- Zang, F.; Ma, Y.; Tu, X.; Huang, P.; Wu, Q.; Li, Z.; Liu, T.; Lin, F.; Pei, S.; Zang, D.; et al. A high-quality chromosome-level genome of wild Rosa rugosa. DNA Res. 2021, 28, dsab017. [Google Scholar] [CrossRef]
- Hadi, K.; Yao, X.; Behr, J.M.; Deshpande, A.; Xanthopoulakis, C.; Tian, H.; Kudman, S.; Rosiene, J.; Darmofal, M.; DeRose, J.; et al. Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs. Cell 2020, 183, 197–210.e32. [Google Scholar] [CrossRef]
- Hollox, E.J.; Zuccherato, L.W.; Tucci, S. Genome structural variation in human evolution. Trends Genet. 2021, 38, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Chintala, S.; Li, W.; Lamoreux, M.L.; Ito, S.; Wakamatsu, K.; Sviderskaya, E.V.; Bennett, D.C.; Park, Y.-M.; Gahl, W.A.; Huizing, M. Slc7a11 gene controls production of pheomelanin pigment and proliferation of cultured cells. Proc. Natl. Acad. Sci. USA 2005, 102, 10964–10969. [Google Scholar] [CrossRef]
- Larimore, J.; Tornieri, K.; Ryder, P.V.; Gokhale, A.; Zlatic, S.A.; Craige, B.; Lee, J.D.; Talbot, K.; Pare, J.F.; Smith, Y.; et al. The schizophrenia susceptibility factor dysbindin and its associated complex sort cargoes from cell bodies to the synapse. Mol. Biol. Cell 2011, 22, 4854–4867. [Google Scholar] [CrossRef]
- Punzeler, S.; Link, S.; Wagner, G.; Keilhauer, E.C.; Kronbeck, N.; Spitzer, R.M.; Leidescher, S.; Markaki, Y.; Mentele, E.; Regnard, C.; et al. Multivalent binding of PWWP2A to H2A.Z regulates mitosis and neural crest differentiation. EMBO J. 2017, 36, 2263–2279. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, N.; Berx, G. From neural crest cells to melanocytes: Cellular plasticity during development and beyond. Cell Mol. Life Sci. 2019, 76, 1919–1934. [Google Scholar] [CrossRef] [PubMed]
- Erickson, C.A.; Goins, T.L. Avian neural crest cells can migrate in the dorsolateral path only if they are specified as melanocytes. Development 1995, 121, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Link, S.; Spitzer, R.M.M.; Sana, M.; Torrado, M.; Volker-Albert, M.C.; Keilhauer, E.C.; Burgold, T.; Punzeler, S.; Low, J.K.K.; Lindstrom, I.; et al. PWWP2A binds distinct chromatin moieties and interacts with an MTA1-specific core NuRD complex. Nat. Commun. 2018, 9, 4300. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, H.E. Genetics, development and evolution of adaptive pigmentation in vertebrates. Heredity 2006, 97, 222–234. [Google Scholar] [CrossRef]
- Setty, S.R.; Tenza, D.; Sviderskaya, E.V.; Bennett, D.C.; Raposo, G.; Marks, M.S. Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature 2008, 454, 1142–1146. [Google Scholar] [CrossRef]
- Chi, A.; Valencia, J.C.; Hu, Z.Z.; Watabe, H.; Yamaguchi, H.; Mangini, N.J.; Huang, H.; Canfield, V.A.; Cheng, K.C.; Yang, F.; et al. Proteomic and bioinformatic characterization of the biogenesis and function of melanosomes. J. Proteome Res. 2006, 5, 3135–3144. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.M.; Cohen-Barak, O.; Hagiwara, N.; Gardner, J.M.; Davisson, M.T.; King, R.A.; Brilliant, M.H. Mutations in the human orthologue of the mouse underwhite gene (uw) underlie a new form of oculocutaneous albinism, OCA4. Am. J. Hum. Genet. 2001, 69, 981–988. [Google Scholar] [CrossRef]
- Camacho-Hubner, A.; Richard, C.; Beermann, F. Genomic structure and evolutionary conservation of the tyrosinase gene family from Fugu. Gene 2002, 285, 59–68. [Google Scholar] [CrossRef]
- Wu, S.; Huang, J.; Li, Y.; Liu, Z.; Zhang, Q.; Pan, Y.; Wang, X. Cloning, sequence analysis, and expression of tyrp1a and tyrp2 genes related to body colour in different developmental stages and tissues of rainbow trout Oncorhynchus mykiss. Aquacult. Int. 2021, 29, 941–961. [Google Scholar] [CrossRef]
- Braasch, I.; Liedtke, D.; Volff, J.N.; Schartl, M. Pigmentary function and evolution of tyrp1 gene duplicates in fish. Pigm. Cell Melanoma R. 2009, 22, 839–850. [Google Scholar] [CrossRef]
- Zhang, X.T.; Wei, K.J.; Chen, Y.Y.; Shi, Z.C.; Liu, L.K.; Li, J.; Zhang, G.R.; Ji, W. Molecular cloning and expression analysis of tyr and tyrp1 genes in normal and albino yellow catfish Tachysurus fulvidraco. J. Fish Biol. 2018, 92, 979–998. [Google Scholar] [CrossRef]
- Yan, W.; Sheng, N.; Seto, M.; Morser, J.; Wu, Q. Corin, a mosaic transmembrane serine protease encoded by a novel cDNA from human heart. J. Biol. Chem. 1999, 274, 14926–14935. [Google Scholar] [CrossRef] [PubMed]
- Enshell-Seijffers, D.; Lindon, C.; Morgan, B.A. The serine protease Corin is a novel modifier of the agouti pathway. Development 2008, 135, 217. [Google Scholar] [CrossRef]
- Xu, X.; Dong, G.X.; Schmidt-Küntzel, A.; Zhang, X.L.; Zhuang, Y.; Fang, R.; Sun, X.; Hu, X.S.; Zhang, T.Y.; Yang, H.D. The genetics of tiger pelage color variations. Cell Res. 2017, 27, 954–957. [Google Scholar] [CrossRef] [PubMed]
- Fukamachi, S.; Shimada, A.; Shima, A. Mutations in the gene encoding B, a novel transporter protein, reduce melanin content in medaka. Nat. Genet. 2001, 28, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Jeong, C.B.; Kang, H.M.; Hong, S.A.; Byeon, E.; Lee, J.S.; Lee, Y.H.; Choi, I.Y.; Bae, S.; Lee, J.S. Generation of albino via SLC45a2 gene targeting by CRISPR/Cas9 in the marine medaka Oryzias melastigma. Mar. Pollut. Bull. 2020, 154, 111038. [Google Scholar] [CrossRef]
- Karawita, A.C.; Cheng, Y.; Chew, K.Y.; Challagulla, A.; Kraus, R.; Mueller, R.C.; Tong, M.Z.; Hulme, K.D.; Bielefeldt-Ohmann, H.; Steele, L.E. The swan genome and transcriptome, it is not all black and white. Genome Biol. 2023, 24, 13. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Hayashi, M.; Nakajima, O.; Kono, M.; Abe, Y.; Hozumi, Y.; Suzuki, T. A 4-bp deletion promoter variant (rs984225803) is associated with mild OCA 4 among Japanese patients. Pigm. Cell Melanoma R. 2019, 32, 79–84. [Google Scholar] [CrossRef]
- Chen, T.; Song, G.; Yang, H.; Mao, L.; Cui, Z.; Huang, K. Development of the Swimbladder Surfactant System and Biogenesis of Lysosome-Related Organelles Is Regulated by BLOS1 in Zebrafish. Genetics 2018, 208, 1131–1146. [Google Scholar] [CrossRef]
- Baxter, L.L.; Watkins-Chow, D.E.; Pavan, W.J.; Loftus, S.K. A curated gene list for expanding the horizons of pigmentation biology. Pigm. Cell Melanoma R. 2019, 32, 348–358. [Google Scholar] [CrossRef]
- Matsuno, S.; Ohue, M.; Akiyama, Y. Multidomain protein structure prediction using information about residues interacting on multimeric protein interfaces. Biophys. Phys. 2020, 17, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Castro-Sowinski, S.; Matan, O.; Star, L.; Okon, Y. Thiol-Redox Potential in Melanin Production and Nitrogen Fixation by Sinorhizobium meliloti. In Current Plant Science and Biotechnology in Agriculture; Springer: Berlin/Heidelberg, Germany, 2008; p. 373. [Google Scholar]
- Hsiao, J.J.; Fisher, D.E. The roles of microphthalmia-associated transcription factor and pigmentation in melanoma. Arch. Biochem. Biophys. 2014, 563, 28–34. [Google Scholar] [CrossRef]
- Bellei, B.; Maresca, V.; Flori, E.; Pitisci, A.; Larue, L.; Picardo, M. p38 Regulates Pigmentation via Proteasomal Degradation of Tyrosinase. J. Bio. Chem. 2010, 285, 7288–7299. [Google Scholar] [CrossRef]
- Kim, Y.M.; Lee, E.C.; Lim, H.M.; Seo, Y.K. Rice Bran Ash Mineral Extract Increases Pigmentation through the p-ERK Pathway in Zebrafish (Danio rerio). Int. J. Mol. Sci. 2019, 20, 2172. [Google Scholar] [CrossRef]
- Moon, H.-R.; Jo, S.Y.; Kim, H.T.; Lee, W.J.; Won, C.H.; Lee, M.W.; Choi, J.H.; Chang, S.E. Loratadine, an H 1 antihistamine, inhibits melanogenesis in human melanocytes. BioMed Res. Int. 2019, 2019, 5971546. [Google Scholar] [CrossRef] [PubMed]
- Su, T.-R.; Lin, J.-J.; Tsai, C.-C.; Huang, T.-K.; Yang, Z.-Y.; Wu, M.-O.; Zheng, Y.-Q.; Su, C.-C.; Wu, Y.-J. Inhibition of melanogenesis by gallic acid: Possible involvement of the PI3K/Akt, MEK/ERK and Wnt/β-catenin signaling pathways in B16F10 cells. Int. J. Mol. Sci. 2013, 14, 20443–20458. [Google Scholar] [CrossRef] [PubMed]
- Wasmeier, C.; Hume, A.N.; Bolasco, G.; Seabra, M.C. Melanosomes at a glance. J. Cell Sci. 2008, 121, 3995–3999. [Google Scholar] [CrossRef]
- Wang, J.Y.; Chen, H.; Wang, Y.Y.; Wang, X.Q.; Chen, H.Y.; Zhang, M.; Tang, Y.; Zhang, B. Network pharmacological mechanisms of Vernonia anthelmintica (L.) in the treatment of vitiligo: Isorhamnetin induction of melanogenesis via up-regulation of melanin-biosynthetic genes. BMC Syst. Biol. 2017, 11, 103. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Ruan, J.; Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21 (Suppl. S1), i351–i358. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef]
- Hoede, C.; Arnoux, S.; Moisset, M.; Chaumier, T.; Inizan, O.; Jamilloux, V.; Quesneville, H. PASTEC: An automatic transposable element classification tool. PLoS ONE 2014, 9, e91929. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. In Current Protocols in Bioinformatics; Wiley Interscience: Hoboken, NJ, USA, 2009; Chapter 4, Unit 4.10; pp. 4.10.1–4.10.14. [Google Scholar]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19 (Suppl. S2), ii215–ii225. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Parra, G.; Guigó, R. Using geneid to Identify Genes. In Current Protocols in Bioinformatics; Wiley Interscience: Hoboken, NJ, USA, 2018; pp. 4.3.1–4.3.28. [Google Scholar]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Jens, K.; Michael, W.; Erickson, J.L.; Schattat, M.H.; Jan, G.; Frank, H. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar]
- Keilwagen, J.; Hartung, F.; Paulini, M.; Twardziok, S.O.; Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinform. 2018, 19, 189. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Wei, Z.; Perte, M. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed]
- Dimmer, E.C.; Huntley, R.P.; Alam-Faruque, Y.; Sawford, T.; O’Donovan, C.; Martin, M.J.; Bely, B.; Browne, P.; Mun Chan, W.; Eberhardt, R.; et al. The UniProt-GO Annotation database in 2011. Nucleic Acids Res. 2012, 40, D565–D570. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.-C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef]
- Nattestad, M.; Schatz, M.C. Assemblytics: A web analytics tool for the detection of variants from an assembly. Bioinformatics 2016, 32, 3021–3023. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J. Miropeats: Graphical DNA sequence comparisons. Bioinformatics 1995, 11, 615–619. [Google Scholar] [CrossRef]
- Chang, N.; Sun, C.; Gao, L.; Zhu, D.; Xu, X.; Zhu, X.; Xiong, J.-W.; Xi, J.J. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013, 23, 465–472. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Contig Number | Contig Length (Mb) | Contig N50 (Mb) | Contig N90 (Mb) | Contig Max (Mb) | GC Content (%) | Complete BUSCOs |
|---|---|---|---|---|---|---|---|
| C. olor | 335 | 1124.00 | 26.82 | 4.90 | 77.28 | 42.04 | 2546 (98.45%) |
| C. atratus | 469 | 1129.04 | 21.91 | 4.55 | 65.49 | 42.06 | 2542 (98.30%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chong, Y.; Tu, X.; Lu, Y.; Gao, Z.; He, X.; Hong, J.; Wu, J.; Wu, D.; Xi, D.; Deng, W. Two High-Quality Cygnus Genome Assemblies Reveal Genomic Variations Associated with Plumage Color. Int. J. Mol. Sci. 2023, 24, 16953. https://doi.org/10.3390/ijms242316953
Chong Y, Tu X, Lu Y, Gao Z, He X, Hong J, Wu J, Wu D, Xi D, Deng W. Two High-Quality Cygnus Genome Assemblies Reveal Genomic Variations Associated with Plumage Color. International Journal of Molecular Sciences. 2023; 24(23):16953. https://doi.org/10.3390/ijms242316953
Chicago/Turabian StyleChong, Yuqing, Xiaolong Tu, Ying Lu, Zhendong Gao, Xiaoming He, Jieyun Hong, Jiao Wu, Dongdong Wu, Dongmei Xi, and Weidong Deng. 2023. "Two High-Quality Cygnus Genome Assemblies Reveal Genomic Variations Associated with Plumage Color" International Journal of Molecular Sciences 24, no. 23: 16953. https://doi.org/10.3390/ijms242316953
APA StyleChong, Y., Tu, X., Lu, Y., Gao, Z., He, X., Hong, J., Wu, J., Wu, D., Xi, D., & Deng, W. (2023). Two High-Quality Cygnus Genome Assemblies Reveal Genomic Variations Associated with Plumage Color. International Journal of Molecular Sciences, 24(23), 16953. https://doi.org/10.3390/ijms242316953