
Gas-Phase vs. Grain-Surface Formation of Interstellar Complex Organic Molecules: A Comprehensive Quantum-Chemical Study



Abstract
1. Introduction
2. Results
2.1. Gas-Phase Reactions
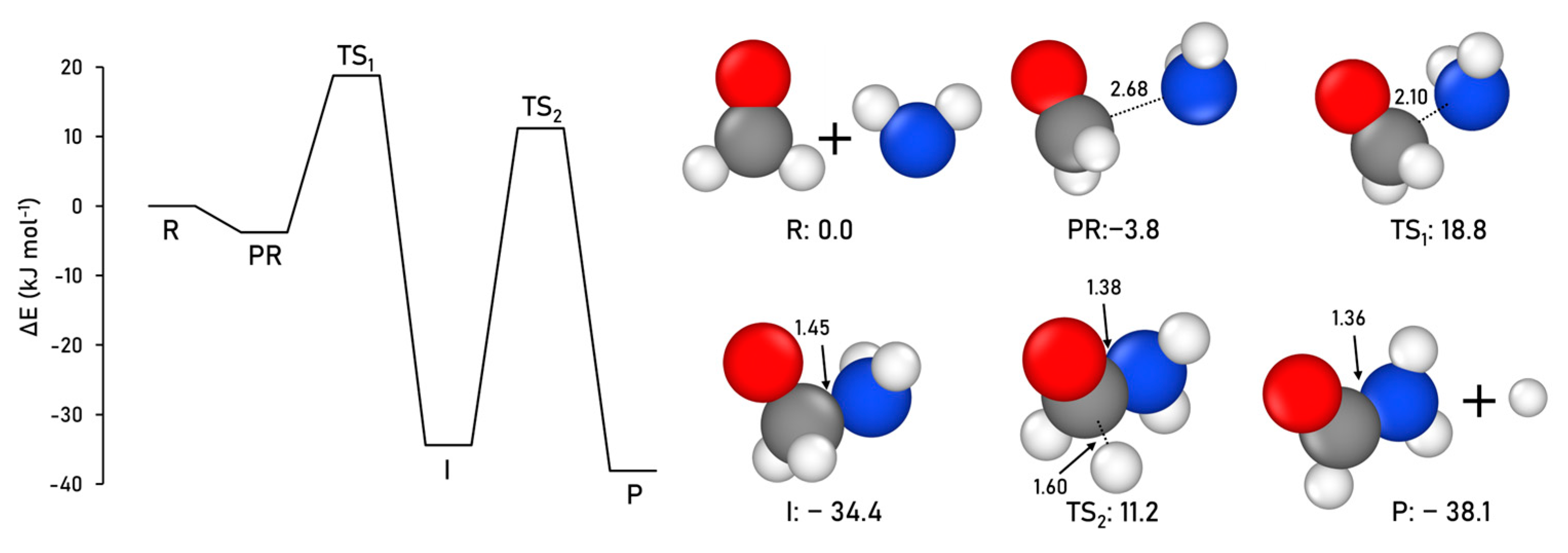
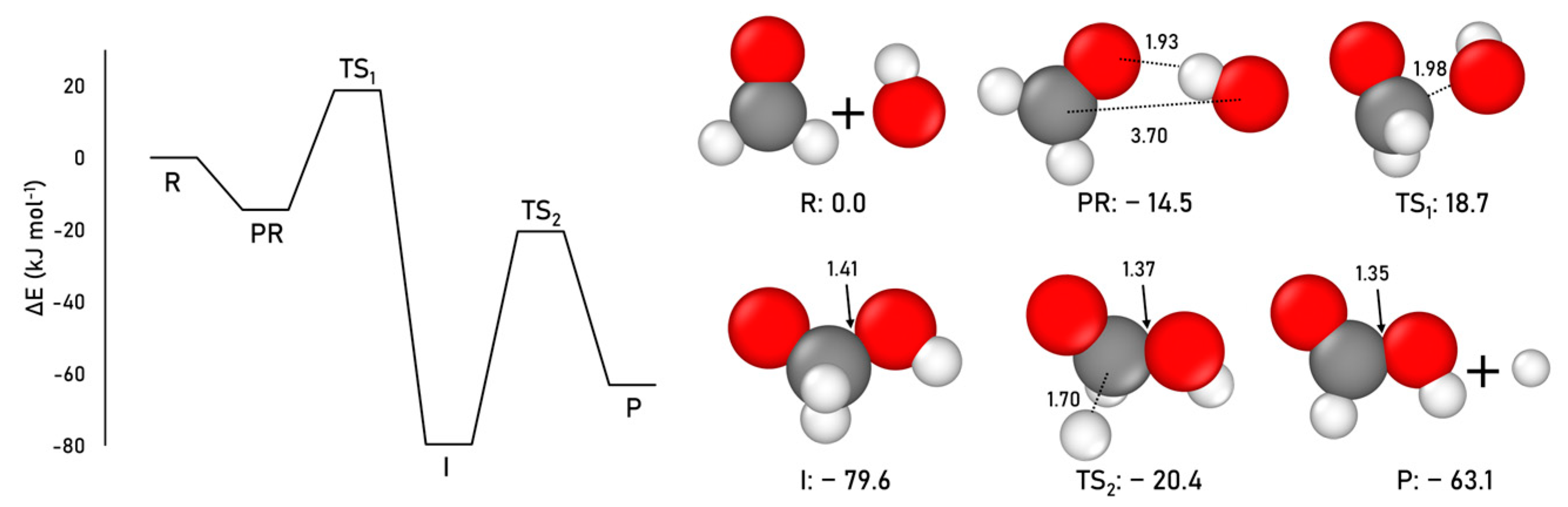
2.1.1. Formamide Formation
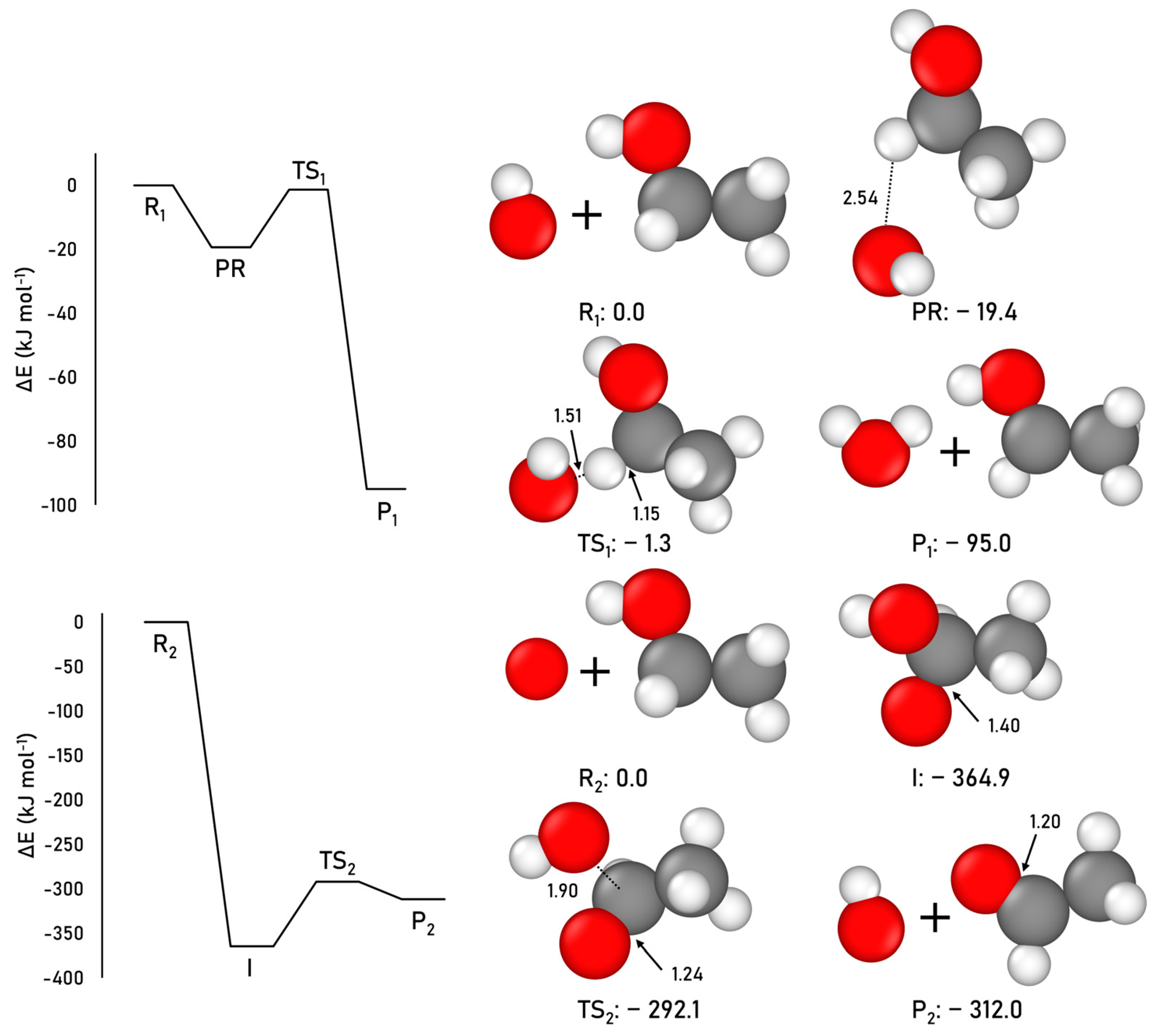
2.1.2. Acetaldehyde Formation

2.1.3. Methyl Formate Formation
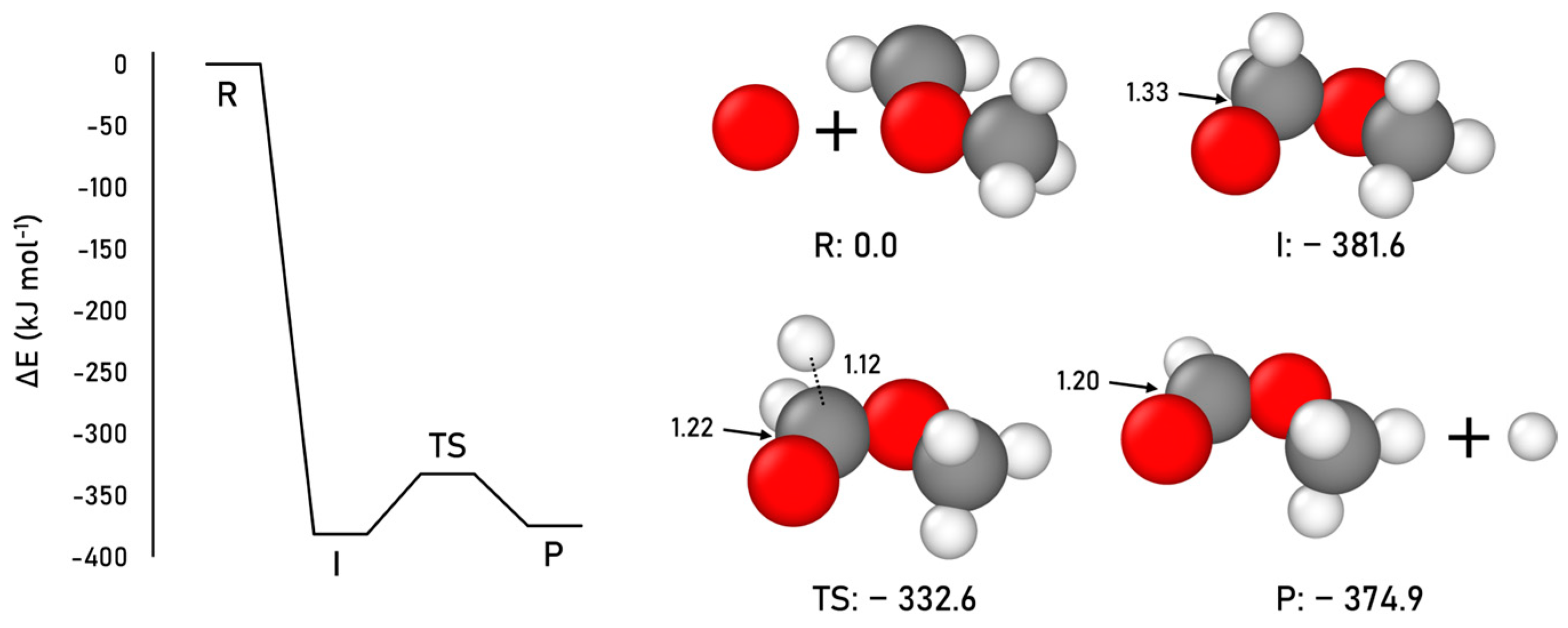
2.1.4. Formic Acid Formation
2.2. Reactivity on the Water Ice Surface Models
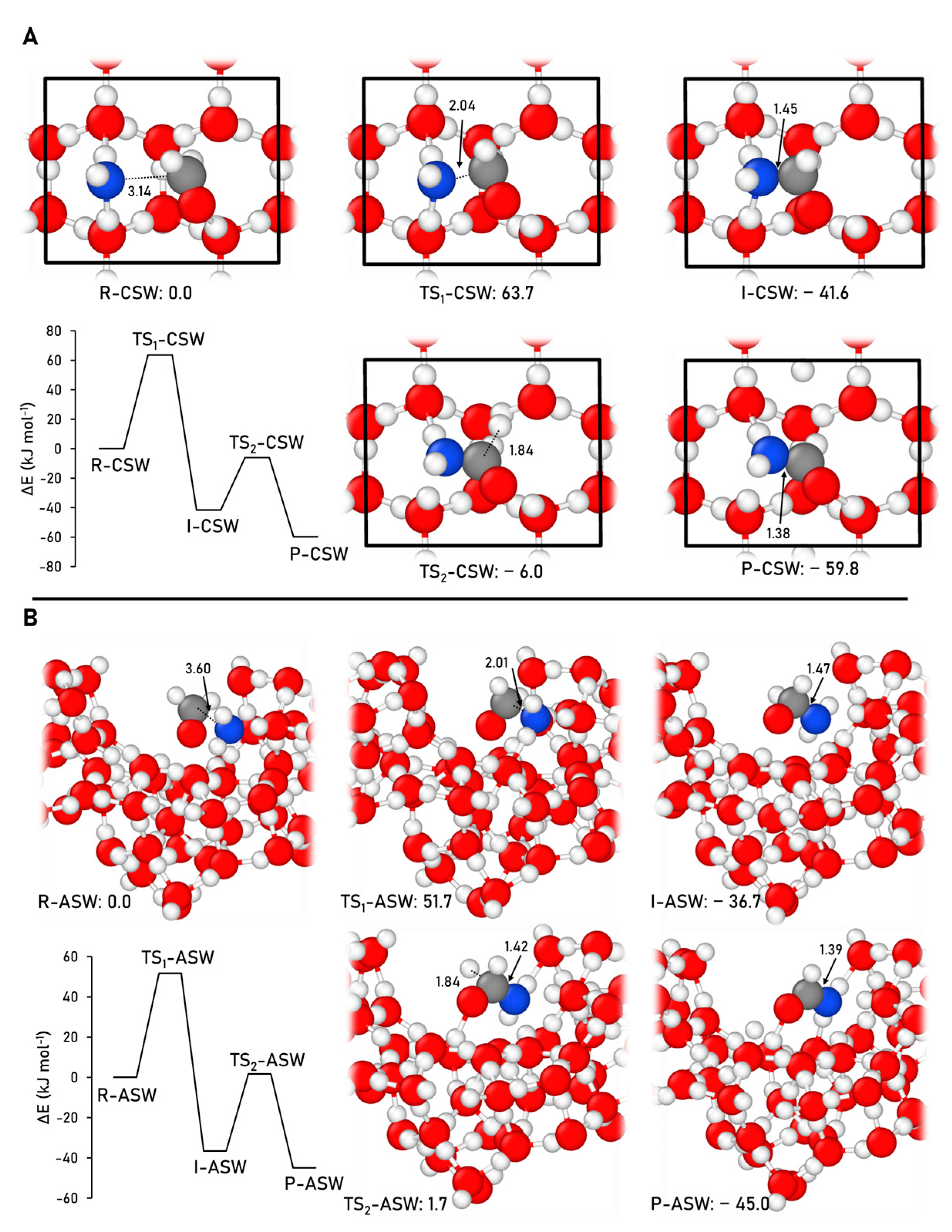
2.2.1. Formamide Formation
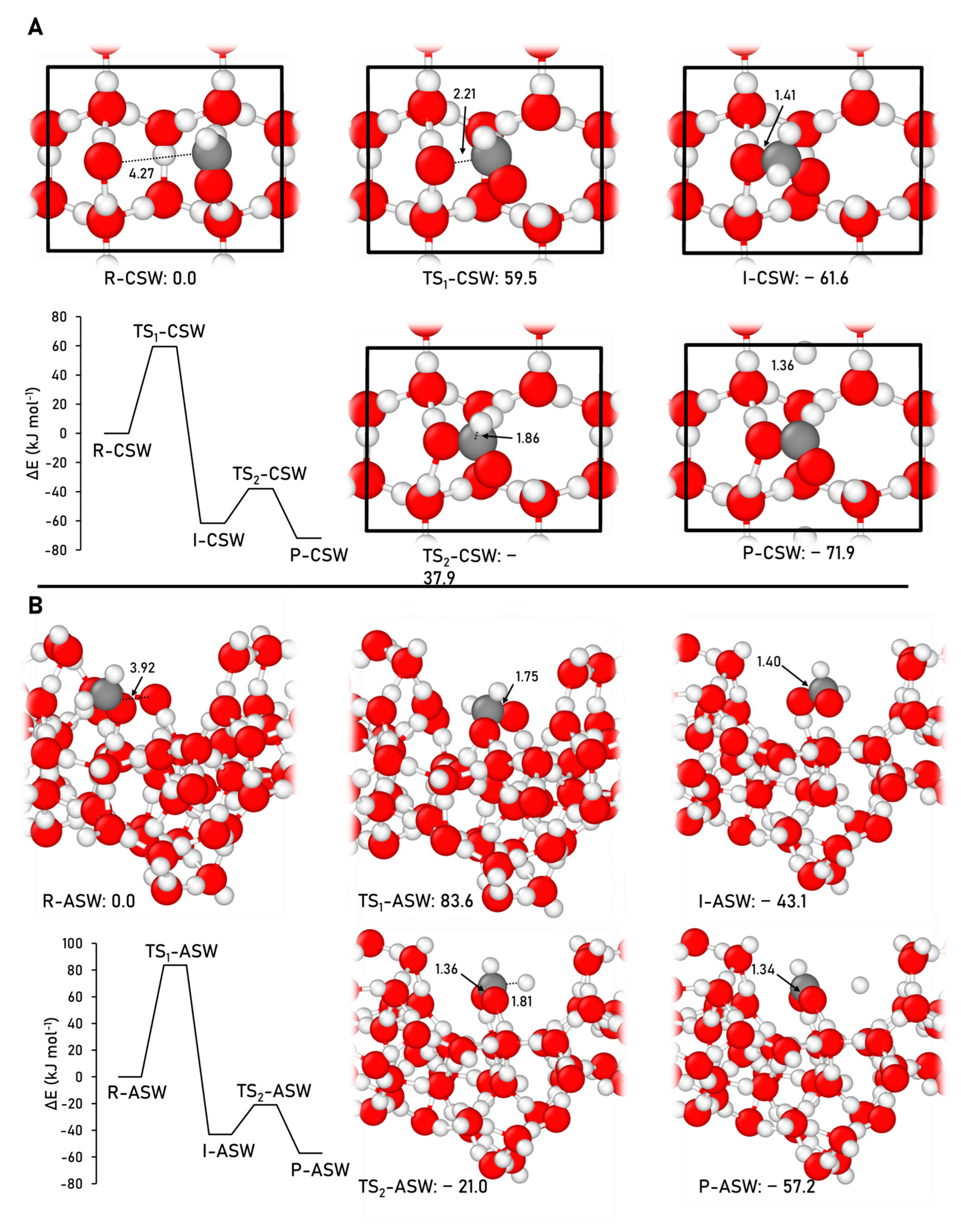
2.2.2. Acetaldehyde Formation
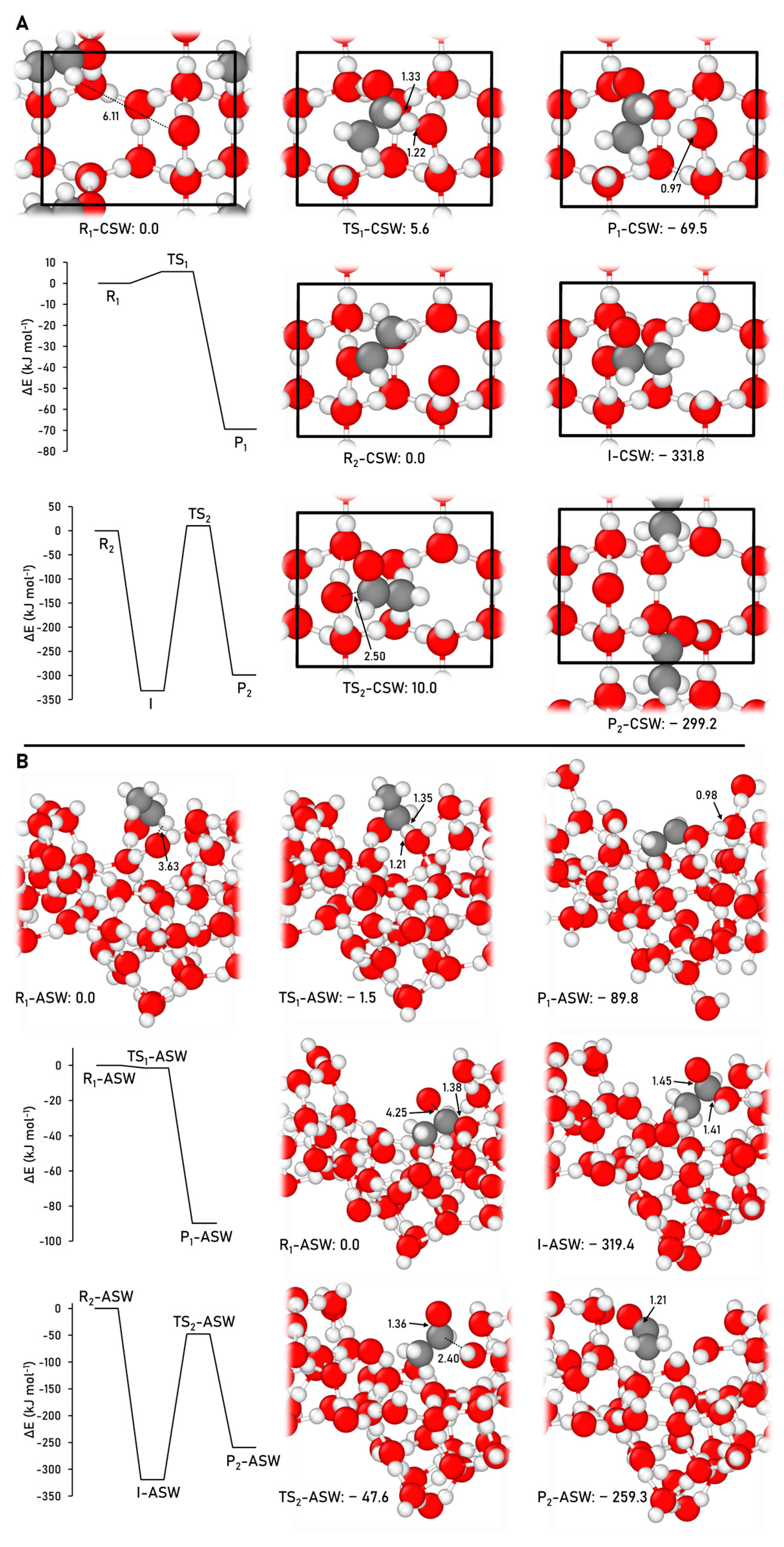
2.2.3. Methyl Formate Formation
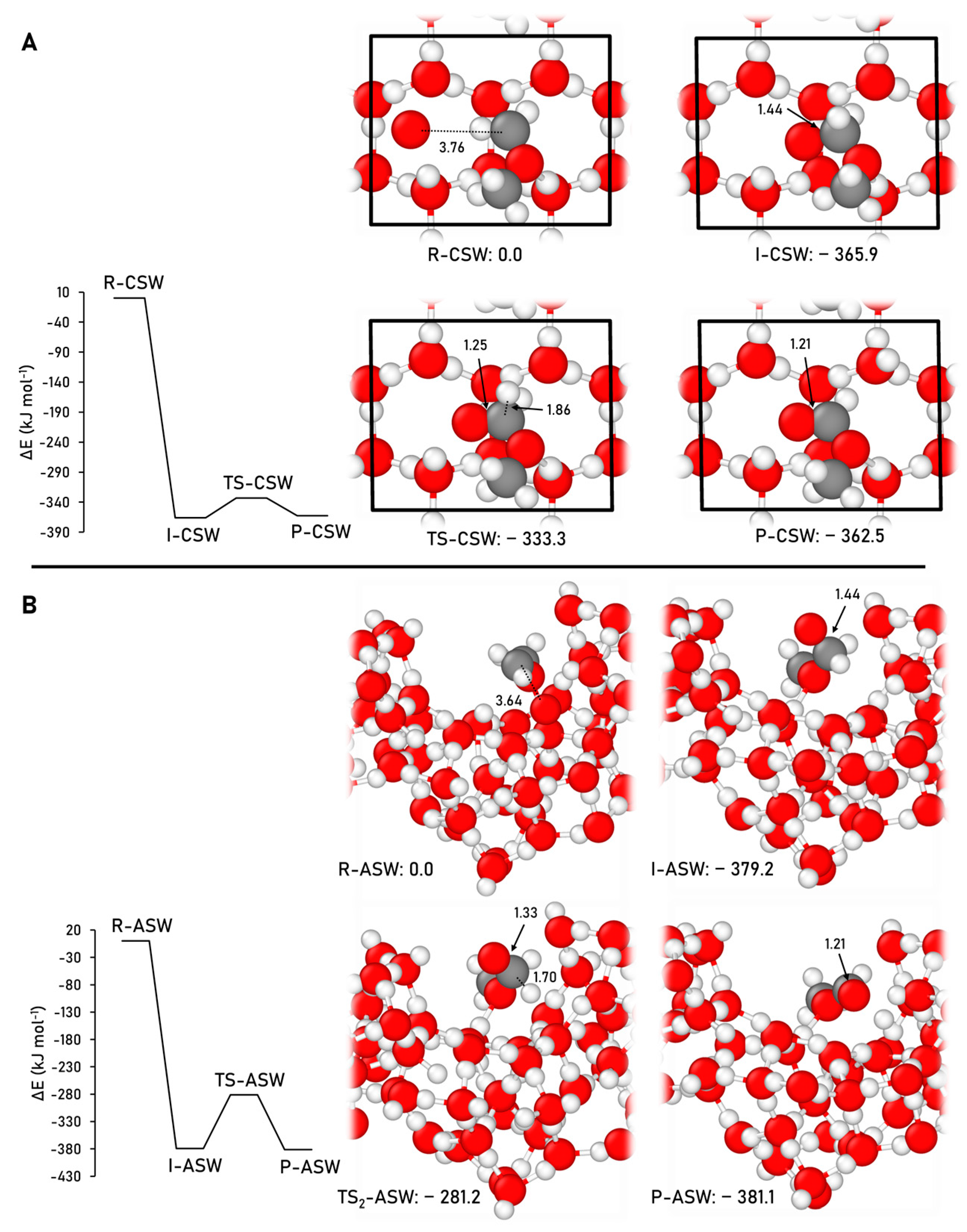
2.2.4. Formic Acid Formation
3. Discussion
4. Materials and Methods
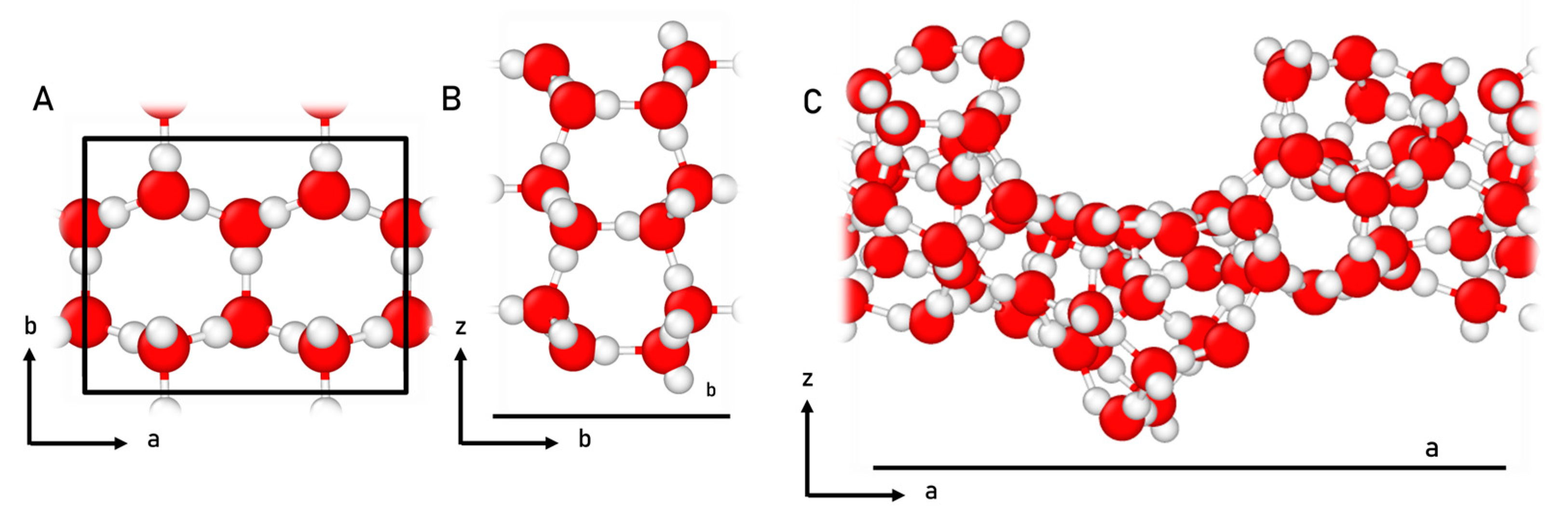
4.1. Surface Modelling
4.2. Computatonal Details
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Benchmark Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Species | PBE-D3(BJ) | B3LYP-D3(BJ) | BHLYP-D3(BJ) | M06−2X-D3 | ||||
|---|---|---|---|---|---|---|---|---|---|
| PBE-D3(BJ) | CCSD(T) | B3LYP-D3(BJ) | CCSD(T) | BHLYP-D3(BJ) | CCSD(T) | M06−2X-D3 | CCSD(T) | ||
| H2CO + NH2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| H2CO − NH2 | – | – | −13.6 | −10.9 | −13.4 | −11.1 | −16.5 | −11.0 | |
| TS1 | – | – | −8.6 | 2.1 | 3.7 | 2.6 | −1.6 | 3.3 | |
| H2CONH2 | −103.2 | −62.6 | −77.4 | −63.2 | −77.9 | −63.7 | −79.5 | −63.7 | |
| HCONH2 | TS2 | −52.9 | 4.3 | −14.9 | 5.4 | 4.3 | 5.1 | −15.0 | 6.4 |
| HCONH2 + H | −75.5 | −37.1 | −42.1 | −37.2 | −31.7 | −37.4 | −57.7 | −37.1 | |
| Error ∆E1‡ (%) | – | – | 61.6 | – | 25.1 | – | 3.8 | – | |
| Error ∆E2‡ (%) | 24.8 | – | 8.9 | – | 19.6 | – | 7.9 | – | |
| Average error (%) | – | – | 35.3 | – | 22.4 | – | 5.9 | – | |
| CH3CH2OH + OH | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| CH3CH2OH − OH | −33.1 | −29.1 | −31.6 | −29.4 | −32.0 | −29.5 | −28.3 | −26.8 | |
| TS1 | −28.1 | −21.7 | −18.9 | −8.4 | 6.7 | 2.0 | −2.9 | 0.4 | |
| CH3CHOH + H2O | −118.5 | −92.6 | −100.4 | −92.4 | −80.3 | −92.2 | −98.2 | −92.5 | |
| CH3CHOH + O | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| CH3CHO | CH3CHOOH | −457.3 | −393.7 | −412.1 | −398.6 | −356.9 | −379.7 | −394.7 | −378.2 |
| TS2 | −348.7 | −323.2 | −315.8 | −297.4 | −261.1 | −297.3 | −296.6 | −296.9 | |
| CH3CHO + OH | −328.9 | −307.1 | −313.9 | −307.1 | −279.4 | −306.1 | −308.4 | −306.8 | |
| Error ∆E1‡ (%) | 32.3 | – | 39.4 | – | 22.8 | – | 6.7 | – | |
| Error ∆E2‡ (%) | 55.3 | – | 4.9 | – | 16.3 | – | 20.8 | – | |
| Average error (%) | 43.8 | – | 22.1 | – | 19.5 | – | 13.8 | – | |
| CH3OCH2 + O | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| H2COOCH3 | −458.4 | −378.6 | −403.4 | −393.5 | −368.5 | −394.2 | −434.1 | −394.5 | |
| HCOOCH3 | TS | −395.6 | −328.7 | −337.1 | −327.3 | −275.3 | −325.3 | −361.8 | −325.7 |
| HCOOCH3 + H | −414.2 | −365.3 | −359.7 | −365.4 | −306.3 | −363.8 | −399.9 | −365.0 | |
| Error ∆E‡ (%) | 25.7 | – | 0.3 | – | 35.4 | – | 5.1 | – | |
| H2CO + OH | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| H2CO − OH | – | – | −23.1 | −22.0 | −23.9 | −23.4 | −23.2 | −23.1 | |
| TS1 | – | – | −5.3 | 7.8 | 12.9 | 7.9 | −17.0 | 8.6 | |
| H2COOH | −154.8 | −100.1 | −109.1 | −95.6 | −116.7 | −107.7 | −126.1 | −107.2 | |
| HCOOH | TS2 | −93.5 | −38.5 | −33.7 | −20.6 | −24.4 | −36.5 | −53.7 | −36.0 |
| HCOOH + H | −113.2 | −76.6 | −56.5 | −58.9 | −55.7 | −75.8 | −92.0 | −76.4 | |
| Error ∆E1‡ (%) | – | – | 40.1 | – | 17.8 | – | 80.5 | – | |
| Error ∆E2‡ (%) | 0.6 | – | 0.6 | – | 29.6 | – | 1.6 | – | |
| Average error (%) | – | – | 20.4 | – | 23.7 | – | 41.1 | – | |
| Species | PBE-D3(BJ) | B3LYP-D3(BJ) | BHLYP-D3(BJ) | M06−2X-D3 | CCSD(T) |
|---|---|---|---|---|---|
| H2CO + NH2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| H2CO − NH2 | −9.0 | −5.6 | −2.7 | −4.9 | −7.2 |
| TS1 | −19.1 | 4.4 | 22.8 | 16.5 | 16.2 |
| H2CONH2 | −80.8 | −61.2 | −68.1 | −67.8 | −50.5 |
| TS2 | −46.3 | −7.8 | 14.0 | −8.2 | 6.8 |
| HCONH2 + H | −74.5 | −41.3 | −30.8 | −57.2 | −36.2 |
| Error ∆E1‡ (%) | 143.0 | 57.1 | 8.7 | 8.5 | – |
| Error ∆E2‡ (%) | 39.9 | 6.8 | 43.4 | 4.0 | – |
| Average error (%) | 91.4 | 32.0 | 26.0 | 6.3 | – |
References
- Boogert, A.A.; Gerakines, P.A.; Whittet, D.C. Observations of the icy universe. Ann. Rev. Astron. Astrophys. 2015, 53, 541–581. [Google Scholar] [CrossRef]
- McClure, M.K.; Rocha, W.R.M.; Pontoppidan, K.M.; Crouzet, N.; Chu, L.E.U.; Dartois, E.; Lamberts, T.; Noble, J.A.; Pendleton, Y.J.; Perotti, G.; et al. An Ice Age JWST inventory of dense molecular cloud ices. Nat. Astron. 2023, 7, 431–443. [Google Scholar] [CrossRef]
- McGuire, B.A. 2021 Census of Interstellar, Circumstellar, Extragalactic, Protoplanetary Disk, and Exoplanetary Molecules. Astrophys. J. Suppl. Ser. 2022, 259, 30. [Google Scholar] [CrossRef]
- Herbst, E.; van Dishoeck, E.F. Complex Organic Interstellar Molecules. Ann. Rev. Astron. Astrophys. 2009, 47, 427–480. [Google Scholar] [CrossRef]
- Herbst, E. The synthesis of large interstellar molecules. Int. Rev. Phys. Chem. 2017, 36, 287–331. [Google Scholar] [CrossRef]
- Ceccarelli, C.; Caselli, P.; Fontani, F.; Neri, R.; López-Sepulcre, A.; Codella, C.; Feng, S.; Jiménez-Serra, I.; Lefloch, B.; Pineda, J.E.; et al. Seeds Of Life In Space (SOLIS): The Organic Composition Diversity at 300–1000 au Scale in Solar-type Star-forming Regions. Astrophys. J. 2017, 850, 176. [Google Scholar] [CrossRef]
- Caselli, P.; Ceccarelli, C. Our astrochemical heritage. Astron. Astrophys. Rev. 2012, 20, 56. [Google Scholar] [CrossRef]
- Rotelli, L.; Trigo-Rodríguez, J.M.; Moyano-Cambero, C.E.; Carota, E.; Botta, L.; Di Mauro, E.; Saladino, R. The key role of meteorites in the formation of relevant prebiotic molecules in a formamide/water environment. Sci. Rep. 2016, 6, 38888. [Google Scholar] [CrossRef]
- Kwok, S. Complex organics in space from Solar System to distant galaxies. Astron. Astrophys. Rev. 2016, 24, 8. [Google Scholar] [CrossRef]
- Kitadai, N.; Maruyama, S. Origins of building blocks of life: A review. Geosci. Front. 2018, 9, 1117–1153. [Google Scholar]
- Garrod, R.T.; Herbst, E. Formation of methyl formate and other organic species in the warm-up phase of hot molecular cores. Astron. Astrophys. 2006, 457, 927–936. [Google Scholar] [CrossRef]
- Garrod, R.T.; Weaver, S.L.W.; Herbst, E. Complex chemistry in star-forming regions: An expanded gas-grain warm-up chemical model. Astrophys. J. 2008, 682, 283. [Google Scholar] [CrossRef]
- Vasyunin, A.I.; Herbst, E. Reactive Desorption and Radiative Association as Possible Drivers of Complex Molecule Formation in the Cold Interstellar Medium. Astrophys. J. 2013, 769, 34. [Google Scholar] [CrossRef]
- Balucani, N.; Ceccarelli, C.; Taquet, V. Formation of complex organic molecules in cold objects: The role of gas-phase reactions. Mon. Not. R. Astron. Soc. 2015, 449, L16–L20. [Google Scholar] [CrossRef]
- Jørgensen, J.K.; Belloche, A.; Garrod, R.T. Astrochemistry during the formation of stars. Ann. Rev. Astron. Astrophys. 2020, 58, 727–778. [Google Scholar] [CrossRef]
- Ceccarelli, C.; Codella, C.; Balucani, N.; Bockelee-Morvan, D.; Herbst, E.; Vastel, C.; Caselli, P.; Favre, C.; Lefloch, B.; Oberg, K.; et al. Organic Chemistry in the First Phases of Solar-Type Protostars. In Proceedings of the Astronomical Society of the Pacific Conference Series; Inutsuka, S., Aikawa, Y., Muto, T., Tomida, K., Tamura, M., Eds.; Cornell University: Ithaca, NY, USA, 2023; Volume 534, p. 379. [Google Scholar]
- Tielens, A.G.G.M.; Hagen, W. Model calculations of the molecular composition of interstellar grain mantles. Astron. Astrophys. 1982, 114, 245–260. [Google Scholar]
- Molpeceres, G.; Rimola, A.; Ceccarelli, C.; Kästner, J.; Ugliengo, P.; Maté, B. Silicate-mediated interstellar water formation: A theoretical study. Mon. Not. R. Astron. Soc. 2019, 482, 5389–5400. [Google Scholar] [CrossRef]
- Ferrero, S.; Pantaleone, S.; Ceccarelli, C.; Ugliengo, P.; Sodupe, M.; Rimola, A. Where does the energy go during the interstellar NH3 formation on water ice? A computational study. Astrophys. J. 2023, 944, 142. [Google Scholar] [CrossRef]
- Rimola, A.; Taquet, V.; Ugliengo, P.; Balucani, N.; Ceccarelli, C. Combined quantum chemical and modeling study of CO hydrogenation on water ice. Astron. Astrophys. 2014, 572, A70. [Google Scholar] [CrossRef]
- Skouteris, D.; Balucani, N.; Ceccarelli, C.; Vazart, F.; Puzzarini, C.; Barone, V.; Codella, C.; Lefloch, B. The genealogical tree of ethanol: Gas-phase formation of glycolaldehyde, acetic acid, and formic acid. Astrophys. J. 2018, 854, 135. [Google Scholar] [CrossRef]
- Vazart, F.; Ceccarelli, C.; Balucani, N.; Bianchi, E.; Skouteris, D. Gas-phase formation of acetaldehyde: Review and new theoretical computations. Mon. Not. R. Astron. Soc. 2020, 499, 5547–5561. [Google Scholar] [CrossRef]
- Heard, D.E. Rapid acceleration of hydrogen atom abstraction reactions of OH at very low temperatures through weakly bound complexes and tunneling. Acc. Chem. Res. 2018, 51, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Latouche, C.; Skouteris, D.; Vazart, F.; Balucani, N.; Ceccarelli, C.; Lefloch, B. Gas-phase formation of the prebiotic molecule formamide: Insights from new quantum computations. Mon. Not. R. Astron. Soc. 2015, 453, L31–L35. [Google Scholar] [CrossRef]
- Skouteris, D.; Vazart, F.; Ceccarelli, C.; Balucani, N.; Puzzarini, C.; Barone, V. New quantum chemical computations of formamide deuteration support gas-phase formation of this prebiotic molecule. Mon. Not. R. Astron. Soc. 2017, 468, L1–L5. [Google Scholar] [CrossRef]
- Enrique-Romero, J.; Rimola, A.; Ceccarelli, C.; Balucani, N. The (impossible?) formation of acetaldehyde on the grain surfaces: Insights from quantum chemical calculations. Mon. Not. R. Astron. Soc. 2016, 459, L6–L10. [Google Scholar] [CrossRef]
- Rimola, A.; Skouteris, D.; Balucani, N.; Ceccarelli, C.; Enrique-Romero, J.; Taquet, V.; Ugliengo, P. Can Formamide Be Formed on In- 697 terstellar Ice? An Atomistic Perspective. ACS Earth Space Chem. 2018, 2, 720–734. [Google Scholar] [CrossRef]
- Enrique-Romero, J.; Ceccarelli, C.; Rimola, A.; Skouteris, D.; Balucani, N.; Ugliengo, P. Theoretical computations on the efficiency of acetaldehyde formation on interstellar icy grains. Astron. Astrophys. 2021, 655, A9. [Google Scholar] [CrossRef]
- Enrique-Romero, J.; Rimola, A.; Ceccarelli, C.; Ugliengo, P.; Balucani, N.; Skouteris, D. Quantum Mechanical Simulations of the Radical–Radical Chemistry on Icy Surfaces. Astrophys. J. Suppl. Ser. 2022, 259, 39. [Google Scholar] [CrossRef]
- Molpeceres, G.; Jiménez-Serra, I.; Oba, Y.; Nguyen, T.; Watanabe, N.; de la Concepción, J.G.; Maté, B.; Oliveira, R.; Kästner, J. Hydrogen abstraction reactions in formic and thioformic acid isomers by hydrogen and deuterium atoms. Astron. Astrophys. 2022, 663, A41. [Google Scholar] [CrossRef]
- Jin, M.; Garrod, R.T. Formation of complex organic molecules in cold interstellar environments through nondiffusive grain-surface and ice-mantle chemistry. Astrophys. J. Suppl. Ser. 2020, 249, 26. [Google Scholar] [CrossRef]
- Garrod, R.T.; Jin, M.; Matis, K.A.; Jones, D.; Willis, E.R.; Herbst, E. Formation of complex organic molecules in hot molecular cores through nondiffusive grain-surface and ice-mantle chemistry. Astrophys. J. Suppl. Ser. 2022, 259, 1. [Google Scholar] [CrossRef]
- Molpeceres, G.; Kastner, J.; Fedoseev, G.; Qasim, D.; Schomig, R.; Linnartz, H.; Lamberts, T. Carbon atom reactivity with amorphous solid water: H2O-catalyzed formation of H2CO. J. Phys. Chem. Lett. 2021, 12, 10854–10860. [Google Scholar] [CrossRef] [PubMed]
- Perrero, J.; Enrique-Romero, J.; Martínez-Bachs, B.; Ceccarelli, C.; Balucani, N.; Ugliengo, P.; Rimola, A. Non-energetic formation of ethanol via CCH reaction with interstellar H2O ices. A computational chemistry study. ACS Earth Space Chem. 2022, 6, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Perrero, J.; Ugliengo, P.; Ceccarelli, C.; Rimola, A. Quantum mechanical modelling of the grain-surface formation of acetaldehyde on H2O: CO dirty ice surfaces. Mon. Not. R. Astron. Soc. 2023, 525, 2654–2667. [Google Scholar] [CrossRef]
- Rubin, R.; Swenson, G., Jr.; Benson, R.; Tigelaar, H.; Flygare, W. Microwave detection of interstellar formamide. Astrophys. J. 1971, 169, L39. [Google Scholar] [CrossRef]
- Kahane, C.; Ceccarelli, C.; Faure, A.; Caux, E. Detection of formamide, the simplest but crucial amide, in a solar-type protostar. Astrophys. J. Lett. 2013, 763, L38. [Google Scholar] [CrossRef]
- Mendoza, E.; Lefloch, B.; López-Sepulcre, A.; Ceccarelli, C.; Codella, C.; Boechat-Roberty, H.; Bachiller, R. Molecules with a peptide link in protostellar shocks: A comprehensive study of L1157. Mon. Not. R. Astron. Soc. 2014, 445, 151–161. [Google Scholar] [CrossRef]
- Jaber, A.A.; Ceccarelli, C.; Kahane, C.; Caux, E. The census of complex organic molecules in the solar-type protostar IRAS16293-2422. Astrophys. J. 2014, 791, 29. [Google Scholar] [CrossRef]
- López-Sepulcre, A.; Jaber, A.A.; Mendoza, E.; Lefloch, B.; Ceccarelli, C.; Vastel, C.; Bachiller, R.; Cernicharo, J.; Codella, C.; Kahane, C.; et al. Shedding light on the formation of the pre-biotic molecule formamide with ASAI. Mon. Not. R. Astron. Soc. 2015, 449, 2438–2458. [Google Scholar] [CrossRef]
- Marcelino, N.; Gerin, M.; Cernicharo, J.; Fuente, A.; Wootten, H.; Chapillon, E.; Pety, J.; Lis, D.C.; Roueff, E.; Commerçon, B.; et al. ALMA observations of the young protostellar system Barnard 1b: Signatures of an incipient hot corino in B1b-S. Astron. Astrophys. 2018, 620, A80. [Google Scholar] [CrossRef]
- Bianchi, E.; Codella, C.; Ceccarelli, C.; Vazart, F.; Bachiller, R.; Balucani, N.; Bouvier, M.; De Simone, M.; Enrique-Romero, J.; Kahane, C.; et al. The census of interstellar complex organic molecules in the Class I hot corino of SVS13-A. Mon. Not. R. Astron. Soc. 2019, 483, 1850–1861. [Google Scholar] [CrossRef]
- López-Sepulcre, A.; Balucani, N.; Ceccarelli, C.; Codella, C.; Dulieu, F.; Theulé, P. Interstellar formamide (NH2CHO), a key prebiotic precursor. ACS Earth Space Chem. 2019, 3, 2122–2137. [Google Scholar] [CrossRef]
- Song, L.; Kästner, J. Formation of the prebiotic molecule NH2CHO on astronomical amorphous solid water surfaces: Accurate tunneling rate calculations. Phys. Chem. Chem. Phys. 2016, 18, 29278–29285. [Google Scholar] [CrossRef] [PubMed]
- Enrique-Romero, J.; Rimola, A.; Ceccarelli, C.; Ugliengo, P.; Balucani, N.; Skouteris, D. Reactivity of HCO with CH3 and NH2 on water ice surfaces. a comprehensive accurate quantum chemistry study. ACS Earth Space Chem. 2019, 3, 2158–2170. [Google Scholar] [CrossRef]
- Vazart, F.; Calderini, D.; Puzzarini, C.; Skouteris, D.; Barone, V. State-of-the-art thermochemical and kinetic computations for astrochemical complex organic molecules: Formamide formation in cold interstellar clouds as a case study. J. Chem. Theory Comput. 2016, 12, 5385–5397. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, C.A. Detection of acetaldehyde in Sagittarius. In Molecules in the Galactic Environment; John Wiley and Sons: Hoboken, NJ, USA, 1973; p. 181. [Google Scholar]
- Pizzarello, S.; Weber, A.L. Prebiotic amino acids as asymmetric catalysts. Science 2004, 303, 1151. [Google Scholar] [CrossRef] [PubMed]
- Córdova, A.; Ibrahem, I.; Casas, J.; Sundén, H.; Engqvist, M.; Reyes, E. Amino acid catalyzed neogenesis of carbohydrates: A plausible ancient transformation. Chem. Eur. J. 2005, 11, 4772–4784. [Google Scholar] [CrossRef]
- Charnley, S. Acetaldehyde in star-forming regions. Adv. Space Res. 2004, 33, 23–30. [Google Scholar] [CrossRef]
- Vasyunin, A.I.; Caselli, P.; Dulieu, F.; Jiménez-Serra, I. Formation of complex molecules in prestellar cores: A multilayer approach. Astrophys. J. 2017, 842, 33. [Google Scholar] [CrossRef]
- Lamberts, T.; Markmeyer, M.N.; Kolb, F.J.; Kastner, J. Formation of acetaldehyde on CO-rich ices. ACS Earth Space Chem. 2019, 3, 958–963. [Google Scholar] [CrossRef]
- Enrique-Romero, J.; Álvarez-Barcia, S.; Kolb, F.; Rimola, A.; Ceccarelli, C.; Balucani, N.; Meisner, J.; Ugliengo, P.; Lamberts, T.; Kästner, J. Revisiting the reactivity between HCO and CH3 on interstellar grain surfaces. Mon. Not. R. Astron. Soc. 2020, 493, 2523–2527. [Google Scholar] [CrossRef]
- Ferrero, S.; Ceccarelli, C.; Ugliengo, P.; Sodupe, M.; Rimola, A. Formation of complex organic molecules on interstellar CO ices? Insights from computational chemistry simulations. Astrophys. J. 2023, 951, 150. [Google Scholar] [CrossRef]
- Xu, S.; Lin, M.C. Theoretical study on the kinetics for OH reactions with CH3OH and C2H5OH. Proc. Combust. Inst. 2007, 31, 159–166. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R.; Bravo-Pérez, G.; Ruiz-Santoyo, M.E. Gas phase reactions of C1–C4 alcohols with the OH radical: A quantum mechanical approach. Phys. Chem. Chem. Phys. 2002, 4, 4648–4662. [Google Scholar] [CrossRef]
- Zheng, J.; Truhlar, D.G. Multi-path variational transition state theory for chemical reaction rates of complex polyatomic species: Ethanol + OH reactions. Faraday Discuss. 2012, 157, 59–88. [Google Scholar] [CrossRef] [PubMed]
- Marinov, N.M. A detailed chemical kinetic model for high temperature ethanol oxidation. Int. J. Chem. Kinet. 1999, 31, 183–220. [Google Scholar] [CrossRef]
- Carr, S.A.; Blitz, M.A.; Seakins, P.W. Site-specific rate coefficients for reaction of OH with ethanol from 298 to 900 K. J. Phys. Chem. A 2011, 115, 3335–3345. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Crofts, J.; Gardner, F.; Godfrey, P.; Robinson, B.; Whiteoak, J. Discovery of interstellar methyl formate. Astrophys. J. 1975, 197, L29–L31. [Google Scholar] [CrossRef]
- Hollis, J.; Vogel, S.; Snyder, L.; Jewell, P.; Lovas, F. The spatial scale of glycolaldehyde in the galactic center. Astrophys. J. 2001, 554, L81. [Google Scholar] [CrossRef]
- Horn, A.; Møllendal, H.; Sekiguchi, O.; Uggerud, E.; Roberts, H.; Herbst, E.; Viggiano, A.; Fridgen, T.D. The gas-phase formation of methyl formate in hot molecular cores. Astrophys. J. 2004, 611, 605. [Google Scholar] [CrossRef]
- Laas, J.C.; Garrod, R.T.; Herbst, E.; Weaver, S.L.W. Contributions from grain surface and gas phase chemistry to the formation of methyl formate and its structural isomers. Astrophys. J. 2011, 728, 71. [Google Scholar] [CrossRef]
- Ruaud, M.; Loison, J.; Hickson, K.C.; Gratier, P.M.; Hersant, F.W.V. Modelling complex organic molecules in dense regions: Eley-Rideal and complex induced reaction. Mon. Not. R. Astron. Soc. 2015, 447, 4004–4017. [Google Scholar] [CrossRef]
- Hoyermann, K.; Nacke, F. Elementary reactions of the methoxymethyl radical in the gas phase: CH3OCH3+F, CH2OCH3+CH2OCH3, CH2OCH3+O2 and CH2OCH3+O. Symp. Int. Combust. Proc. 1996, 26, 505–512. [Google Scholar] [CrossRef]
- Song, X.; Hou, H.; Wang, B. Mechanistic and kinetic study of the O + CH3OCH2 reaction and the unimolecular decomposition of CH3OCH2O. Phys. Chem. Chem. Phys. 2005, 7, 3980–3988. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, B.; Ball, J.A.; Gottlieb, C.A. Microwave detection of interstellar formic acid. Astrophys. J. 1971, 163, L41. [Google Scholar] [CrossRef]
- Pilling, S.; Baptista, L.; Boechat-Roberty, H.M.; Andrade, D.P. Formation routes of interstellar glycine involving carboxylic acids: Possible favoritism between gas and solid phase. Astrobiology 2011, 11, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Swiderek, P.; Bredehoft, J.H. Formation of formic acid, formaldehyde, and carbon dioxide by electron-induced chemistry in ices of water and carbon monoxide. ACS Earth Space Chem. 2019, 3, 1974–1986. [Google Scholar] [CrossRef]
- Woon, D.E. Quantum Chemical Cluster Studies of Cation–Ice Reactions for Astrochemical Applications: Seeking Experimental Confirmation. Acc. Chem. Res. 2021, 54, 490–497. [Google Scholar] [CrossRef]
- McElroy, D.; Walsh, C.; Markwick, A.; Cordiner, M.; Smith, K.; Millar, T. The UMIST database for astrochemistry 2012. Astron. Astrophys. 2013, 550, A36. [Google Scholar] [CrossRef]
- Wakelam, V.; Loison, J.C.; Herbst, E.; Pavone, B.; Bergeat, A.; Béroff, K.; Chabot, M.; Faure, A.; Galli, D.; Geppert, W.D.; et al. The 2014 KIDA network for interstellar chemistry. Astrophys. J. Suppl. Ser. 2015, 217, 20. [Google Scholar] [CrossRef]
- Yetter, R.A.; Rabitz, H.; Dryer, F.L.; Maki, R.G.; Klemm, R.B. Evaluation of the rate constant for the reaction OH+H2CO: Application of modeling and sensitivity analysis techniques for determination of the product branching ratio. J. Chem. Phys. 1989, 91, 4088–4097. [Google Scholar] [CrossRef]
- D’Anna, B.; Bakken, V.; Beukes, J.A.; Nielsen, C.J.; Brudnik, K.; Jodkowski, J.T. Experimental and theoretical studies of gas phase NO3 and OH radical reactions with formaldehyde, acetaldehyde and their isotopomers. Phys. Chem. Chem. Phys. 2003, 5, 1790–1805. [Google Scholar] [CrossRef]
- Xu, S.; Zhu, R.; Lin, M.C. Ab initio study of the OH+ CH2O reaction: The effect of the OH···OCH2 complex on the H-abstraction kinetics. Int. J. Chem. Kinet. 2006, 38, 322–326. [Google Scholar] [CrossRef]
- Garrod, R.T. A three-phase chemical model of hot cores: The formation of glycine. Astrophys. J. 2013, 765, 60. [Google Scholar] [CrossRef]
- Collings, M.P.; Frankland, V.L.; Lasne, J.; Marchione, D.; Rosu-Finsen, A.; McCoustra, M.R.S. Probing model interstellar grain surfaces with small molecules. Mon. Not. R. Astron. Soc. 2015, 449, 1826–1833. [Google Scholar] [CrossRef]
- Yu, A.; Tatiana, F.; Drebushchak, N.; Tantardini, C. Seeking the best model for non-covalent interactions within the crystal structure of meloxicam. Comput. Theor. Chem. 2019, 1157, 47–53. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Significance of hydrogen bonding and other noncovalent interactions in determining octahedral tilting in the CH3NH3PbI3 hybrid organic-inorganic halide perovskite solar cell semiconductor. Sci. Rep. 2019, 9, 50. [Google Scholar] [CrossRef]
- Chaudhari, S.; Nikam, S.; Khatri, N.; Wakde, S. Co-crystals: A review. J. Drug Deliv. Ther. 2018, 8, 350–358. [Google Scholar] [CrossRef]
- Fucke, K.; Myz, S.A.; Shakhtshneider, T.P.; Boldyreva, E.V.; Griesser, U.J. How good are the crystallisation methods for co-crystals? A comparative study of piroxicam. New J. Chem. 2012, 36, 1969–1977. [Google Scholar] [CrossRef]
- Minissale, M.; Aikawa, Y.; Bergin, E.; Bertin, M.; Brown, W.A.; Cazaux, S.; Charnley, S.B.; Coutens, A.; Cuppen, H.M.; Guzman, V.; et al. Thermal desorption of interstellar ices: A review on the controlling parameters and their implications from snowlines to chemical complexity. ACS Earth Space Chem. 2022, 6, 597–630. [Google Scholar] [CrossRef]
- Congiu, E.; Minissale, M.; Baouche, S.; Chaabouni, H.; Moudens, A.; Cazaux, S.; Manicò, G.; Pirronello, V.; Dulieu, F. Efficient diffusive mechanisms of O atoms at very low temperatures on surfaces of astrophysical interest. Faraday Discuss. 2014, 168, 151. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, M.W.; Meuwly, M. Diffusion of atomic oxygen relevant to water formation in amorphous interstellar ices. Faraday Discuss. 2014, 168, 205. [Google Scholar] [CrossRef] [PubMed]
- Perrero, J.; Enrique-Romero, J.; Ferrero, S.; Ceccarelli, C.; Podio, L.; Codella, C.; Rimola, A.; Ugliengo, P. Binding Energies of Interstellar Relevant S-bearing Species on Water Ice Mantles: A Quantum Mechanical Investigation. Astrophys. J. 2022, 938, 158. [Google Scholar] [CrossRef]
- Pantaleone, S.; Enrique-Romero, J.; Ceccarelli, C.; Ugliengo, P.; Balucani, N.; Rimola, A. Chemical desorption versus energy dissipation: Insights from ab initio molecular dynamics of HCO· formation. Astrophys. J. 2020, 897, 56. [Google Scholar] [CrossRef]
- Pantaleone, S.; Enrique-Romero, J.; Ceccarelli, C.; Ferrero, S.; Balucani, N.; Rimola, A.; Ugliengo, P. H2 formation on interstellar grains and the fate of reaction energy. Astrophys. J. 2021, 917, 49. [Google Scholar] [CrossRef]
- Balakrishnan, N. Quantum mechanical investigation of the O + H2 → OH + H reaction. J. Chem. Phys. 2003, 119, 195–199. [Google Scholar] [CrossRef]
- Meisner, J.; Kamp, I.; Thi, W.F.; Kästner, J. The role of atom tunneling in gas-phase reactions in planet-forming disks. Astron. Astrophys. 2019, 627, A45. [Google Scholar] [CrossRef]
- Fermann, J.T.; Auerbach, S. Modeling proton mobility in acidic zeolite clusters: II. Room temperature tunneling effects from semiclassical rate theory. J. Chem. Phys. 2000, 112, 6787–6794. [Google Scholar] [CrossRef]
- Bao, J.L.; Truhlar, D.G. Variational transition state theory: Theoretical framework and recent developments. Chem. Soc. Rev. 2017, 46, 7548–7596. [Google Scholar] [CrossRef]
- Kästner, J. Theory and simulation of atom tunneling in chemical reactions. WIREs Comput. Mol. Sci. 2014, 4, 158–168. [Google Scholar] [CrossRef]
- Pisani, C.; Casassa, S.; Ugliengo, P. Proton-ordered ice structures at zero pressure. A quantum-mechanical investigation. Chem. Phys. Lett. 1996, 253, 201–208. [Google Scholar] [CrossRef]
- Casassa, S.; Ugliengo, P.; Pisani, C. Proton-ordered models of ordinary ice for quantum-mechanical studies. J. Chem. Phys. 1997, 106, 8030–8040. [Google Scholar] [CrossRef]
- Zamirri, L.; Casassa, S.; Rimola, A.; Segado-Centellas, M.; Ceccarelli, C.; Ugliengo, P. IR spectral fingerprint of carbon monoxide in interstellar water–ice models. Mon. Not. R. Astron. Soc. 2018, 480, 1427–1444. [Google Scholar] [CrossRef]
- Watanabe, N.; Kouchi, A. Ice surface reactions: A key to chemical evolution in space. Prog. Surf. Sci. 2008, 83, 439–489. [Google Scholar] [CrossRef]
- Ferrero, S.; Zamirri, L.; Ceccarelli, C.; Witzel, A.; Rimola, A.; Ugliengo, P. Binding Energies of Interstellar Molecules on Crystalline and Amorphous Models of Water Ice by Ab Initio Calculations. Astrophys. J. 2020, 904, 11. [Google Scholar] [CrossRef]
- Shimonishi, T.; Nakatani, N.; Furuya, K.; Hama, T. Adsorption Energies of Carbon, Nitrogen, and Oxygen Atoms on the Low-temperature Amorphous Water Ice: A Systematic Estimation from Quantum Chemistry Calculations. Astrophys. J. 2018, 732, 27. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Sure, R.; Grimme, S. Corrected small basis set Hartree-Fock method for large systems. J. Comput. Chem. 2013, 34, 1672. [Google Scholar] [CrossRef] [PubMed]
- Kruse, H.; Grimme, S. A geometrical correction for the inter-and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101. [Google Scholar] [CrossRef]
- Tatewaki, H.; Huzinaga, S. A systematic preparation of new contracted Gaussian-type orbital sets. III. Second-row atoms from Li through Ne. J. Comput. Chem. 1980, 1, 205. [Google Scholar] [CrossRef]
- Liu, B.; McLean, A. Accurate calculation of the attractive interaction of two ground state helium atoms. J. Chem. Phys. 1973, 59, 4557. [Google Scholar] [CrossRef]
- Jansen, H.; Ros, P. Non-empirical molecular orbital calculations of the protonation of carbon monoxide. Chem. Phys. Lett. 1969, 3, 140. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Pascale, F.; Zicovich-Wilson, C.M.; López Gejo, F.; Civalleri, B.; Orlando, R.; Dovesi, R. The calculation of the vibrational frequencies of crystalline compounds and its implementation in the CRYSTAL code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef]
- Zicovich-Wilson, C.M.; Pascale, F.; Roetti, C.; Saunders, V.R.; Orlando, R.; Dovesi, R. Calculation of the vibration frequencies of quartz: The effect of Hamiltonian and basis set. J. Comput. Chem. 2004, 25, 1873–1881. [Google Scholar] [CrossRef]








| Reaction | Conditions | Energy Terms | |||
|---|---|---|---|---|---|
| ∆E1‡ | ∆E2‡ | ∆EI−R | ∆EP−R | ||
| Gas phase | 22.6 | 45.6 | −34.4 | −38.1 | |
| Reaction (1) | CWS | 63.7 | 35.6 | −41.6 | −59.8 |
| ASW | 51.7 | 38.4 | −36.7 | −45.0 | |
| ∆E‡ | ∆EP1−R1 | ||||
| Gas phase | 18.1 | −95.0 | |||
| Reaction (2) | CWS | 5.6 | −69.5 | ||
| ASW | Barrierless | −89.8 | |||
| ∆E1‡ | ∆E2‡ | ∆EI−R2 | ∆EP2−R2 | ||
| Gas phase | Barrierless | 72.8 | −364.9 | −312.0 | |
| Reaction (3) | CWS | Barrierless | 341.8 | −331.8 | −299.2 |
| ASW | Barrierless | 271.8 | −319.4 | −259.3 | |
| ∆E1‡ | ∆E2‡ | ∆EI−R | ∆EP−R | ||
| Gas phase | Barrierless | 49.0 | −381.6 | −374.9 | |
| Reaction (4) | CWS | Barrierless | 32.6 | −365.9 | −362.5 |
| ASW | Barrierless | 98.0 | −379.2 | −381.1 | |
| ∆E1‡ | ∆E2‡ | ∆EI−R | ∆EP−R | ||
| Gas phase | 33.1 | 59.2 | −79.6 | −63.1 | |
| Reaction (5) | CWS | 59.5 | 23.7 | −61.6 | −71.9 |
| ASW | 83.6 | 22.1 | −43.1 | −57.2 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Bachs, B.; Rimola, A. Gas-Phase vs. Grain-Surface Formation of Interstellar Complex Organic Molecules: A Comprehensive Quantum-Chemical Study. Int. J. Mol. Sci. 2023, 24, 16824. https://doi.org/10.3390/ijms242316824
Martínez-Bachs B, Rimola A. Gas-Phase vs. Grain-Surface Formation of Interstellar Complex Organic Molecules: A Comprehensive Quantum-Chemical Study. International Journal of Molecular Sciences. 2023; 24(23):16824. https://doi.org/10.3390/ijms242316824
Chicago/Turabian StyleMartínez-Bachs, Berta, and Albert Rimola. 2023. "Gas-Phase vs. Grain-Surface Formation of Interstellar Complex Organic Molecules: A Comprehensive Quantum-Chemical Study" International Journal of Molecular Sciences 24, no. 23: 16824. https://doi.org/10.3390/ijms242316824
APA StyleMartínez-Bachs, B., & Rimola, A. (2023). Gas-Phase vs. Grain-Surface Formation of Interstellar Complex Organic Molecules: A Comprehensive Quantum-Chemical Study. International Journal of Molecular Sciences, 24(23), 16824. https://doi.org/10.3390/ijms242316824