
A Kinetic Investigation of the Supramolecular Chiral Self-Assembling Process of Cationic Organometallic (2,2′:6′,2″-terpyridine)methylplatinum(II) Complexes with Poly(L-glutamic Acid)

 ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
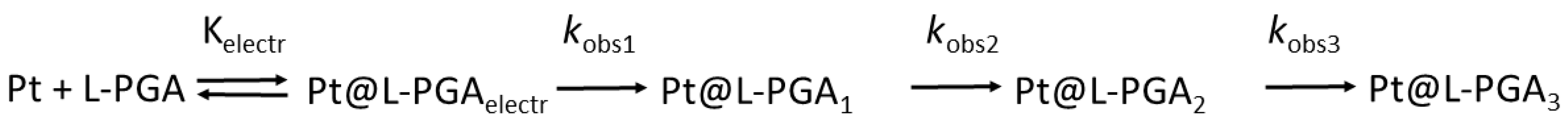{kind=link}
Abstract
1. Introduction
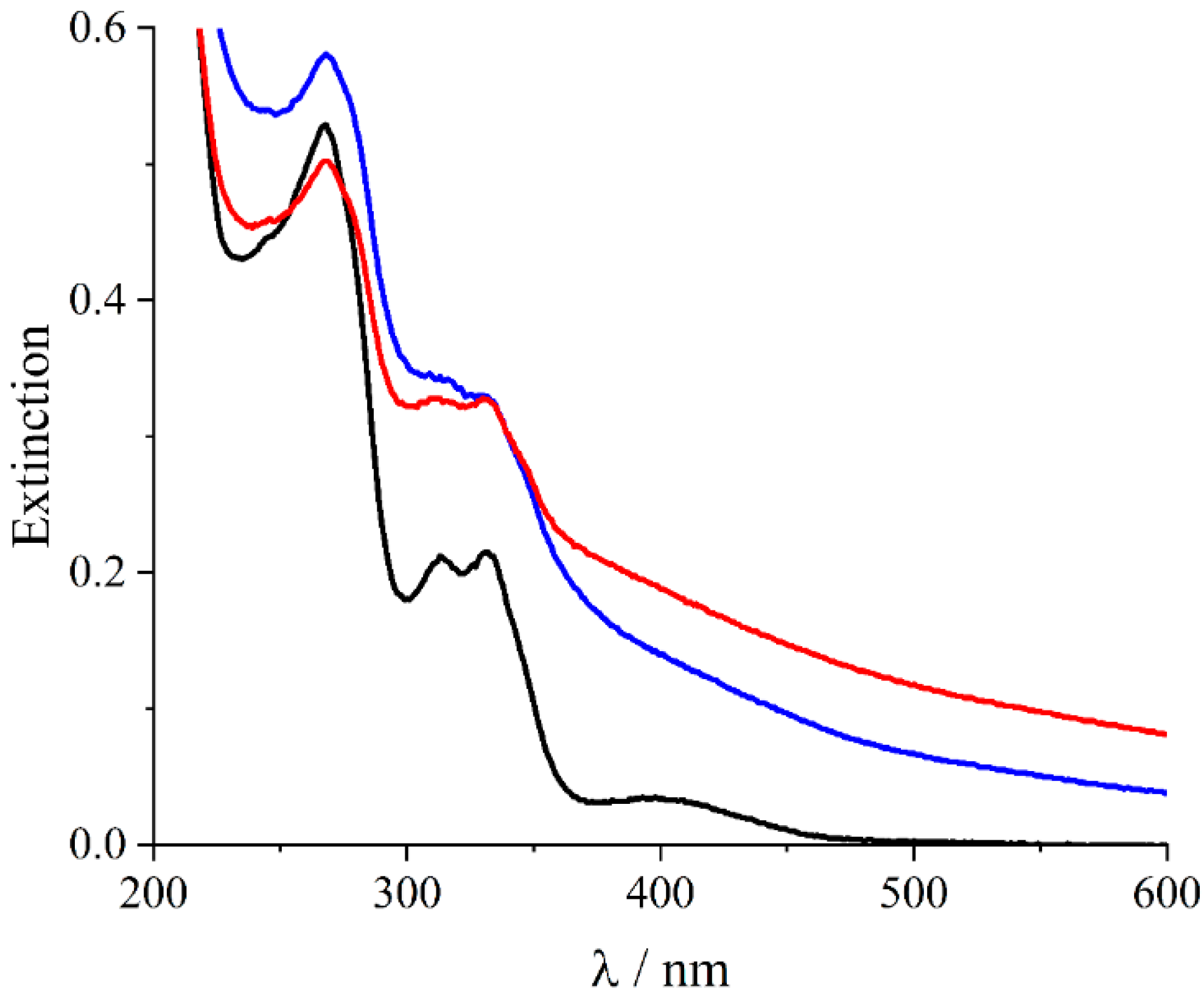
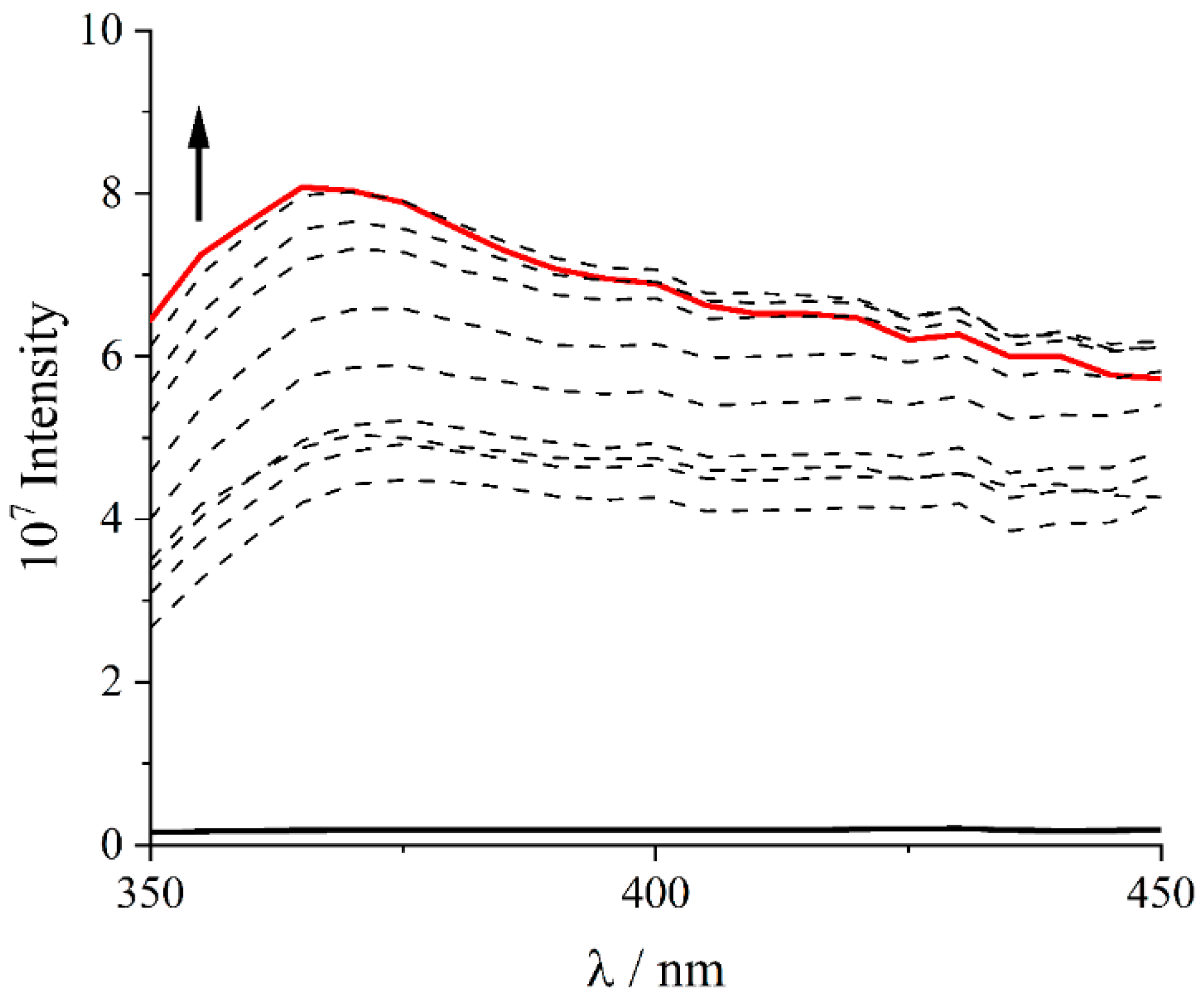
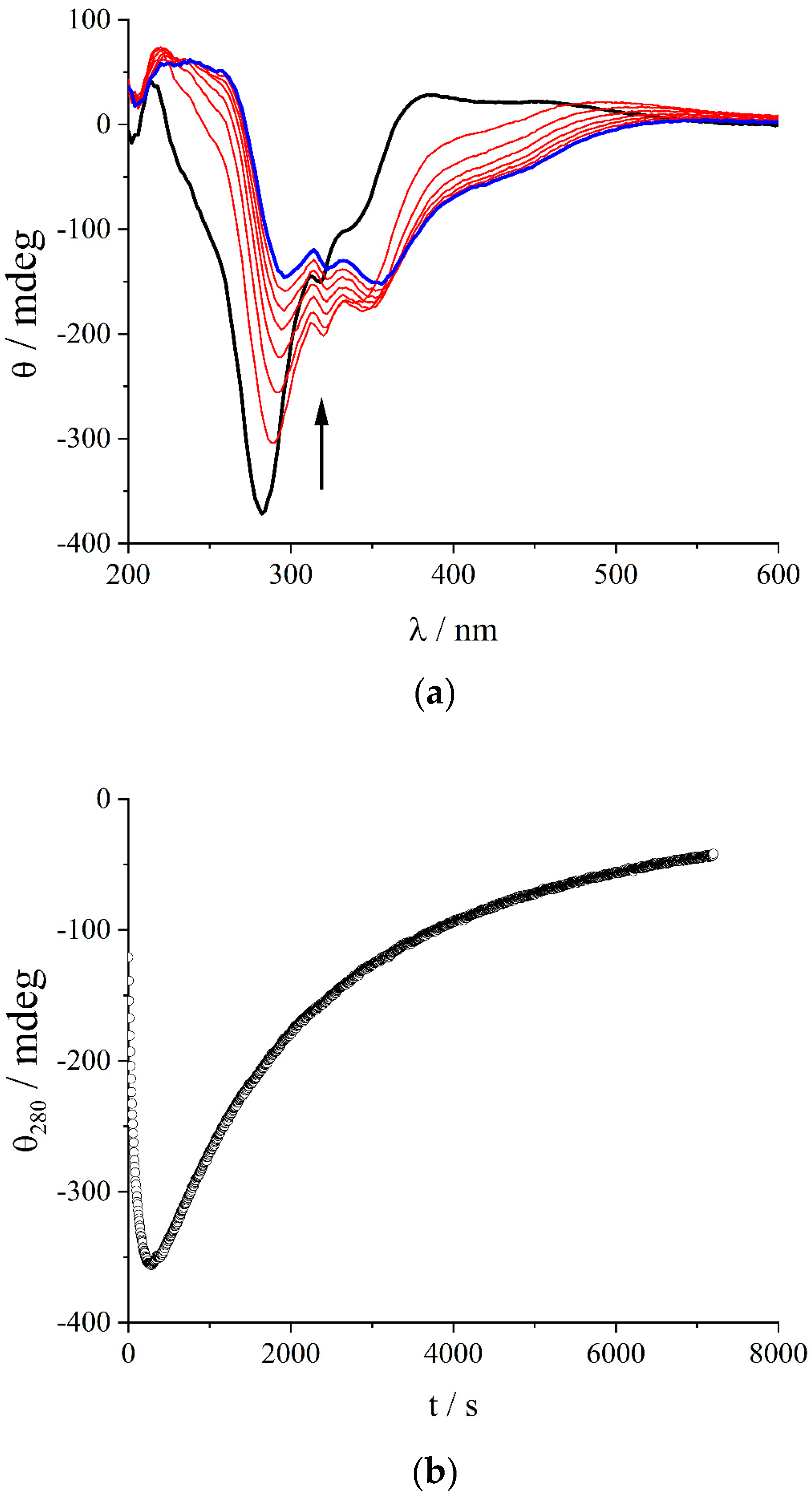
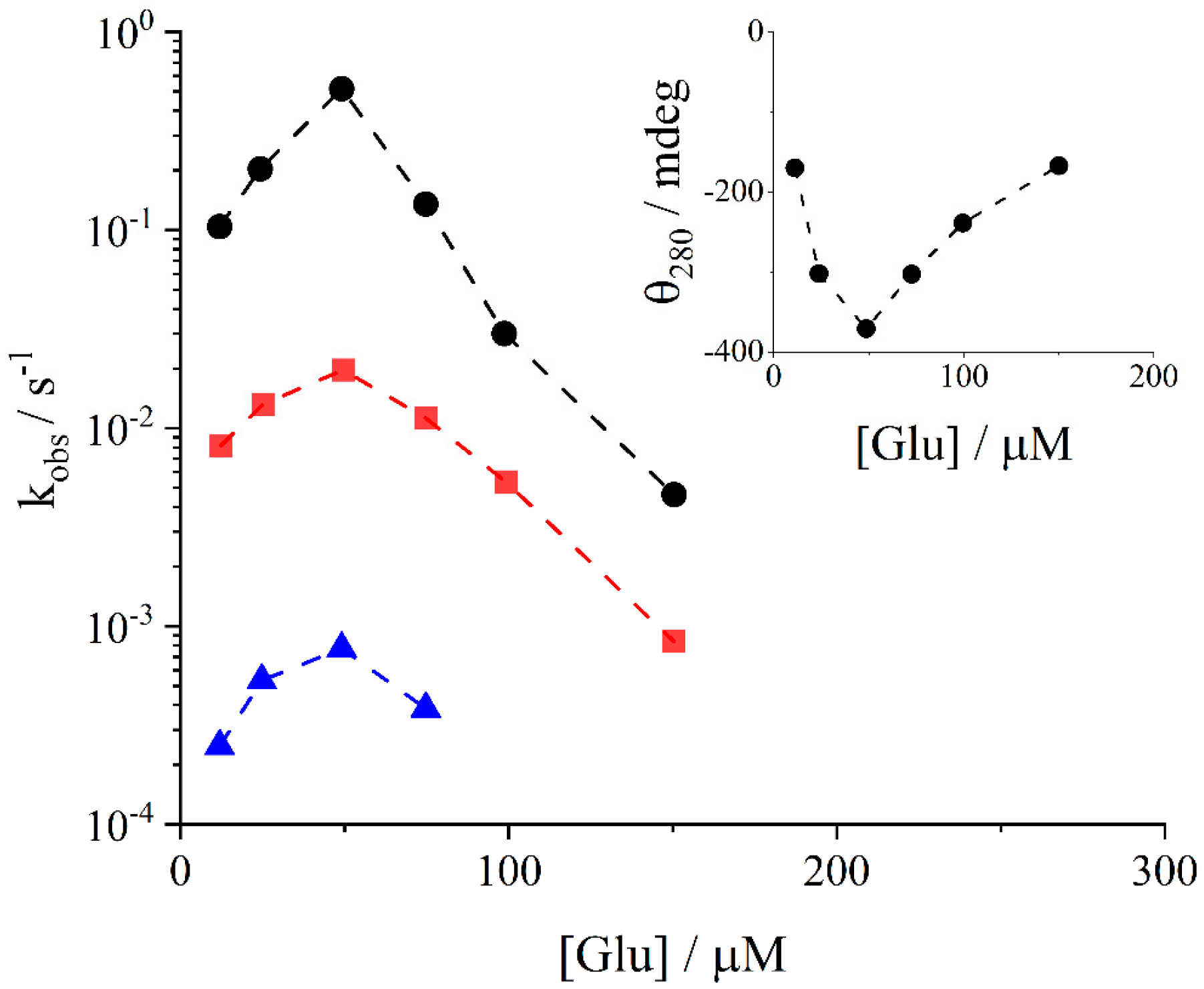
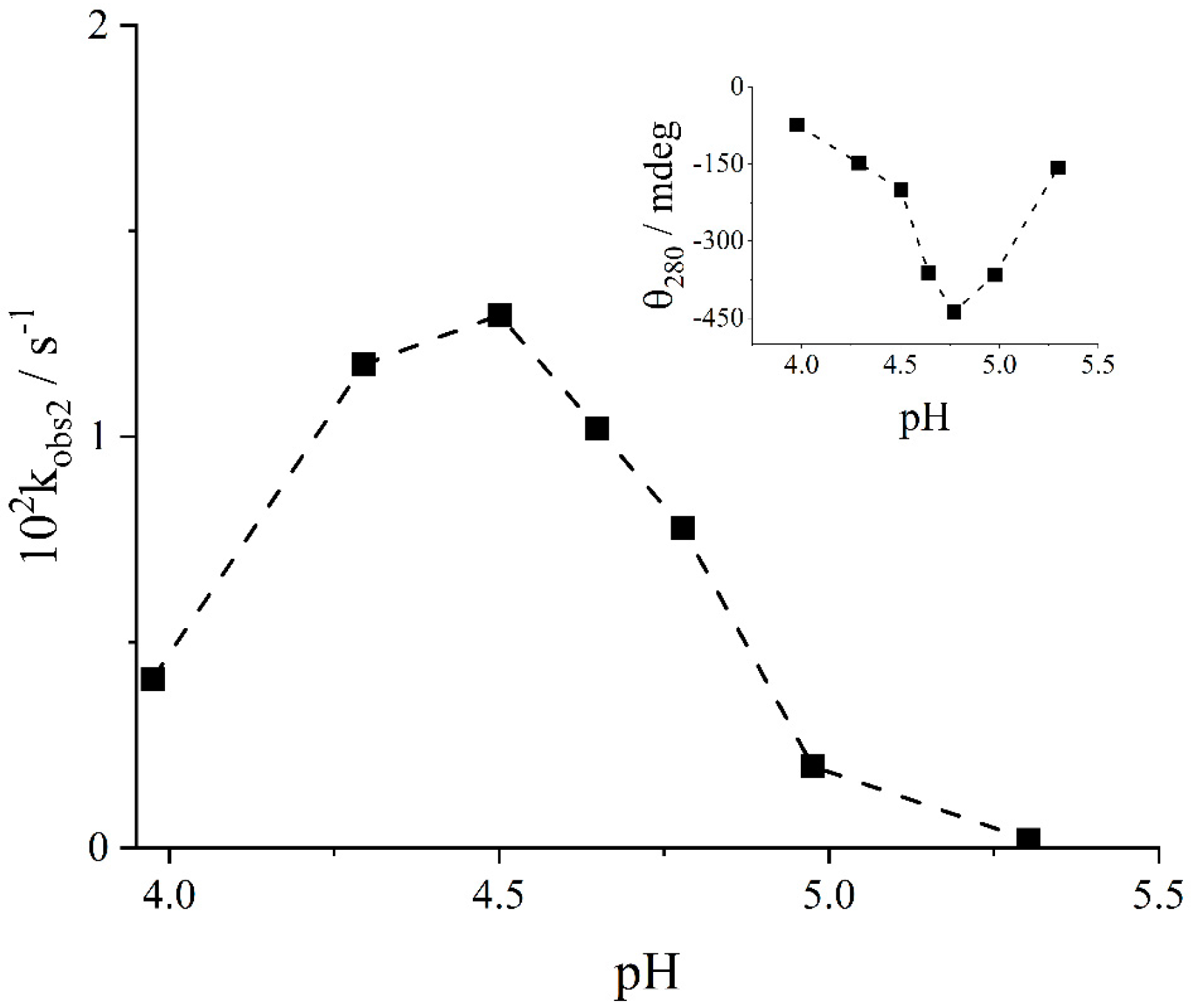
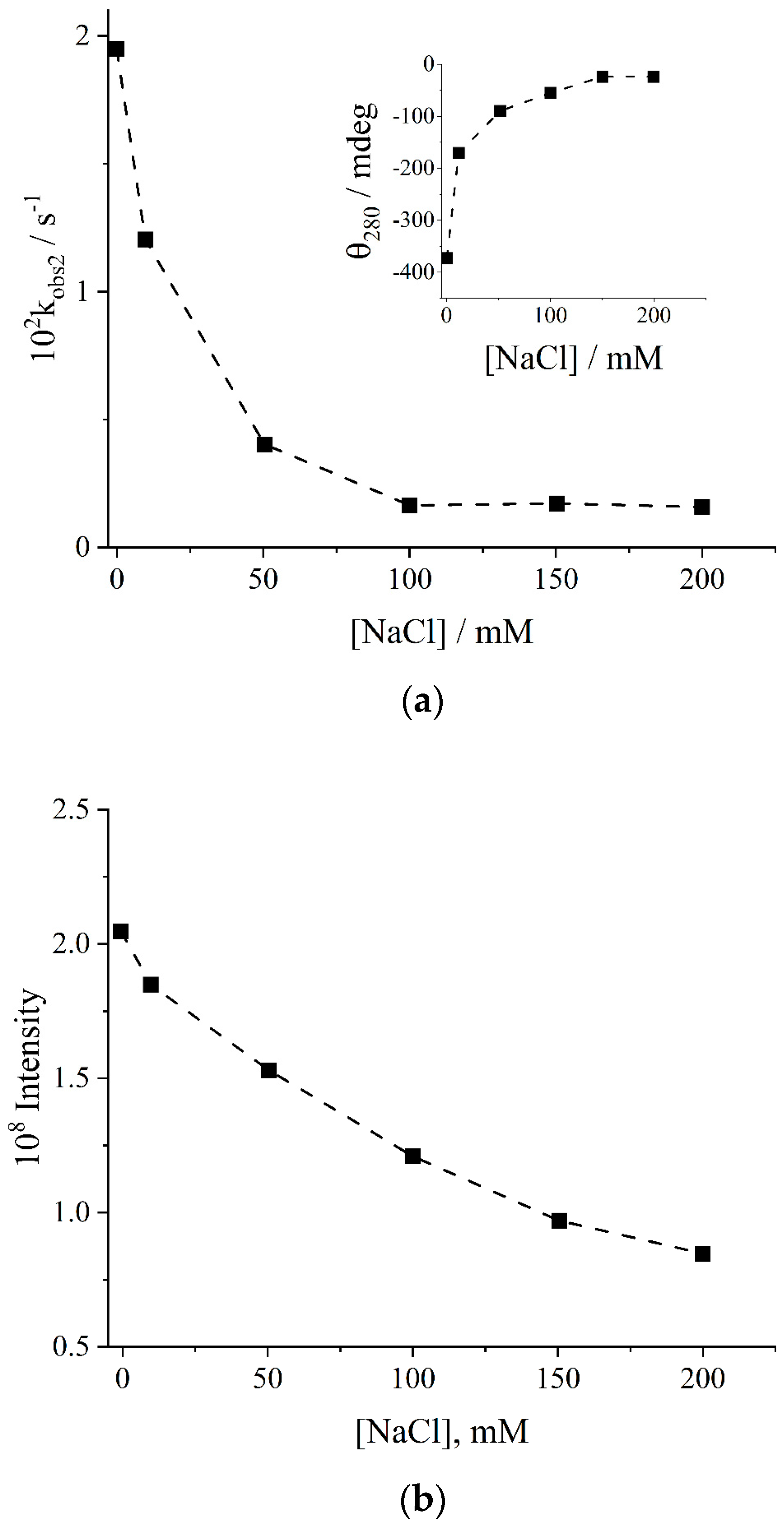
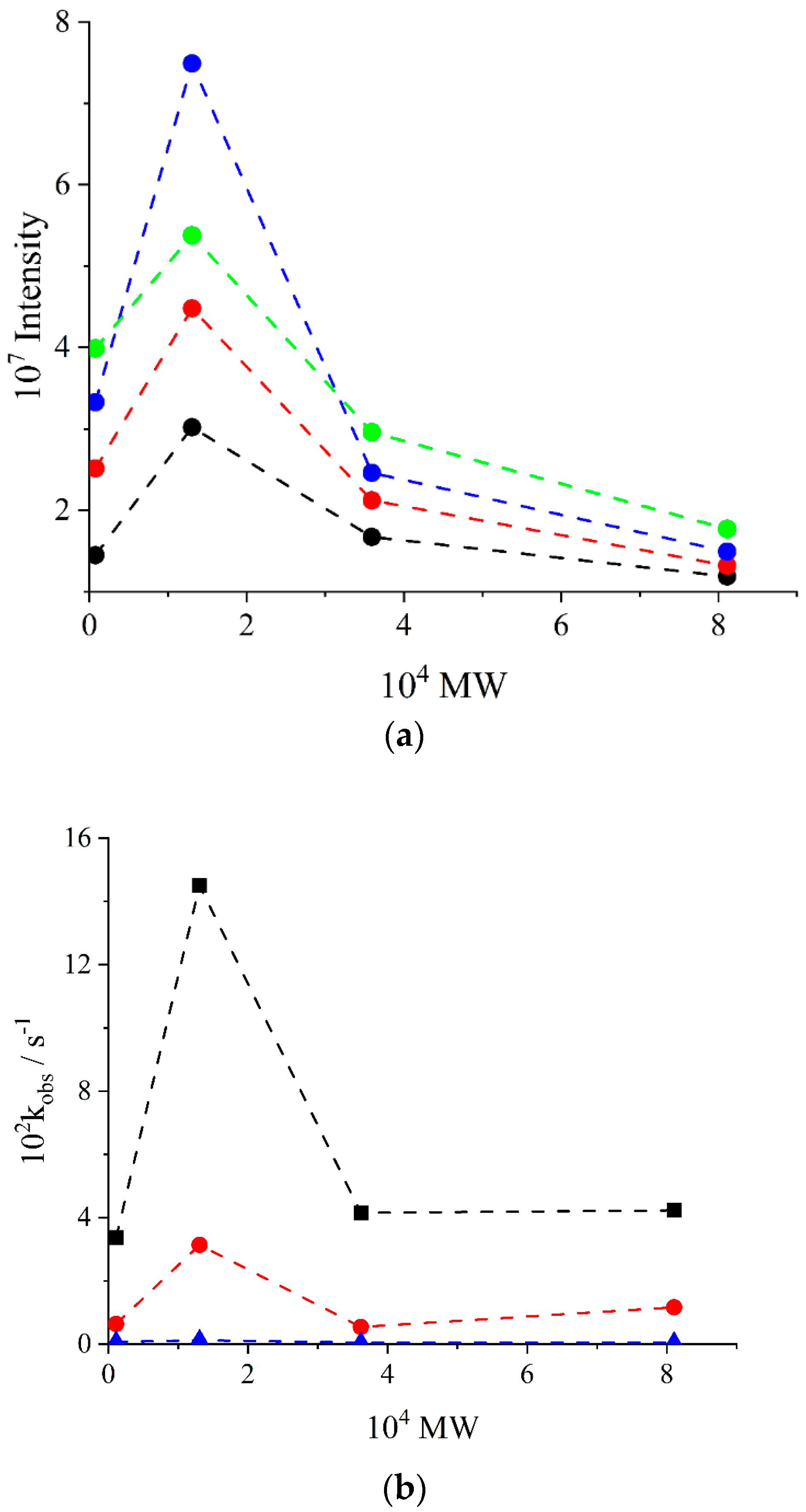
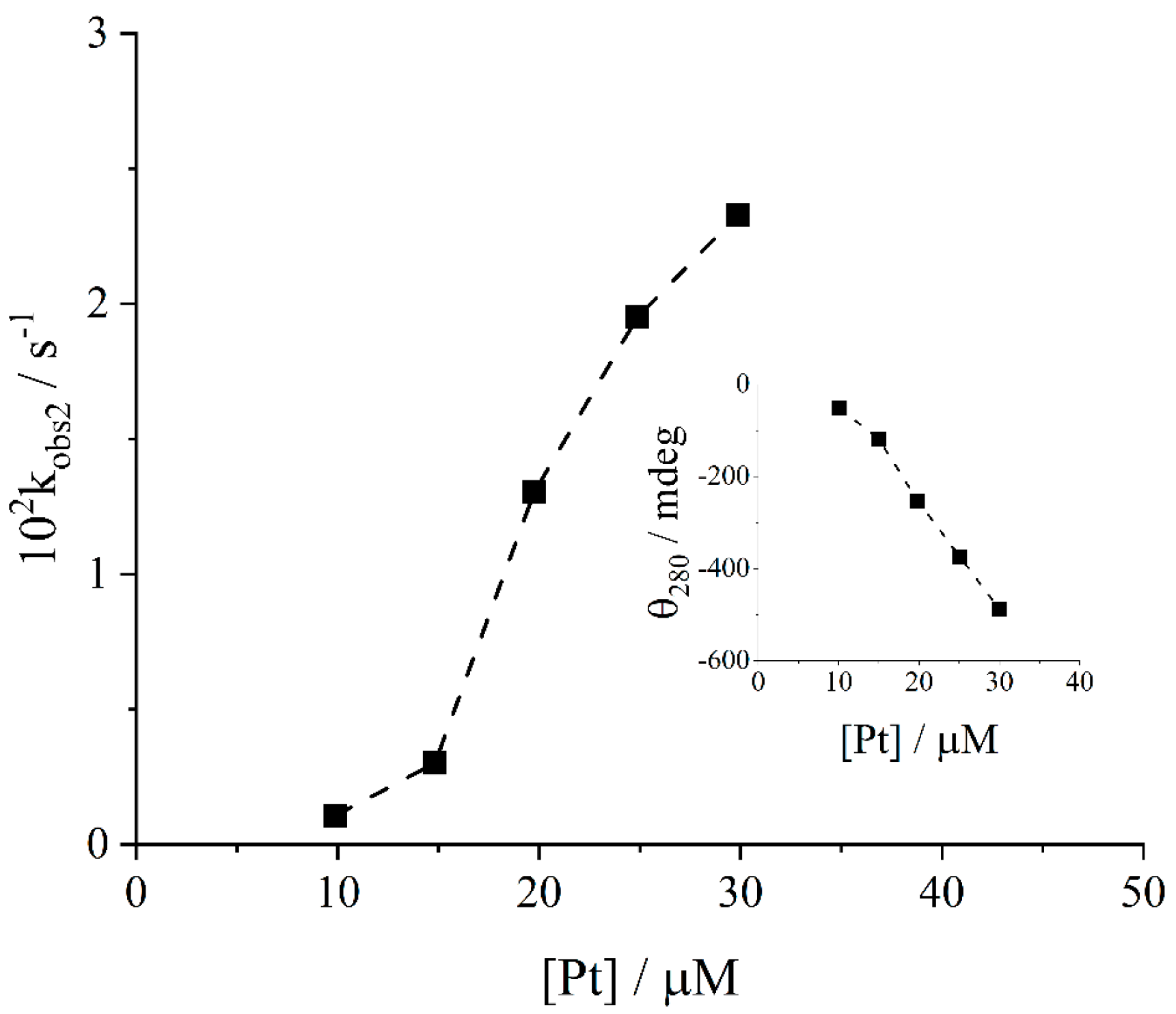
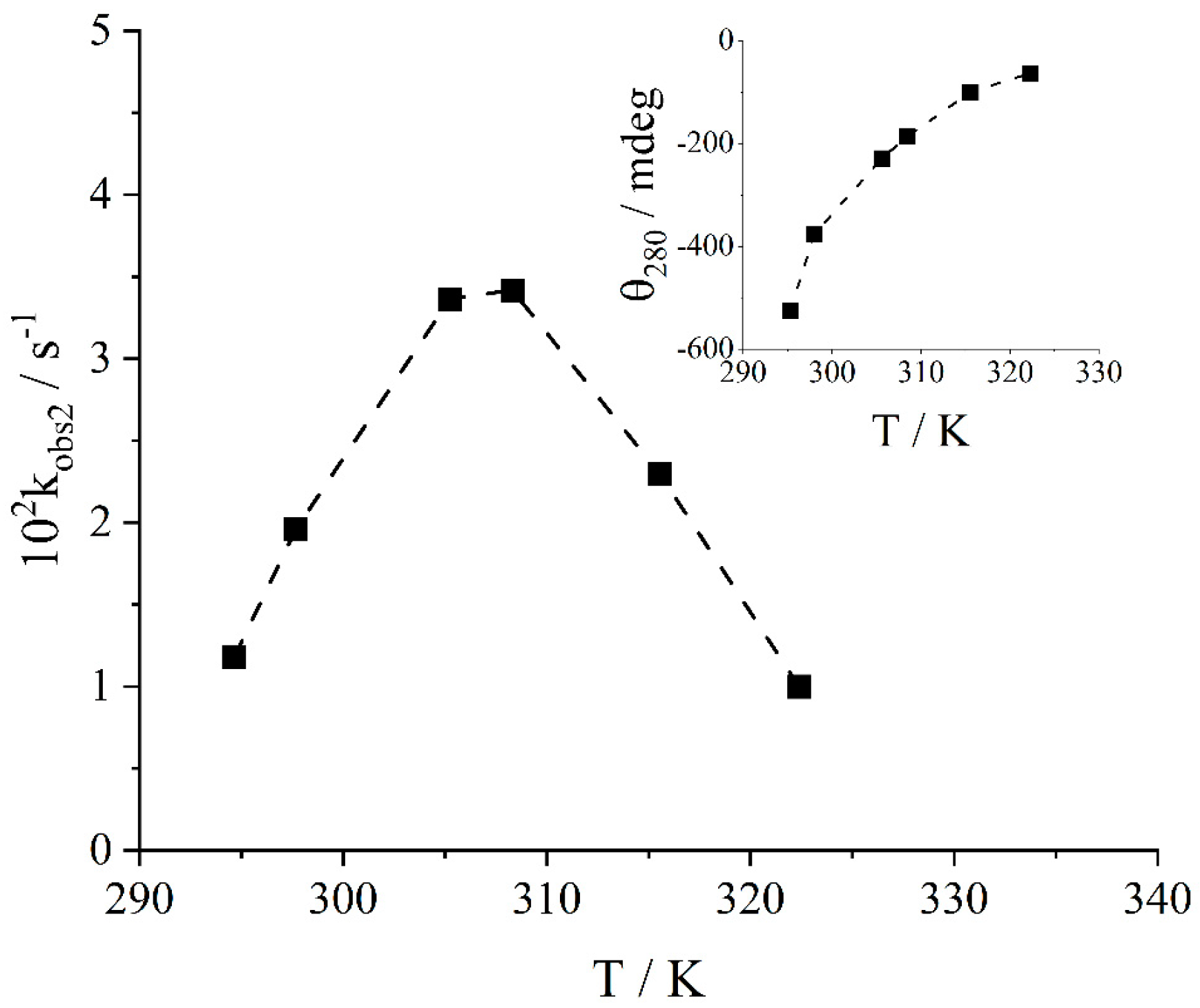
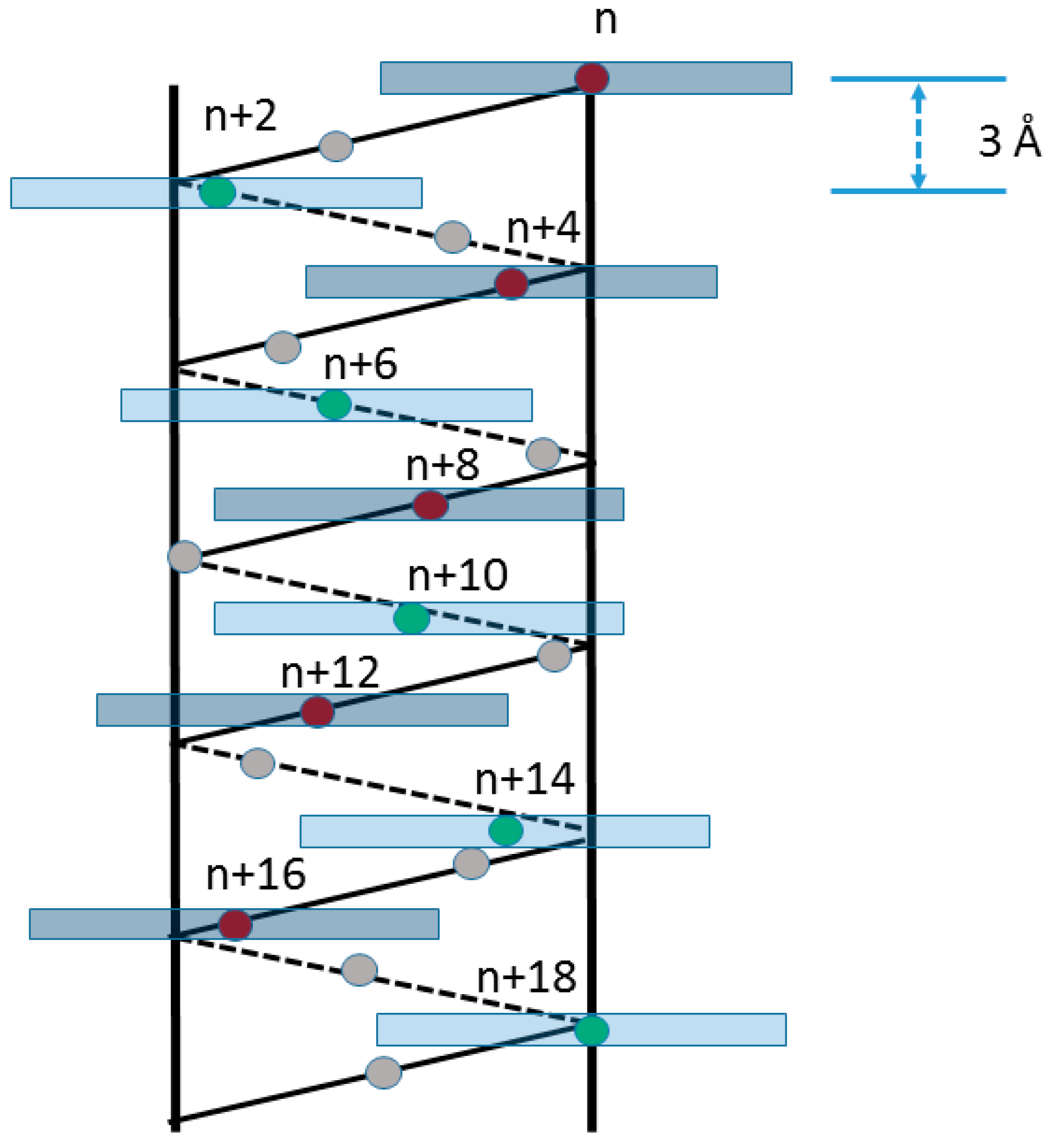
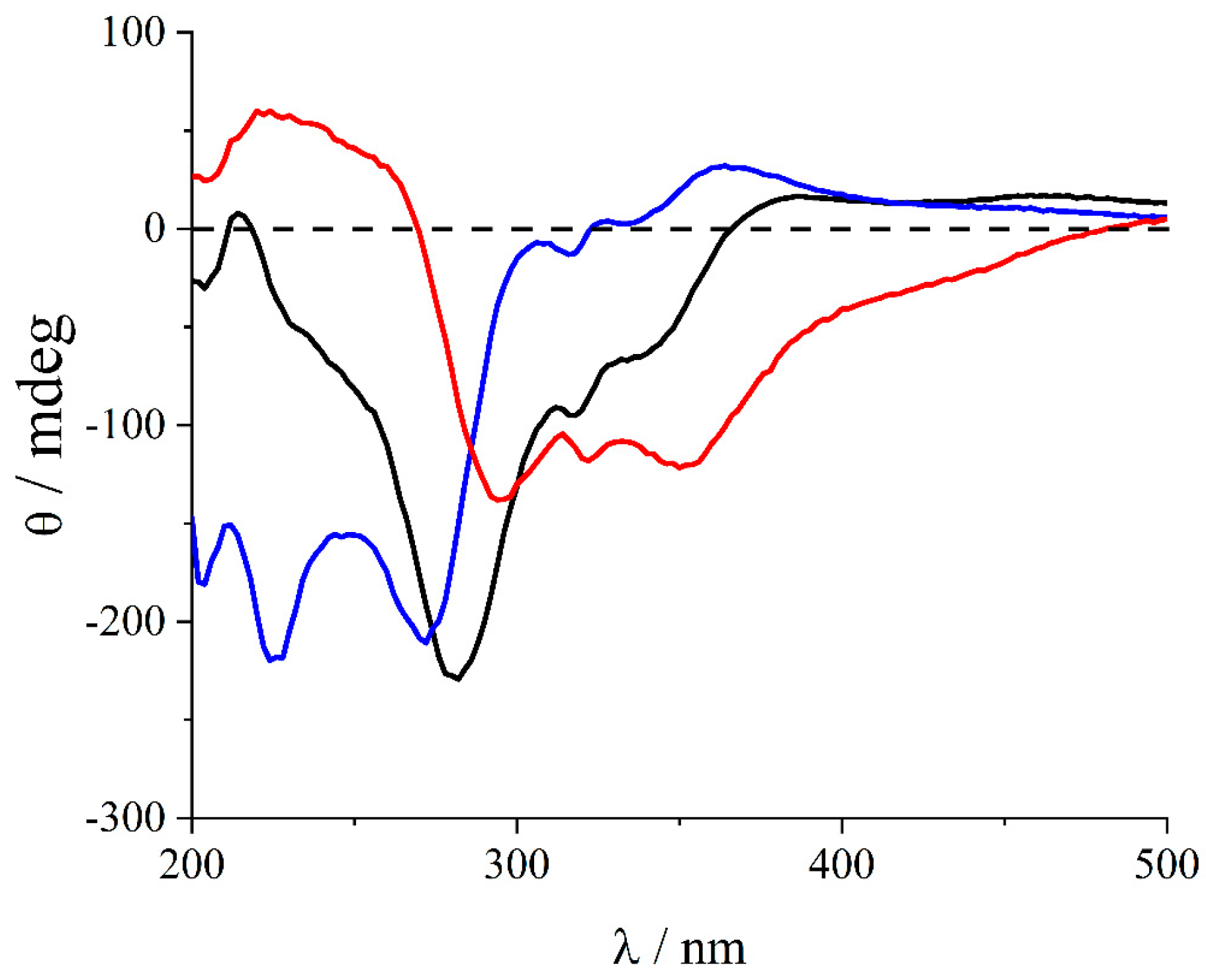
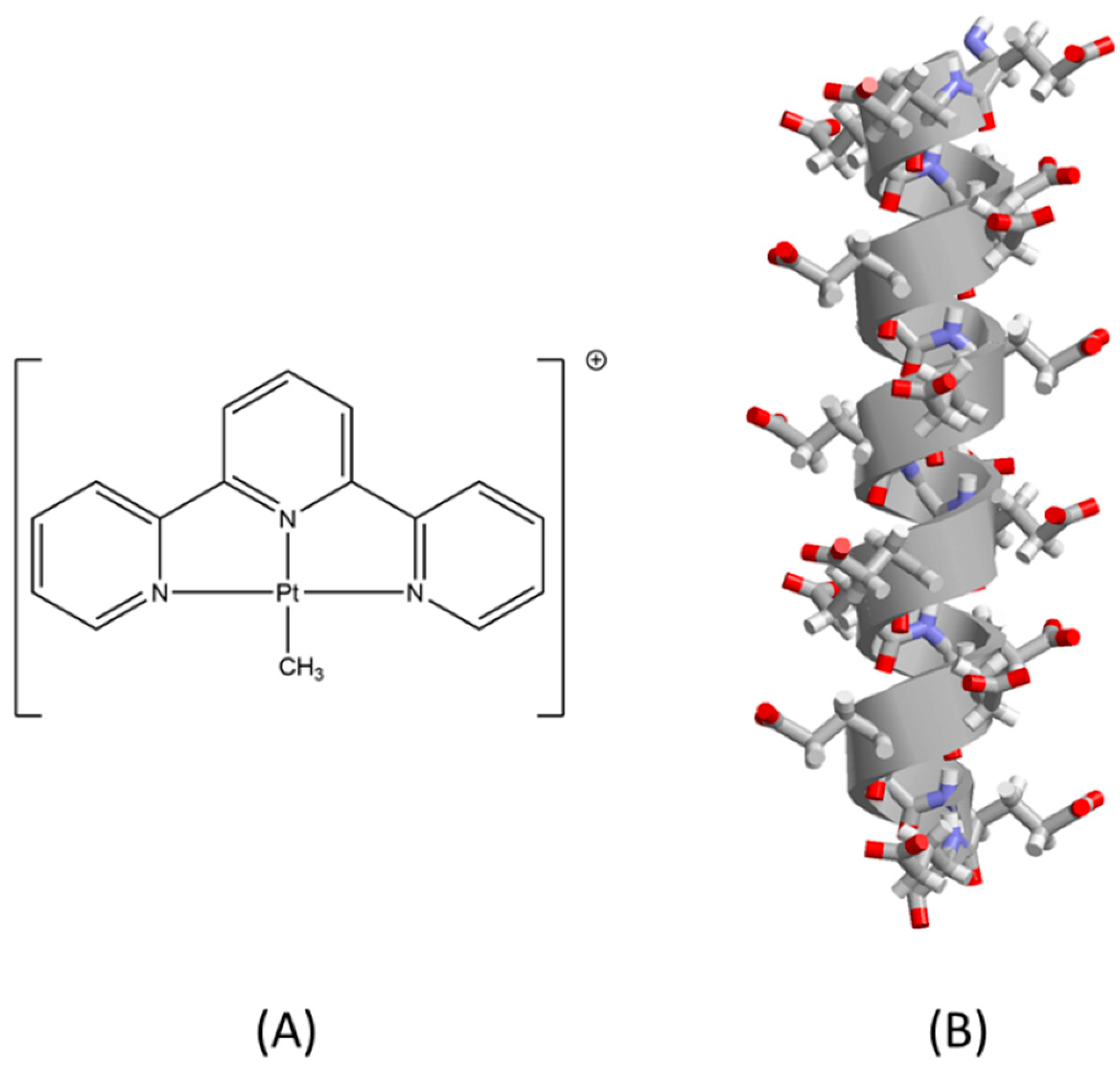
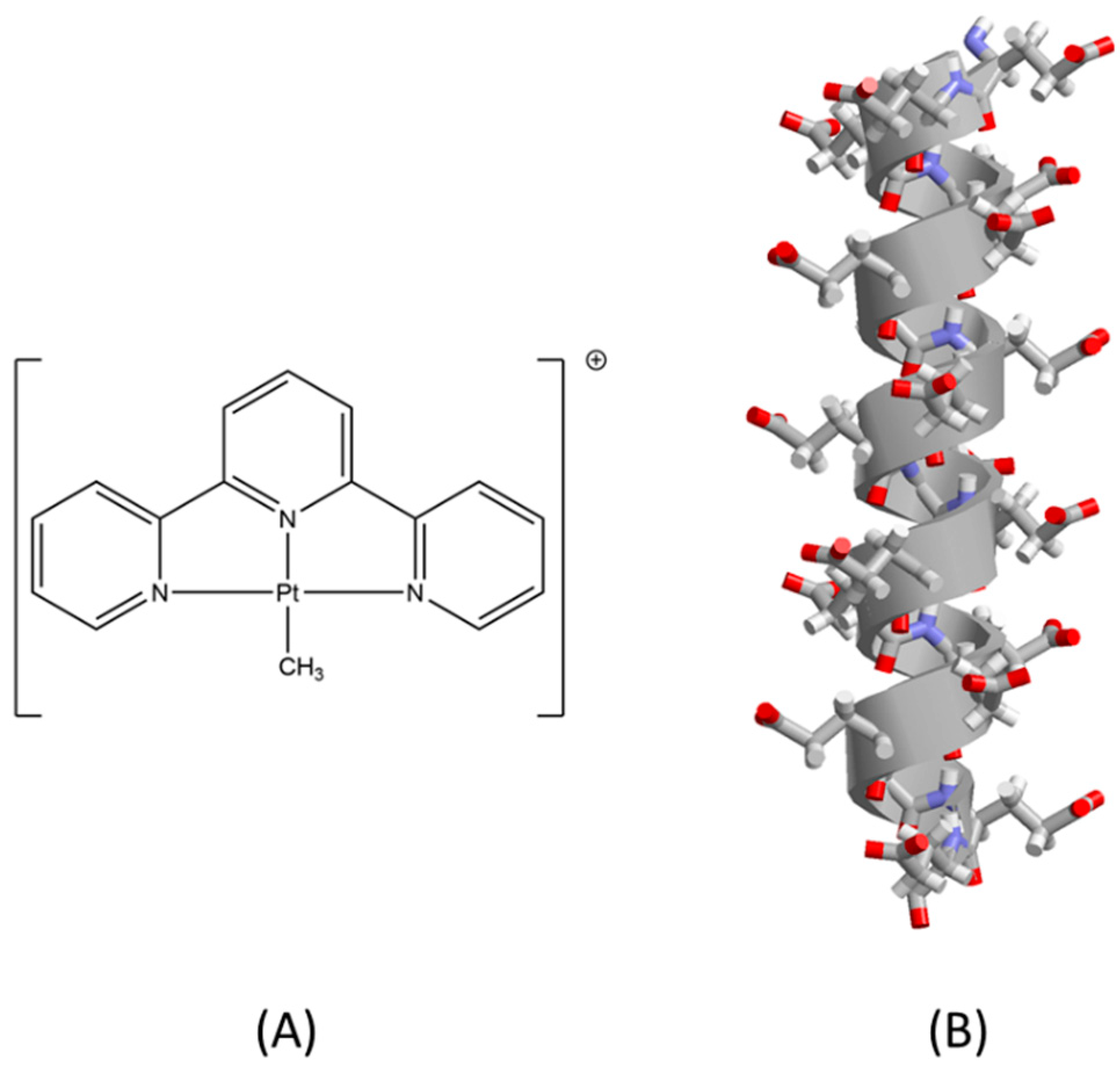
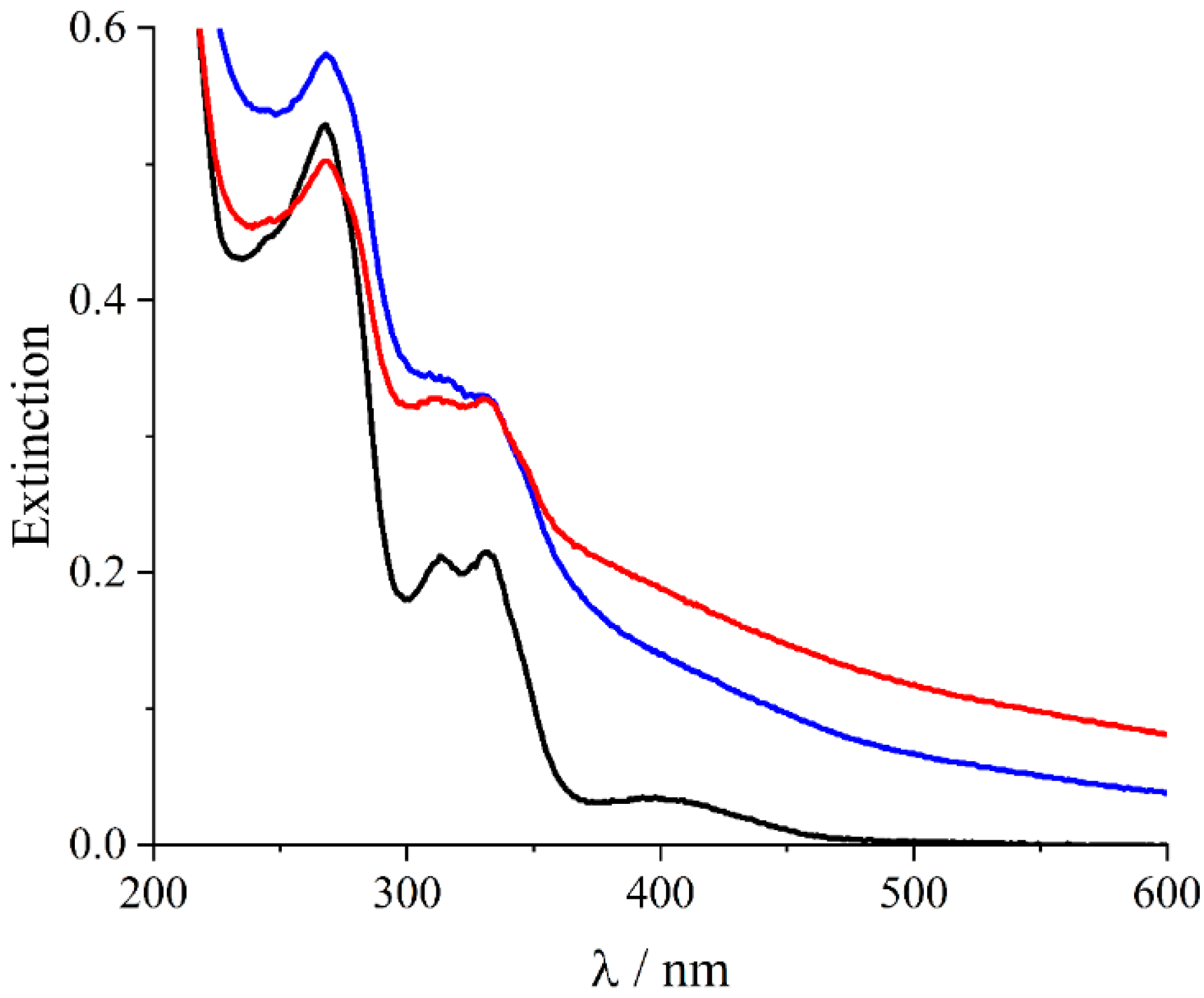
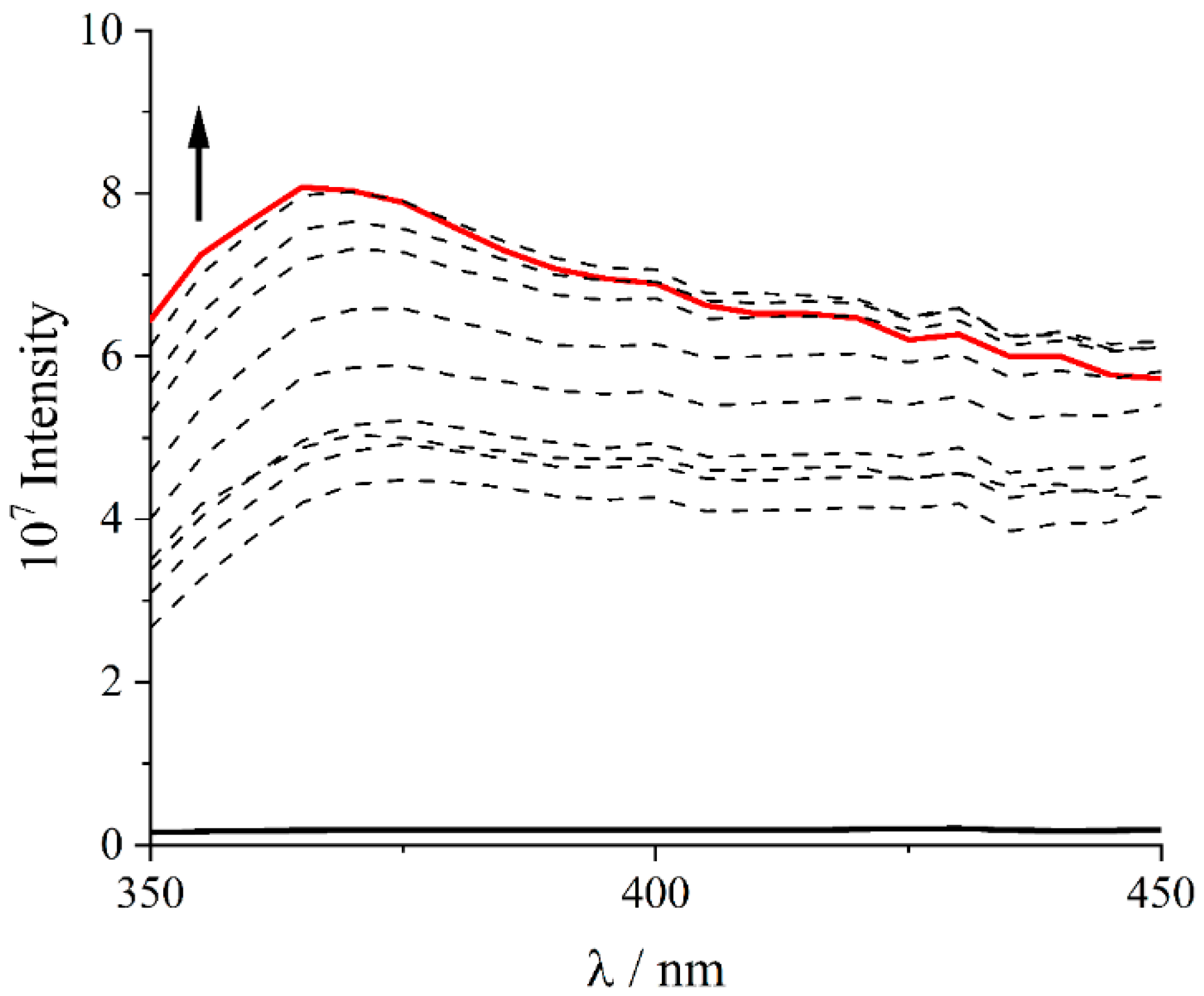
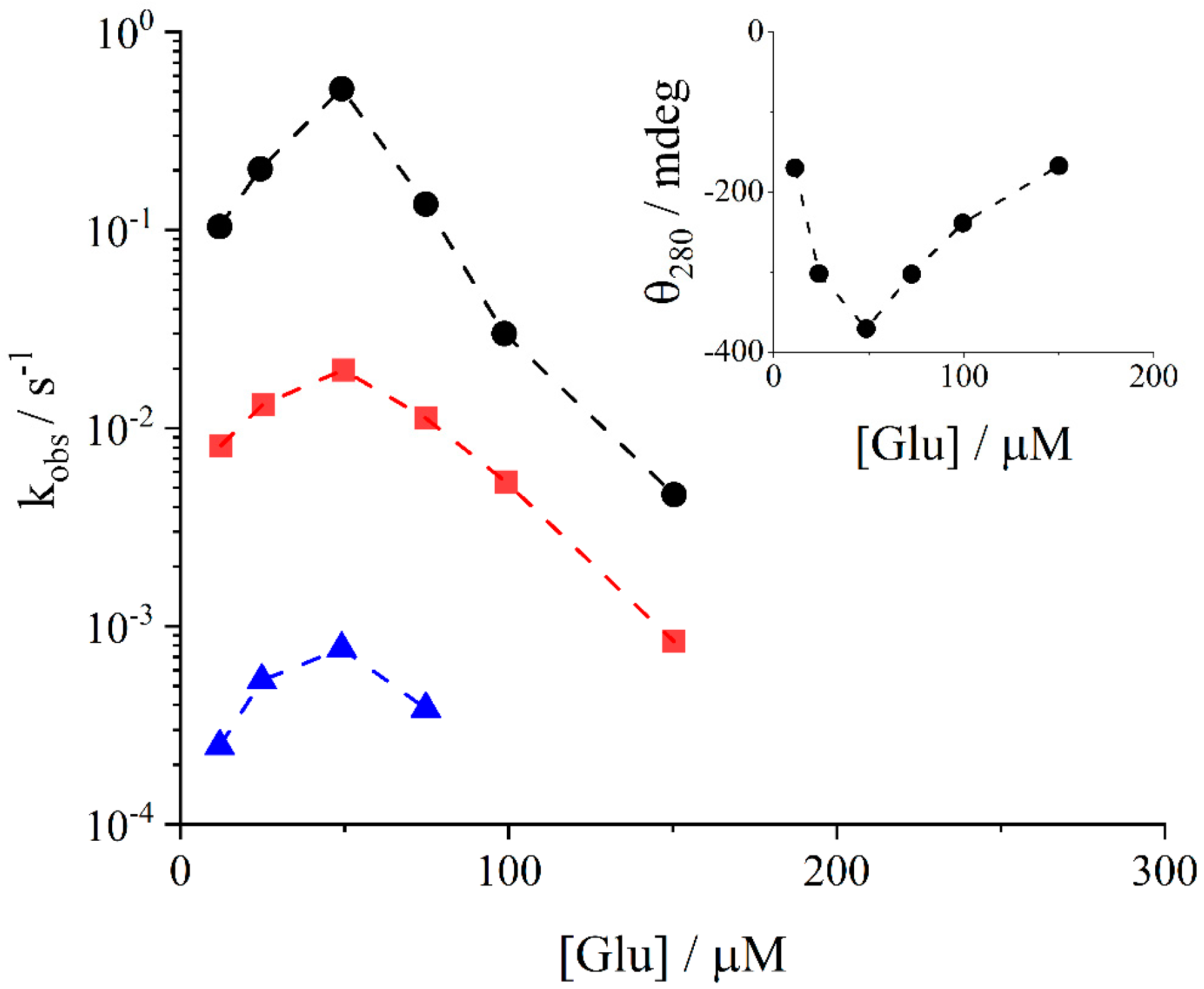
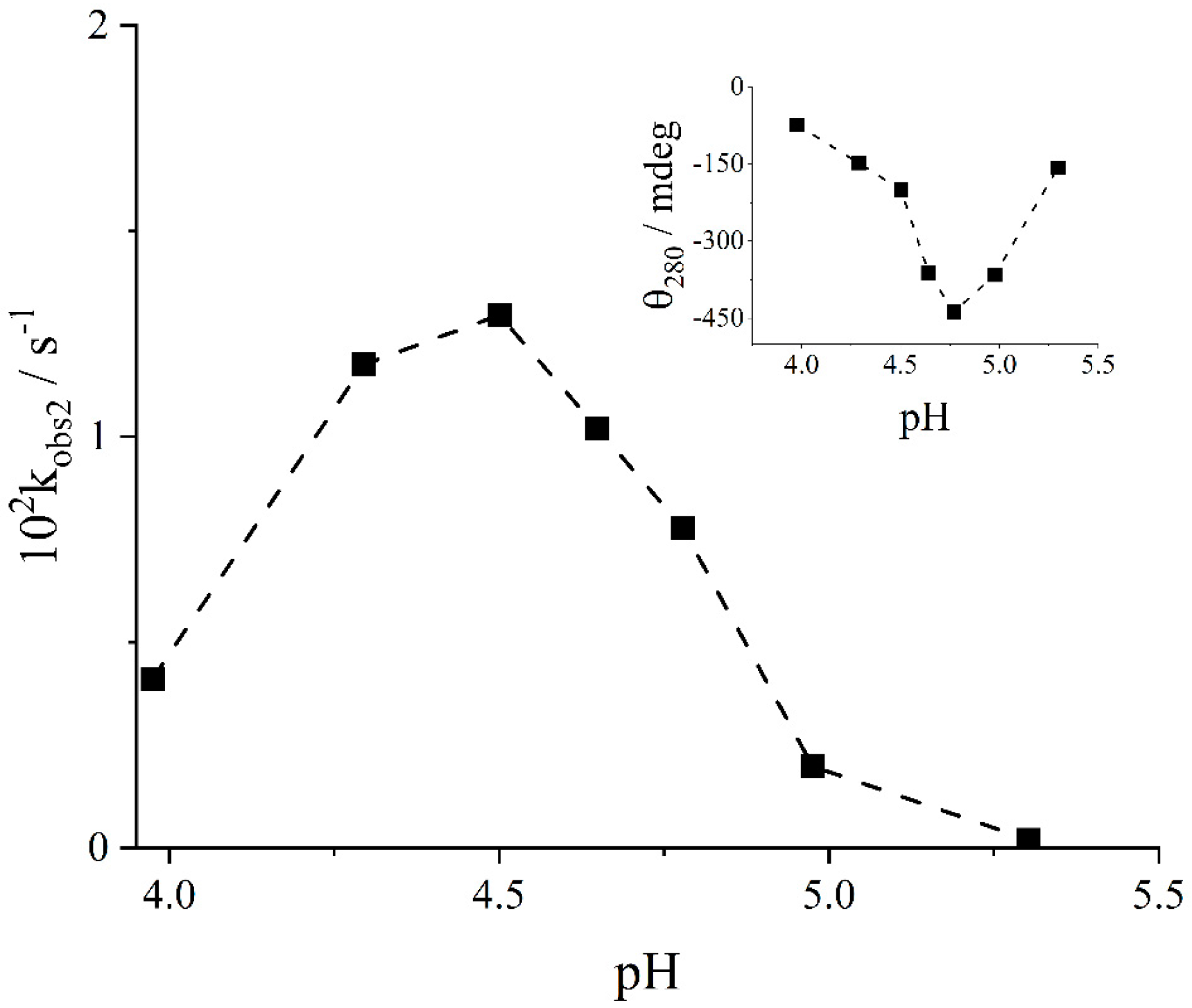
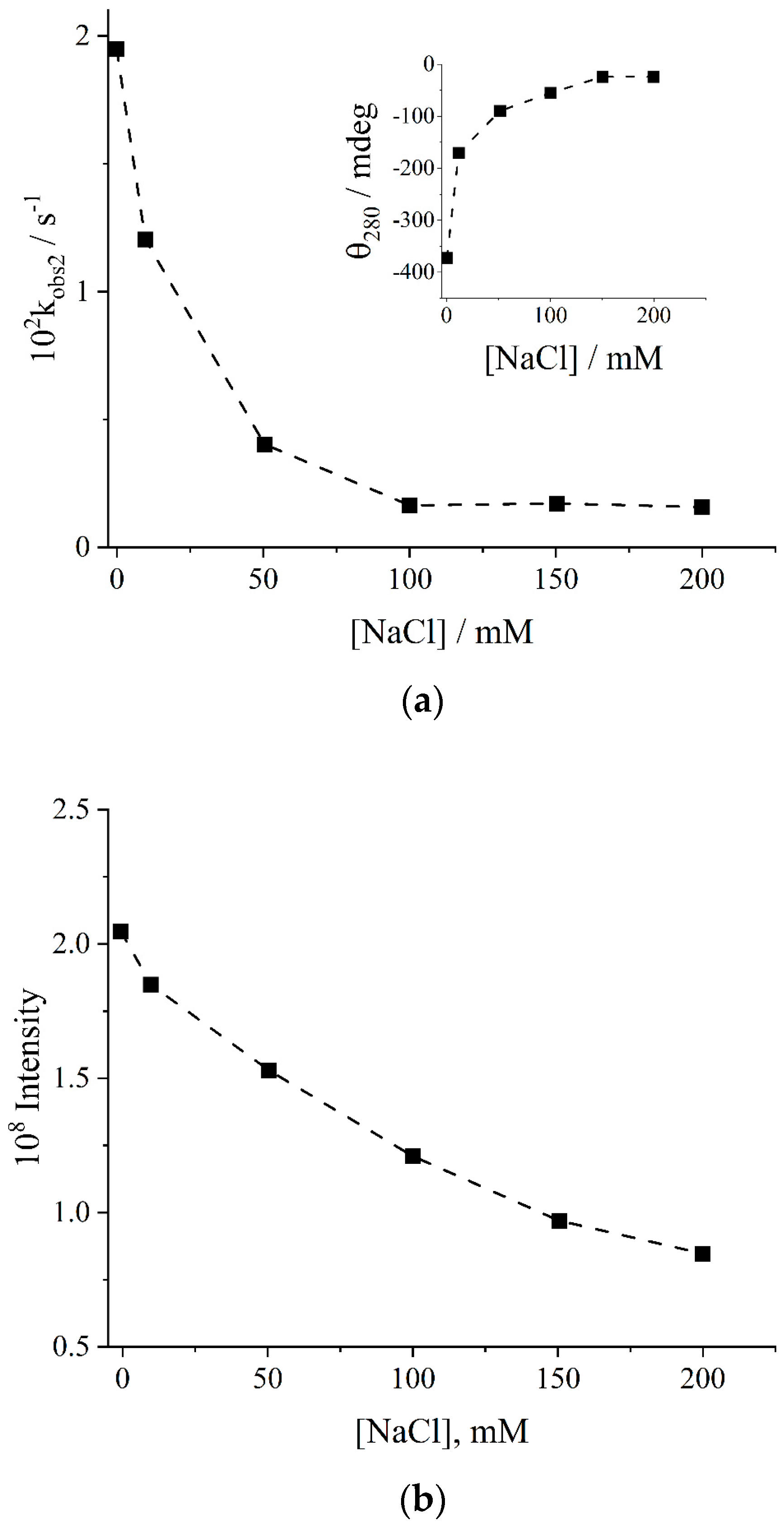
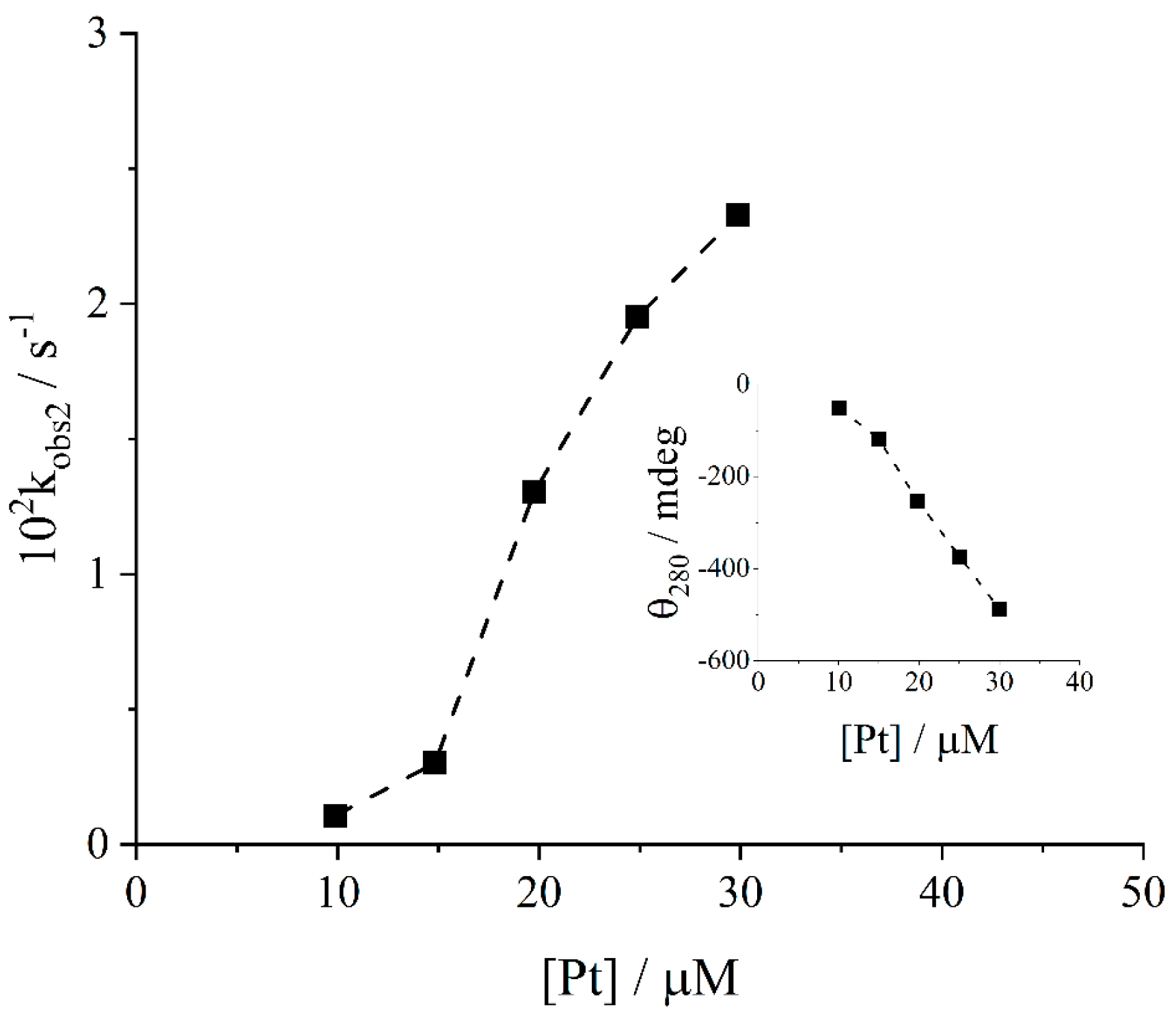
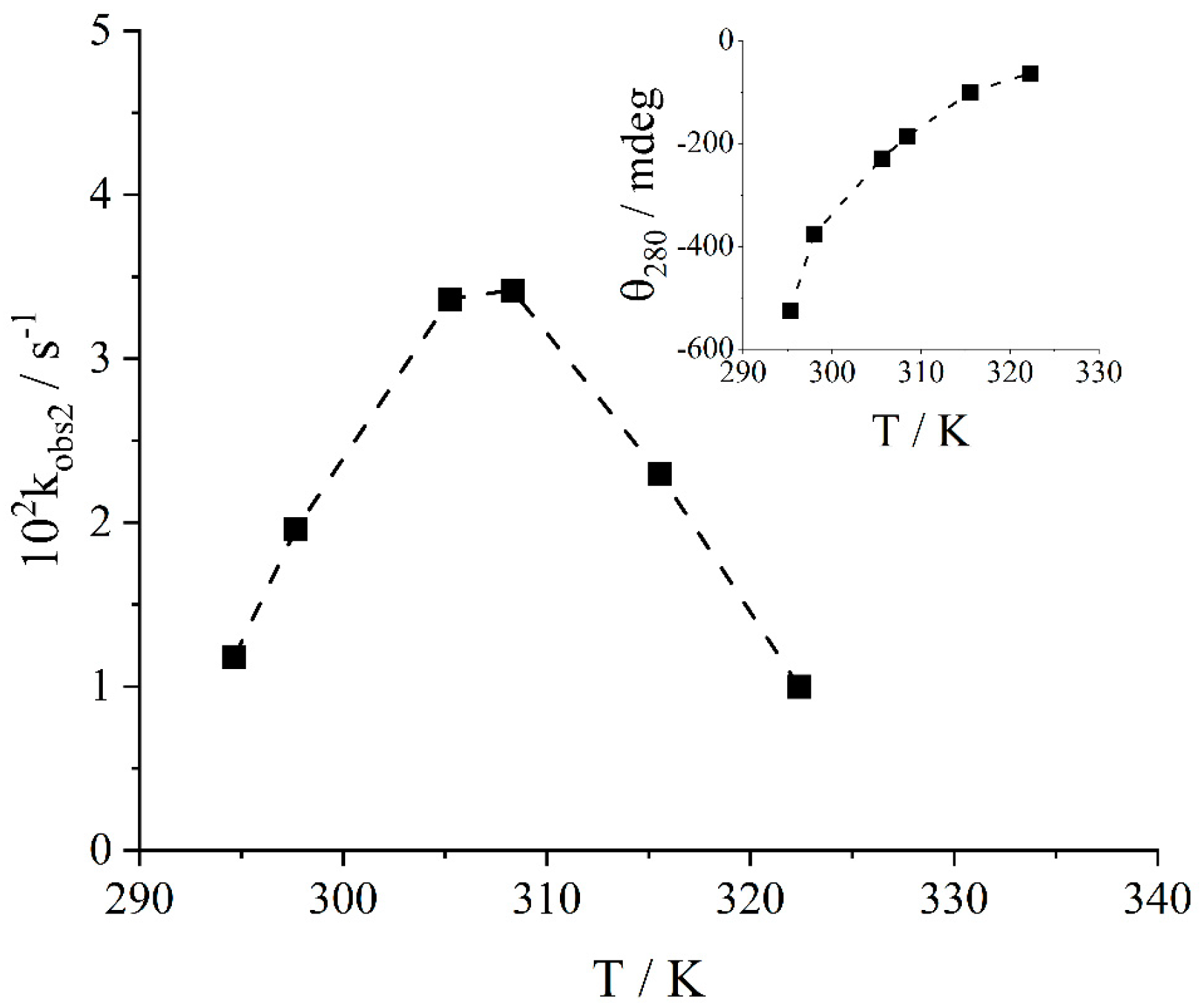
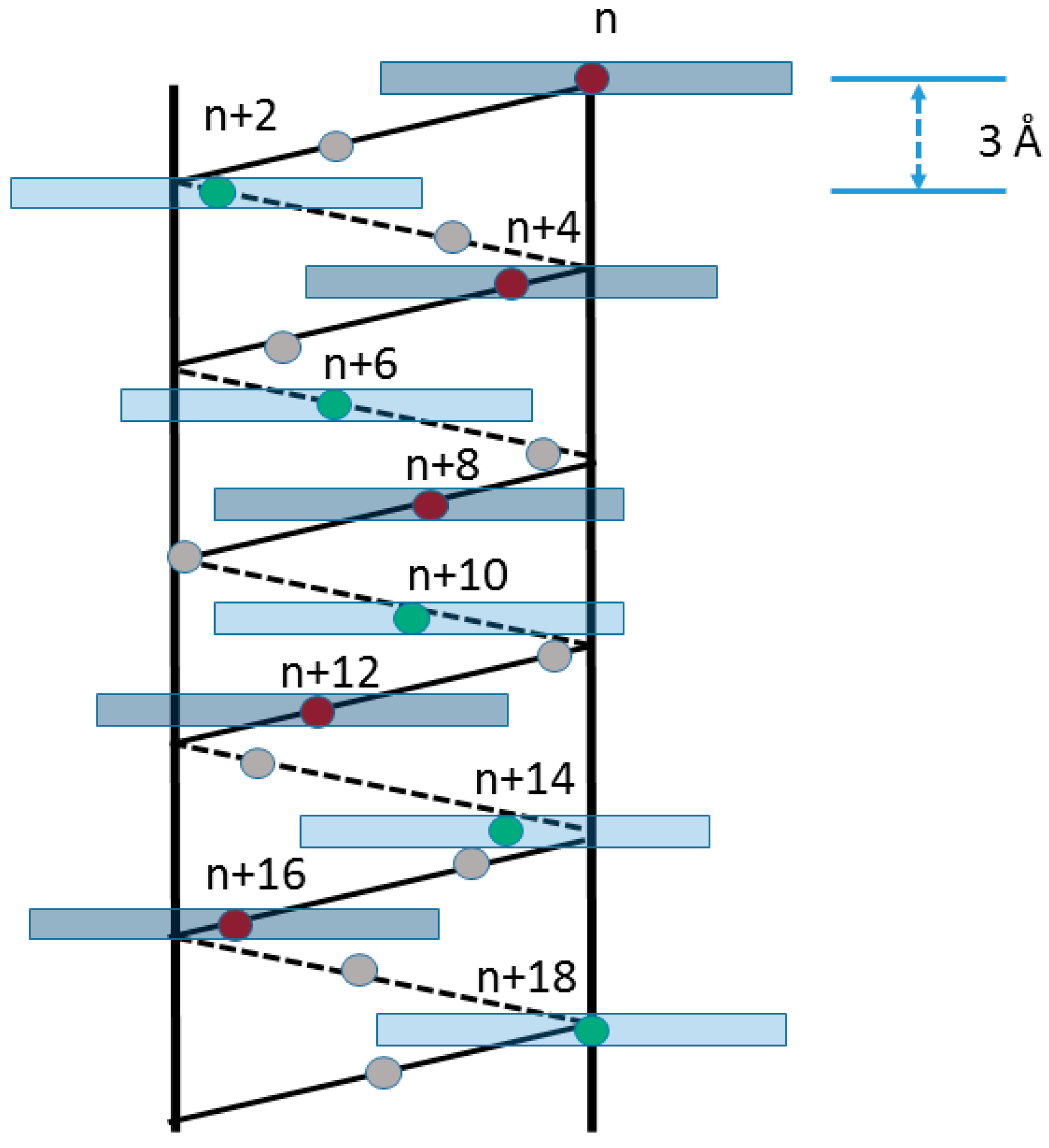
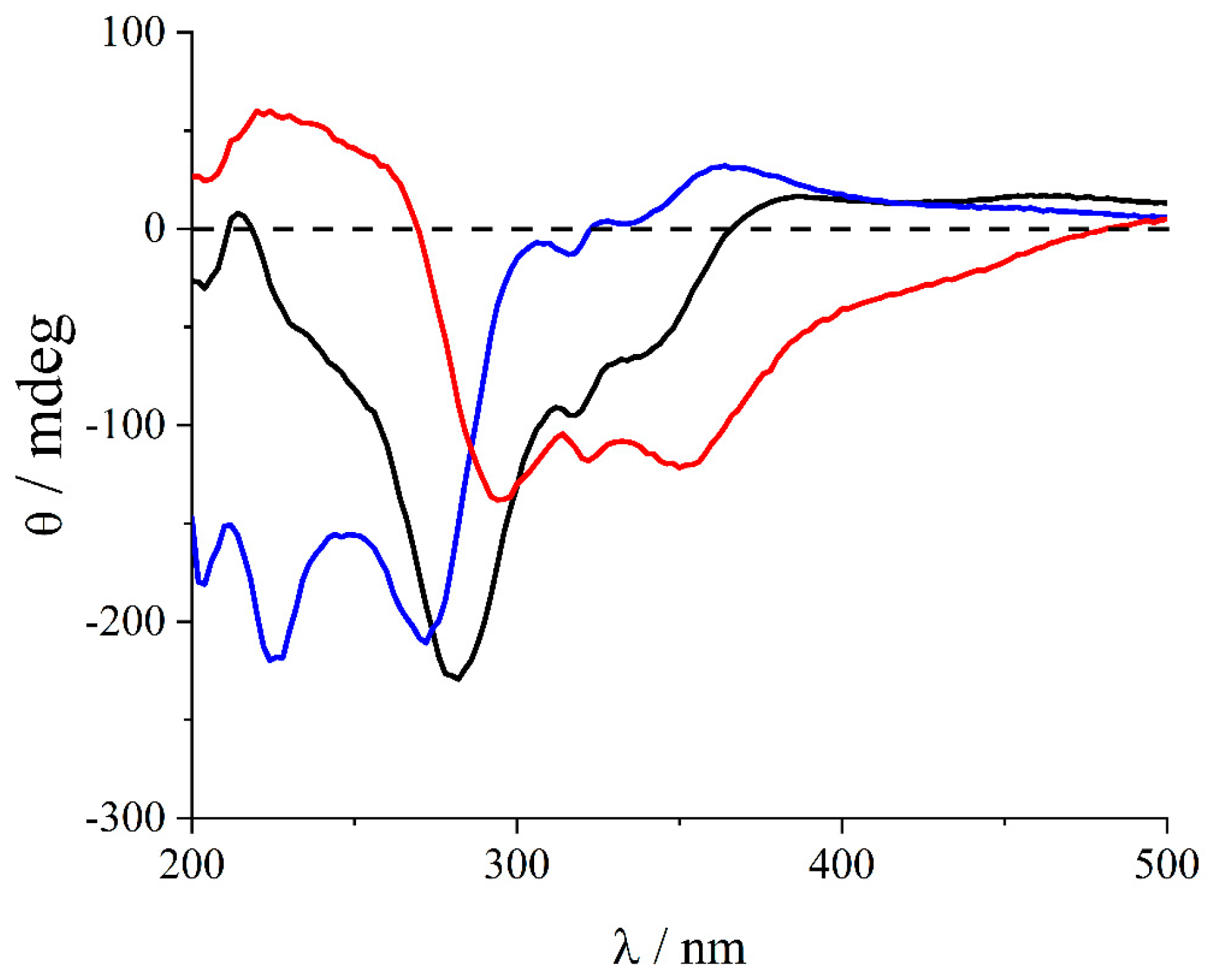
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Badjic, J.D.; Nelson, A.; Cantrill, S.J.; Turnbull, W.B.; Stoddart, J.F. Multivalency and cooperativity in supramolecular chemistry. Acc. Chem. Res. 2005, 38, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Lehn, J.M. Perspectives in Chemistry—Steps towards Complex Matter. Angew. Chem. Int. Ed. 2013, 52, 2836–2850. [Google Scholar] [CrossRef]
- Oshovsky, G.V.; Reinhoudt, D.N.; Verboom, W. Supramolecular chemistry in water. Angew. Chem. Int. Ed. 2007, 46, 2366–2393. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhang, H.; Zhao, Y.; Zhao, Y. Pillararene/Calixarene-based systems for battery and supercapacitor applications. eScience 2021, 1, 28–43. [Google Scholar] [CrossRef]
- Tong, F.; Zhou, Y.; Xu, Y.; Chen, Y.; Yudintceva, N.; Shevtsov, M.; Gao, H. Supramolecular nanomedicines based on host–guest interactions of cyclodextrins. Exploration 2023, 3, 20210111. [Google Scholar] [CrossRef]
- Yan, M.; Wang, Y.; Chen, J.; Zhou, J. Potential of nonporous adaptive crystals for hydrocarbon separation. Chem. Soc. Rev. 2023, 52, 6075–6119. [Google Scholar] [CrossRef]
- Yan, M.; Wu, S.; Wang, Y.; Liang, M.; Wang, M.; Hu, W.; Yu, G.; Mao, Z.; Huang, F.; Zhou, J. Recent Progress of Supramolecular Chemotherapy Based on Host–Guest Interactions. Adv. Mat. 2023, 35, 2304249. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, L.; Wang, T. Supramolecular Chirality in Self-Assembled Systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef]
- Mateos-Timoneda, M.A.; Crego-Calama, M.; Reinhoudt, D.N. Supramolecular chirality of self-assembled systems in solution. Chem. Soc. Rev. 2004, 33, 363–372. [Google Scholar] [CrossRef]
- Sommerdijk, N.; Buynsters, P.; Akdemir, H.; Geurts, D.G.; Pistorius, A.M.A.; Feiters, M.C.; Nolte, R.J.M.; Zwanenburg, B. Expression of supramolecular chirality in aggregates of chiral amide-containing surfactants. Chem. Eur. J. 1998, 4, 127–136. [Google Scholar] [CrossRef]
- Besenius, P.; Portale, G.; Bomans, P.H.H.; Janssen, H.M.; Palmans, A.R.A.; Meijer, E.W. Controlling the growth and shape of chiral supramolecular polymers in water. Proc. Natl. Acad. Sci. USA 2010, 107, 17888–17893. [Google Scholar] [CrossRef]
- Akeroyd, N.; Nolte, R.J.M.; Rowan, A.E. Polyisocyanides. In Isocyanide Chemistry; Wiley-VCH Verlag: Weinheim, Germany; GmbH Co. KGaA: Frankfurt, Germany, 2012; pp. 551–585. [Google Scholar]
- Castriciano, M.A.; Zagami, R.; Trapani, M.; Romeo, A.; Patane, S.; Scolaro, L.M. Investigation of the Aggregation Properties of a Chiral Porphyrin Bearing Citronellal Meso Substituent Groups. Chirality 2015, 27, 900–906. [Google Scholar] [CrossRef]
- Palmans, A.R.A.; Meijer, E.W. Amplification of Chirality in Dynamic Supramolecular Aggregates. Angew. Chem. Int. Ed. 2007, 46, 8948–8968. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.Z.; Tao, T.T.; Xie, Y.J.; Yu, L.Y.; Kang, X.; Zhang, Y.; Tang, W.W.; Gong, J.B. Chirality Supramolecular Systems: Helical Assemblies, Structure Designs, and Functions. Small 2023, 2307874. [Google Scholar] [CrossRef]
- Dou, X.Q.; Mehwish, N.; Zhao, C.L.; Liu, J.Y.; Xing, C.; Feng, C.L. Supramolecular Hydrogels with Tunable Chirality for Promising Biomedical Applications. Acc. Chem. Res. 2020, 53, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Xing, P.Y.; Zhao, Y.L. Controlling Supramolecular Chirality in Multicomponent Self-Assembled Systems. Acc. Chem. Res. 2018, 51, 2324–2334. [Google Scholar] [CrossRef]
- Lv, Z.Y.; Chen, Z.H.; Shao, K.N.; Qing, G.Y.; Sun, T.L. Stimuli-Directed Helical Chirality Inversion and Bio-Applications. Polymers 2016, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Travagliante, G.; Gaeta, M.; Purrello, R.; D’Urso, A. Supramolecular Chirality in Porphyrin Self-Assembly Systems in Aqueous Solution. Curr. Org. Chem. 2022, 26, 563–579. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Bustamante, C.; Collings, P.J.; Giannetto, A.; Gibbs, E.J. Porphyrin Assemblies on DNA as Studied by a Resonance Light-Scattering Technique. J. Am. Chem. Soc. 1993, 115, 5393–5399. [Google Scholar] [CrossRef]
- D’Urso, A.; Nardis, S.; Pomarico, G.; Fragalà, M.E.; Paolesse, R.; Purrello, R. Interaction of Tricationic Corroles with Single/Double Helix of Homopolymeric Nucleic Acids and DNA. J. Am. Chem. Soc. 2013, 135, 8632–8638. [Google Scholar] [CrossRef]
- Scolaro, L.M.; Romeo, A.; Pasternack, R.F. Tuning porphyrin/DNA supramolecular assemblies by competitive binding. J. Am. Chem. Soc. 2004, 126, 7178–7179. [Google Scholar] [CrossRef]
- Ghazaryan, A.A.; Dalyan, Y.B.; Haroutiunian, S.G.; Tikhomirova, A.; Taulier, N.; Wells, J.W.; Chalikian, T.V. Thermodynamics of interactions of water-soluble porphyrins with RNA duplexes. J. Am. Chem. Soc. 2006, 128, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.; Gurrieri, S.; Pasternack, R.F.; Purrello, R.; Rizzarelli, E. Interaction of Water-Soluble Porphyrins with Single-Stranded And Double-Stranded Polyribonucleotides. Biopolymers 1994, 340, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Pasternack, R.F.; Giannetto, A.; Pagano, P.; Gibbs, E.J. Self-Assembly of Porphyrins on Nucleic-Acids and Polypeptides. J. Am. Chem. Soc. 1991, 113, 7799–7800. [Google Scholar] [CrossRef]
- Ribo, J.M.; Crusats, J.; Sagues, F.; Claret, J.; Rubires, R. Chiral Sign Induction by Vortices During the Formation of Mesophases in Stirred Solutions. Science 2001, 292, 2063. [Google Scholar] [CrossRef] [PubMed]
- Escudero, C.; Crusat, J.; Diez-Perez, I.; El-Hachemi, Z.; Ribo, J.M. Folding and hydrodynamic forces in J-aggregates of 5-phenyl-10,15,20-tris-(4-sulfo-phenyl)porphyrin. Angew. Chem. Int. Ed. 2006, 45, 8032–8035. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, O.; Canillas, A.; Purrello, R.; Ribo, J.M. Evidence of induced chirality in stirred solutions of supramolecular nanofibers. Opt. Lett. 2009, 34, 2177–2179. [Google Scholar] [CrossRef]
- El-Hachemi, Z.; Balaban, T.S.; Campos, J.L.; Cespedes, S.; Crusats, J.; Escudero, C.; Kamma-Lorger, C.S.; Llorens, J.; Malfois, M.; Mitchell, G.R.; et al. Effect of Hydrodynamic Forces on meso-(4-Sulfonatophenyl)-Substituted Porphyrin J-Aggregate Nanoparticles: Elasticity, Plasticity and Breaking. Chem. Eur. J. 2016, 22, 9740–9749. [Google Scholar] [CrossRef]
- Crusats, J.; El-Hachemi, Z.; Ribo, J.M. Hydrodynamic effects on chiral induction. Chem. Soc. Rev. 2010, 39, 569. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, A.; Randazzo, R.; Lo Faro, L.; Purrello, R. Vortexes and Nanoscale Chirality. Angew. Chem. Int. Ed. 2010, 49, 108–112. [Google Scholar] [CrossRef]
- Sun, J.S.; Li, Y.K.; Yan, F.S.; Liu, C.; Sang, Y.T.; Tian, F.; Feng, Q.; Duan, P.F.; Zhang, L.; Shi, X.H.; et al. Control over the emerging chirality in supramolecular gels and solutions by chiral microvortices in milliseconds. Nat. Commun. 2018, 9, 2599. [Google Scholar] [CrossRef]
- Nicosia, A.; Vento, F.; Marletta, G.; Messina, G.M.L.; Satriano, C.; Villari, V.; Micali, N.; De Martino, M.T.; Schotman, M.J.G.; Mineo, P.G. Porphyrin-Based Supramolecular Flags in the Thermal Gradients’ Wind: What Breaks the Symmetry, How and Why. Nanomaterials 2021, 11, 1673. [Google Scholar] [CrossRef]
- Mineo, P.; Villari, V.; Scamporrino, E.; Micali, N. New Evidence about the Spontaneous Symmetry Breaking: Action of an Asymmetric Weak Heat Source. J. Phys. Chem. B 2015, 119, 12345–12353. [Google Scholar] [CrossRef] [PubMed]
- Po, C.; Tam, A.Y.Y.; Wong, K.M.C.; Yam, V.W.W. Supramolecular Self-Assembly of Amphiphilic Anionic Platinum(II) Complexes: A Correlation between Spectroscopic and Morphological Properties. J. Am. Chem. Soc. 2011, 133, 12136–12143. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.F.; Gao, Z.C.; Wang, C.; Zhong, R.L.; Wang, F. Recent progress on supramolecular assembly of organoplatinum(II) complexes into long-range ordered nanostructures. Coord. Chem. Rev. 2020, 414, 213300. [Google Scholar] [CrossRef]
- Zhang, K.K.; Yeung, M.C.L.; Leung, S.Y.L.; Yam, V.W.W. Living supramolecular polymerization achieved by collaborative assembly of platinum(II) complexes and block copolymers. Proc. Natl. Acad. Sci. USA 2017, 114, 11844–11849. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.Y.L.; Wong, K.M.C.; Yam, V.W.W. Self-assembly of alkynylplatinum(II) terpyridine amphiphiles into nanostructures via steric control and metal-metal interactions. Proc. Natl. Acad. Sci. USA 2016, 113, 2845–2850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Ching-LamYeung, M.; Yu-LutLeung, S.; Yam, V.-W. Manipulation of Nanostructures in the Coassembly of Platinum(II) Complexes and Block Copolymers. Chem 2017, 2, 825–839. [Google Scholar] [CrossRef]
- Zheng, X.Y.; Chan, M.H.Y.; Chan, A.K.W.; Cao, S.Q.; Ng, M.; Sheong, F.K.; Li, C.; Goonetilleke, E.C.; Lam, W.W.Y.; Lau, T.C.; et al. Elucidation of the key role of Pt-Pt interactions in the directional self-assembly of platinum(II) complexes. Proc. Natl. Acad. Sci. USA 2022, 119, 2307874. [Google Scholar] [CrossRef]
- Zhang, K.K.; Yeung, M.C.L.; Leung, S.Y.L.; Yam, V.W.W. Energy Landscape in Supramolecular Coassembly of Platinum(II) Complexes and Polymers: Morphological Diversity, Transformation, and Dilution Stability of Nanostructures. J. Am. Chem. Soc. 2018, 140, 9594–9605. [Google Scholar] [CrossRef]
- Eryazici, I.; Moorefield, C.N.; Newkome, G.R. Square-planar Pd(II), Pt(II), and Au(III) terpyridine complexes: Their syntheses, physical properties, supramolecular constructs, and biomedical activities. Chem. Rev. 2008, 108, 1834–1895. [Google Scholar] [CrossRef]
- Cummings, S.D. Platinum complexes of terpyridine: Interaction and reactivity with biomolecules. Coord. Chem. Rev. 2009, 253, 1495–1516. [Google Scholar] [CrossRef]
- Kang, H.W.; Lee, J.H.; Seo, M.L.; Jung, S.H. Platinum(II) terpyridine-based supramolecular polymer gels with induced chirality. Soft Matter 2023, 19, 9365–9368. [Google Scholar] [CrossRef]
- Arena, G.; Monsù Scolaro, L.; Pasternack, R.F.; Romeo, R. Synthesis, Characterization, and Interaction with DNA of the Novel Metallointercalator Cationic Complex (2,2′6′,2″-Terpyridine)Methylplatinum(II). Inorg. Chem. 1995, 34, 2994–3002. [Google Scholar] [CrossRef]
- Casamento, M.; Arena, G.E.; Lo Passo, C.; Pernice, I.; Romeo, A.; Scolaro, L.M. Interaction of organometallic cationic complex ions containing terpyridine ligands with nucleic acids: An investigation on aggregative phenomena. Inorgica Chim. Acta 1998, 276, 242–249. [Google Scholar] [CrossRef]
- Sato, Y.; Hatano, M.; Yoneyama, M. Neutral Salt Effect on Interaction of Poly-Alpha, L-Glutamic Acid With Acridine-Orange. Bull. Chem. Soc. Jpn. 1973, 46, 1980–1983. [Google Scholar] [CrossRef]
- Stryer, L.; Blout, E.R. Optical Rotatory Dispersion of Dyes Bound to Macromolecules. Cationic Dyes: Polyglutamic Acid Complexes. J. Am. Chem. Soc. 1961, 83, 1411–1418. [Google Scholar] [CrossRef]
- De Luca, G.; Romeo, A.; Scolaro, L.M.; Pasternack, R.F. Conformations of a model protein revealed by an aggregating Cu(II) porphyrin: Sensing the difference. Chem. Commun. 2010, 46, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Occhiuto, I.; De Luca, G.; Villari, V.; Romeo, A.; Micali, N.; Pasternack, R.F.; Scolaro, L.M. Supramolecular chirality transfer to large random aggregates of porphyrins. Chem. Commun. 2011, 47, 6045–6047. [Google Scholar] [CrossRef]
- Scolaro, L.M.; Romeo, A.; Terracina, A. Supramolecular assembling of the cationic complex (2,2′:6′,2″-terpyridine)methylplatinum(II) ion on alpha-helical poly(L-glutamic acid). Chem. Commun. 1997, 1451–1452. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Collings, P.J. Resonance Light-Scattering—A New Technique for Studying Chromophore Aggregation. Science 1995, 269, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Cantor, C.R.; Schimmel, P.R. Biophysical Chemistry Part I; W.H.F.A. Co.: New York, NY, USA, 1980. [Google Scholar]
- Bailey, J.A.; Hill, M.G.; Marsh, R.E.; Miskowski, V.M.; Schaefer, W.P.; Gray, H.B. Electronic Spectroscopy of Chloro(terpyridine)platinum(II). Inorg. Chem. 1995, 34, 4591–4599. [Google Scholar] [CrossRef]
- Romeo, R.; Scolaro, L.M.; Plutino, M.R.; Albinati, A. Structural properties of the metallointercalator cationic complex (2,2′:6′,2″-terpyridine)methylplatinum(II) ion. J. Organomet. Chem. 2000, 593, 403–408. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castriciano, M.A.; Zagami, R.; Mazzaglia, A.; Romeo, A.; Monsù Scolaro, L. A Kinetic Investigation of the Supramolecular Chiral Self-Assembling Process of Cationic Organometallic (2,2′:6′,2″-terpyridine)methylplatinum(II) Complexes with Poly(L-glutamic Acid). Int. J. Mol. Sci. 2024, 25, 1176. https://doi.org/10.3390/ijms25021176
Castriciano MA, Zagami R, Mazzaglia A, Romeo A, Monsù Scolaro L. A Kinetic Investigation of the Supramolecular Chiral Self-Assembling Process of Cationic Organometallic (2,2′:6′,2″-terpyridine)methylplatinum(II) Complexes with Poly(L-glutamic Acid). International Journal of Molecular Sciences. 2024; 25(2):1176. https://doi.org/10.3390/ijms25021176
Chicago/Turabian StyleCastriciano, Maria Angela, Roberto Zagami, Antonino Mazzaglia, Andrea Romeo, and Luigi Monsù Scolaro. 2024. "A Kinetic Investigation of the Supramolecular Chiral Self-Assembling Process of Cationic Organometallic (2,2′:6′,2″-terpyridine)methylplatinum(II) Complexes with Poly(L-glutamic Acid)" International Journal of Molecular Sciences 25, no. 2: 1176. https://doi.org/10.3390/ijms25021176
APA StyleCastriciano, M. A., Zagami, R., Mazzaglia, A., Romeo, A., & Monsù Scolaro, L. (2024). A Kinetic Investigation of the Supramolecular Chiral Self-Assembling Process of Cationic Organometallic (2,2′:6′,2″-terpyridine)methylplatinum(II) Complexes with Poly(L-glutamic Acid). International Journal of Molecular Sciences, 25(2), 1176. https://doi.org/10.3390/ijms25021176