
Regulatory T Cells in the Pathogenesis of Graves’ Disease

 , ,
, , 
 and
and 
Abstract
:1. Introduction
2. Methods
3. Results and Discussion
3.1. Regulatory T Cell (Treg)
3.2. Regulatory T Cells in Autoimmunity
3.3. Graves’ Disease
3.4. Graves’ Disease Pathogenesis
3.5. Mouse Models of Graves’ Disease
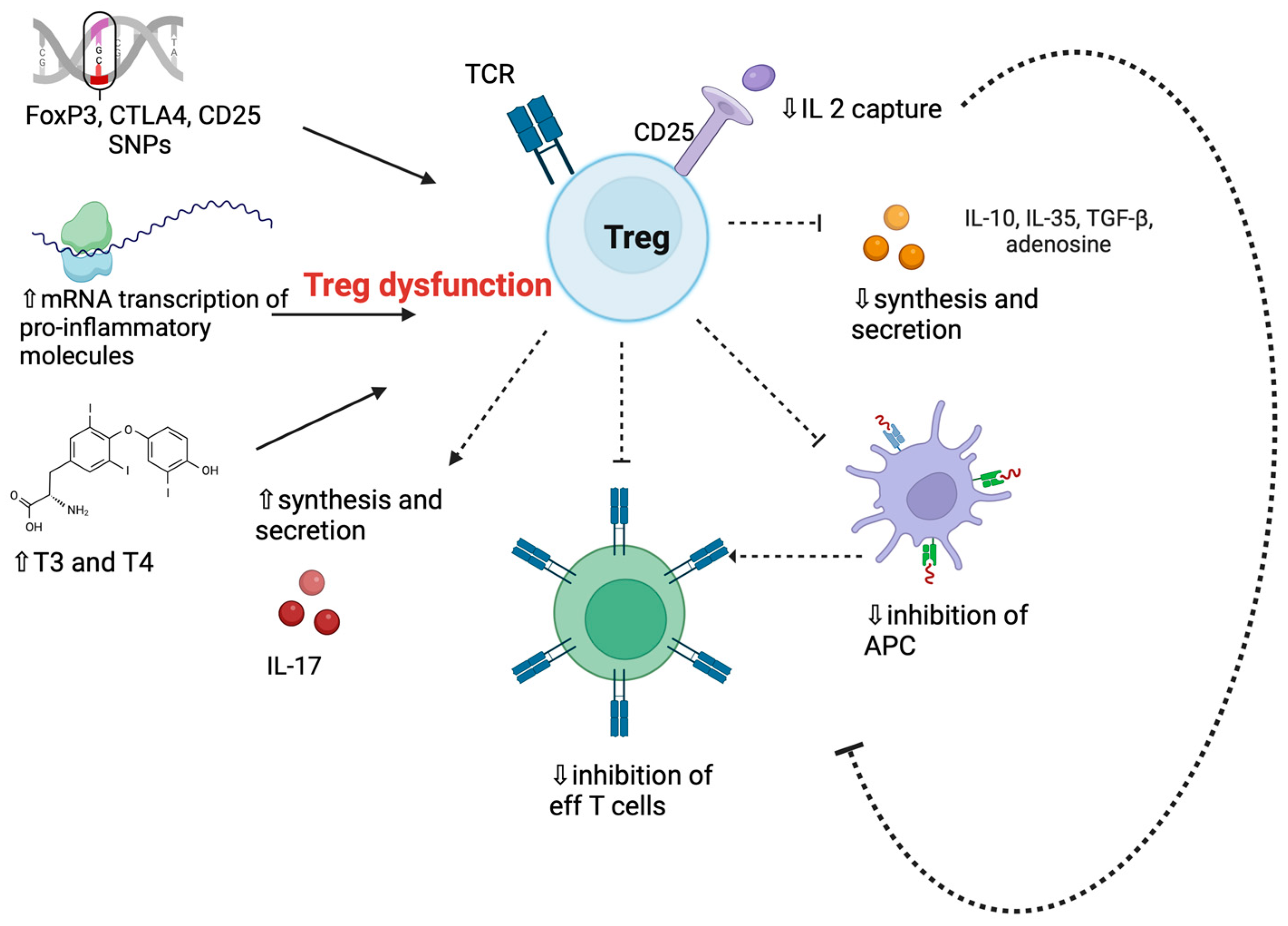
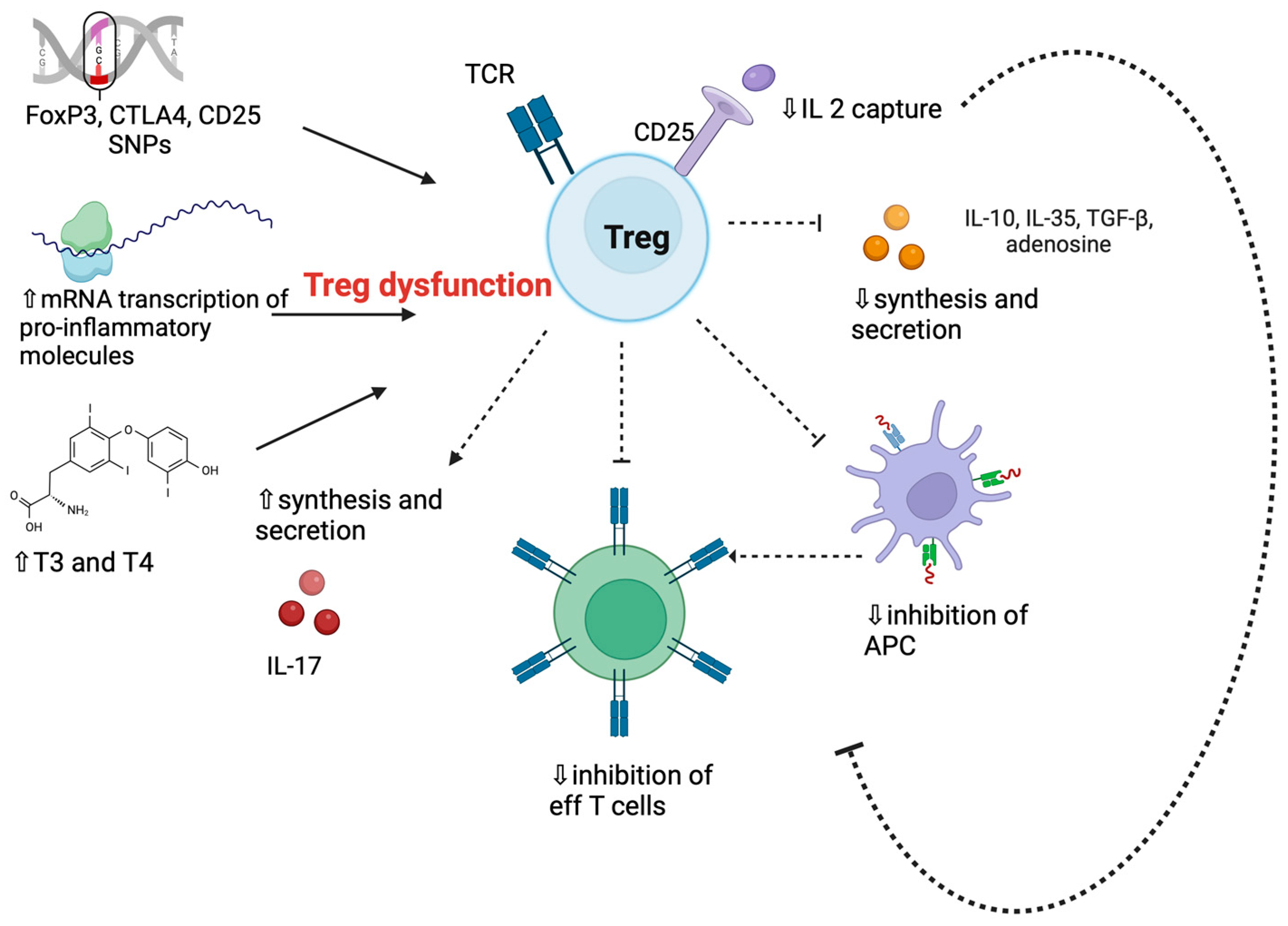
3.6. Regulatory T Cells in Graves’ Disease

{kind=link}
{kind=link}
| First Author, Year [Ref] | Main Findings in the Study in GD Patients Compared to HC |
|---|---|
| Marazuela et al., 2006 [75] |
|
| Wang et al., 2006 [63] |
|
| Nakano et al., 2007 [66] |
|
| Fountoulakis et al., 2008 [83] |
|
| Pan et al., 2009 [64] |
|
| Mao et al., 2011 [65] |
|
| Glick et al., 2013 [76] |
|
| Bossowski et al., 2013 [68] |
|
| Klatka et al., 2014 [72] |
|
| Rodríguez-Muñoz et al., 2015 [74] |
|
| Rodríguez-Muñoz et al., 2016 [73] |
|
| Li et al., 2016 [84] |
|
| Qin et al., 2017 [85] |
|
| Teniente-Serra et al., 2019 [70] |
|
| Chen et al., 2020 [71] |
|
3.7. T Regulatory and T Helper 17 Cells’ Interplay in Graves’ Disease
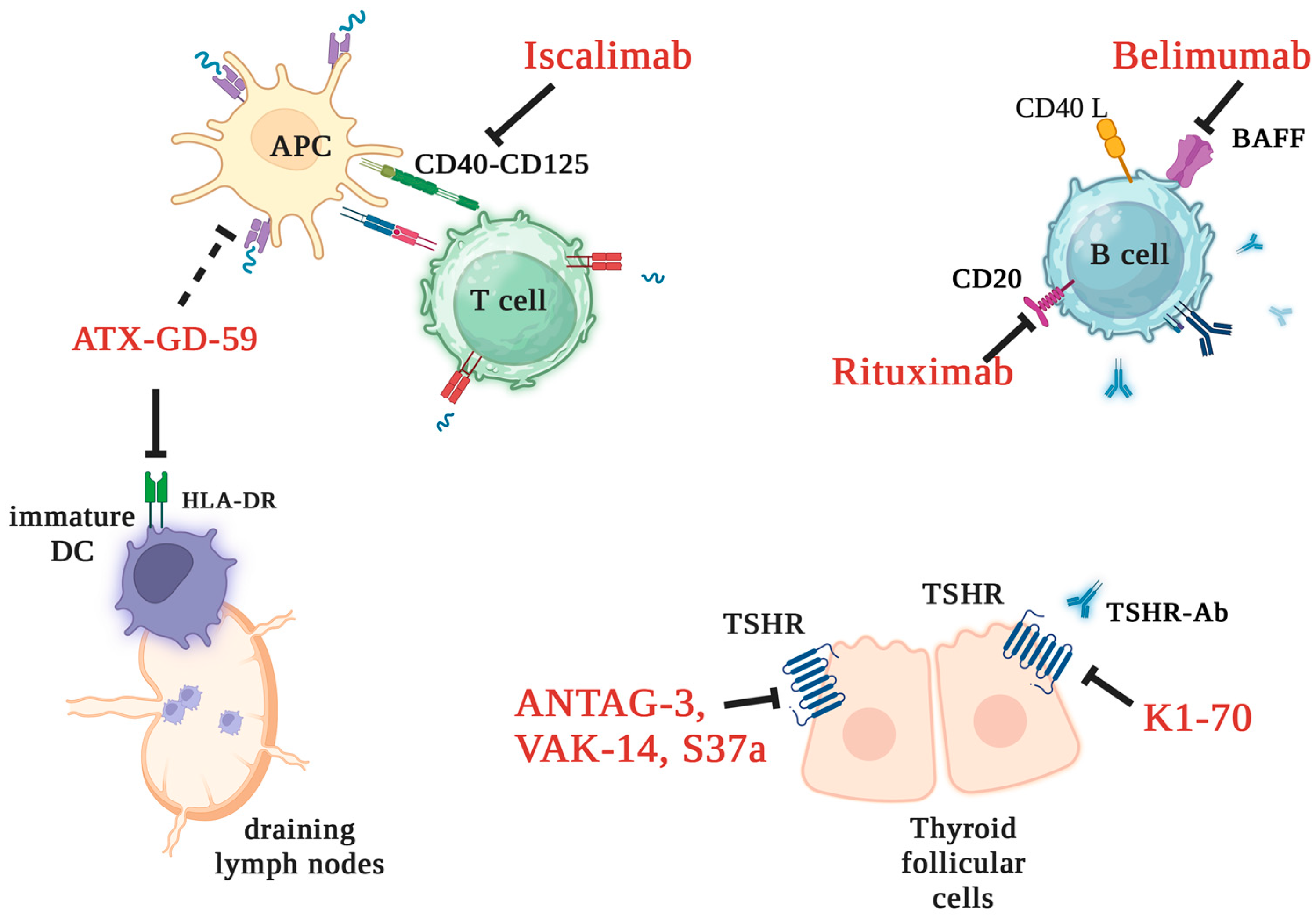
3.8. New Treatment Strategies for Graves’ Disease
3.9. Regulatory T Cells as Target Therapy for Autoimmune Disorders: Brief Insights
3.10. Regulatory T Cell-Based Strategies in Graves’ Disease
4. Conclusions
- Loss of central and peripheral immune tolerance is crucial in GD pathogenesis;
- Tregs mediate immune tolerance, acting as negative regulators of inflammation;
- Reduced Tregs’ number/function impairs immunoregulation and exposes susceptible subjects to autoimmunity development/propagation;
- Targeting Treg may emerge as a prospective strategy for ameliorating or even curing hyperthyroidism;
- Diverse markers characterize human Tregs, posing a challenge to the comparability of studies in autoimmune patients.
Author Contributions
Funding
Conflicts of Interest
References
- Shimizu, J.; Yamazaki, S.; Sakaguchi, S. Induction of tumor immunity by removing CD25+CD4+ T cells: A common basis between tumor immunity and autoimmunity. J. Immunol. 1999, 163, 5211–5218. [Google Scholar] [CrossRef]
- Iglesias-Escudero, M.; Arias-González, N.; Martínez-Cáceres, E. Regulatory cells and the effect of cancer immunotherapy. Mol. Cancer. 2023, 22, 26. [Google Scholar] [CrossRef]
- Taylor, P.A.; Noelle, R.J.; Blazar, B.R. CD4+CD25+ immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J. Exp. Med. 2001, 193, 1311–1318. [Google Scholar] [CrossRef]
- Qi, J.; Liu, C.; Bai, Z.; Li, X.; Yao, G. T follicular helper cells and T follicular regulatory cells in autoimmune diseases. Front. Immunol. 2023, 14, 1178792. [Google Scholar] [CrossRef] [PubMed]
- Laukova, M.; Glatman Zaretsky, A. Regulatory T cells as a therapeutic approach for inflammatory bowel disease. Eur. J. Immunol. 2023, 53, e2250007. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, S.M.; Nagayama, Y.; Pichurin, P.N.; Mizutori, Y.; Chen, C.R.; Misharin, A.; Aliesky, H.A.; Rapoport, B. The link between Graves’ disease and Hashimoto’s thyroiditis: A role for regulatory T cells. Endocrinology 2007, 148, 5724–5733. [Google Scholar] [CrossRef]
- Ramchandani, R.; Hossenbaccus, L.; Ellis, A.K. Immunoregulatory T cell epitope peptides for the treatment of allergic disease. Immunotherapy 2021, 13, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Eggenhuizen, P.J.; Ng, B.H.; Ooi, J.D. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int. J. Mol. Sci. 2020, 21, 7015. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef]
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; Fazekas de St Groth, B.; et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Bailey-Bucktrout, S.L.; Jeker, L.T.; Penaranda, C.; Martínez-Llordella, M.; Ashby, M.; Nakayama, M.; Rosenthal, W.; Bluestone, J.A. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 2009, 10, 1000–1007. [Google Scholar] [CrossRef]
- Barzaghi, F.; Passerini, L. IPEX Syndrome: Improved Knowledge of Immune Pathogenesis Empowers Diagnosis. Front. Pediatr. 2021, 9, 612760. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol. 2005, 6, 345–352. [Google Scholar] [CrossRef]
- Roncarolo, M.G.; Gregori, S.; Bacchetta, R.; Battaglia, M. Tr1 cells and the counter-regulation of immunity: Natural mechanisms and therapeutic applications. Curr. Top. Microbiol. Immunol. 2014, 380, 39–68. [Google Scholar]
- Vitales-Noyola, M.; Doníz-Padilla, L.; Álvarez-Quiroga, C.; Monsiváis-Urenda, A.; Portillo-Salazar, H.; González-Amaro, R. Quantitative and functional analysis of CD69+ NKG2D+ T regulatory cells in healthy subjects. Hum. Immunol. 2015, 76, 511–518. [Google Scholar] [CrossRef]
- Vieyra-Lobato, M.R.; Vela-Ojeda, J.; Montiel-Cervantes, L.; López-Santiago, R.; Moreno-Lafont, M.C. Description of CD8+ Regulatory T Lymphocytes and Their Specific Intervention in Graft-versus-Host and Infectious Diseases, Autoimmunity, and Cancer. J. Immunol. Res. 2018, 2018, 3758713. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Ono, M.; Setoguchi, R.; Yagi, H.; Hori, S.; Fehervari, Z.; Shimizu, J.; Takahashi, T.; Nomura, T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 2006, 212, 8–27. [Google Scholar] [CrossRef]
- Askenasy, N.; Kaminitz, A.; Yarkoni, S. Mechanisms of T regulatory cell function. Autoimmun. Rev. 2008, 7, 370–375. [Google Scholar] [CrossRef]
- Povoleri, G.A.; Scottà, C.; Nova-Lamperti, E.A.; John, S.; Lombardi, G.; Afzali, B. Thymic versus induced regulatory T cells—Who regulates the regulators? Front. Immunol. 2013, 4, 169. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, N.; Kitagawa, Y.; Sakaguchi, S. Development and maintenance of regulatory T cells. Immunity 2013, 38, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.Y.; Low, J.S.; Tanimine, N.; Finn, K.K.; Priyadharshini, B.; Germana, S.K.; Kaech, S.M.; Turka, L.A. Differential Roles of IL-2 Signaling in Developing versus Mature Tregs. Cell Rep. 2018, 25, 1204–1213.e4. [Google Scholar] [CrossRef]
- Takahashi, T.; Kuniyasu, Y.; Toda, M.; Sakaguchi, N.; Itoh, M.; Iwata, M.; Shimizu, J.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: Induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 1998, 10, 1969–1980. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Lider, O.; Weiner, H.L. Antigen-driven bystander suppression after oral administration of antigens. J. Exp. Med. 1991, 174, 791–798. [Google Scholar] [CrossRef]
- Kitz, A.; Dominguez-Villar, M. Molecular mechanisms underlying Th1-like Treg generation and function. Cell. Mol. Life Sci. 2017, 74, 4059–4075. [Google Scholar] [CrossRef]
- Koch, M.A.; Tucker-Heard, G.; Perdue, N.R.; Killebrew, J.R.; Urdahl, K.B.; Campbell, D.J. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 2009, 10, 595–602. [Google Scholar] [CrossRef]
- Levine, A.G.; Mendoza, A.; Hemmers, S.; Moltedo, B.; Niec, R.E.; Schizas, M.; Hoyos, B.E.; Putintseva, E.V.; Chaudhry, A.; Dikiy, S.; et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 2017, 546, 421–425. [Google Scholar] [CrossRef]
- Noval Rivas, M.; Burton, O.T.; Wise, P.; Charbonnier, L.M.; Georgiev, P.; Oettgen, H.C.; Rachid, R.; Chatila, T.A. Regulatory T cell reprogramming toward a Th2-cell-like lineage impairs oral tolerance and promotes food allergy. Immunity 2015, 42, 512–523. [Google Scholar] [CrossRef]
- Zheng, Y.; Chaudhry, A.; Kas, A.; deRoos, P.; Kim, J.M.; Chu, T.T.; Corcoran, L.; Treuting, P.; Klein, U.; Rudensky, A.Y. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 2009, 458, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders—Advances and challenges. Hum. Vaccin. Immunother. 2022, 18, 2035117. [Google Scholar] [CrossRef]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Rasmussen, J.P.; Rudensky, A.Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 2007, 8, 191–197. [Google Scholar] [CrossRef]
- Hatzioannou, A.; Boumpas, A.; Papadopoulou, M.; Papafragkos, I.; Varveri, A.; Alissafi, T.; Verginis, P. Regulatory T Cells in Autoimmunity and Cancer: A Duplicitous Lifestyle. Front. Immunol. 2021, 12, 731947. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Hegedüs, L. Graves’ Disease. N. Engl. J. Med. 2016, 375, 1552–1565. [Google Scholar] [CrossRef] [PubMed]
- Boelaert, K.; Newby, P.R.; Simmonds, M.J.; Holder, R.L.; Carr-Smith, J.D.; Heward, J.M.; Manji, N.; Allahabadia, A.; Armitage, M.; Chatterjee, K.V.; et al. Prevalence and relative risk of other autoimmune diseases in subjects with autoimmune thyroid disease. Am. J. Med. 2010, 123, 183.e1–183.e9. [Google Scholar] [CrossRef]
- Wiersinga, W.M.; Poppe, K.G.; Effraimidis, G. Hyperthyroidism: Aetiology, pathogenesis, diagnosis, management, complications, and prognosis. Lancet Diabetes Endocrinol. 2023, 11, 282–298. [Google Scholar] [CrossRef]
- Bartalena, L.; Tanda, M.L. Current concepts regarding Graves’ orbitopathy. J. Int. Med. 2022, 5, 692–716. [Google Scholar] [CrossRef]
- Bartalena, L.; Piantanida, E.; Gallo, D.; Ippolito, S.; Tanda, M.L. Management of Graves’ hyperthyroidism: Present and future. Exp. Rev. Endocrinol. Metab. 2022, 17, 153–166. [Google Scholar] [CrossRef]
- Struja, T.; Kutz, A.; Fischli, S.; Meier, C.; Mueller, B.; Recher, M.; Schuetz, P. Is Graves’ disease a primary immunodeficiency? New immunological perspectives on an endocrine disease. BMC Med. 2017, 15, 174. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Pan, C.M.; Zhao, S.X.; Liang, J.; Gao, G.Q.; Zhang, X.M.; Yuan, G.Y.; Li, C.G.; Xue, L.Q.; Shen, M.; et al. China Consortium for Genetics of Autoimmune Thyroid Disease. A genome-wide association study identifies two new risk loci for Graves’ disease. Nat. Genet. 2011, 43, 897–901. [Google Scholar] [PubMed]
- Gallo, D.; Bruno, A.; Gallazzi, M.; Cattaneo, S.A.M.; Veronesi, G.; Genoni, A.; Tanda, M.L.; Bartalena, L.; Passi, A.; Piantanida, E.; et al. Immunomodulatory role of vitamin D and selenium supplementation in newly diagnosed Graves’ disease patients during methimazole treatment. Front. Endocrinol. 2023, 14, 1145811. [Google Scholar] [CrossRef] [PubMed]
- Bottazzo, G.F.; Pujol-Borrell, R.; Hanafusa, T.; Feldmann, M. Role of aberrant HLA-DR expression and antigen presentation in induction of endocrine autoimmunity. Lancet 1983, 2, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.F.; Andersen, S.; Latif, R.; Nagayama, Y.; Barbesino, G.; Brito, M.; Eckstein, A.K.; Stagnaro-Green, A.; Kahaly, G.J. Graves’ disease. Nat. Rev. Dis. Primers 2020, 6, 52. [Google Scholar] [CrossRef]
- Gallo, D.; Piantanida, E.; Gallazzi, M.; Bartalena, L.; Tanda, M.L.; Bruno, A.; Mortara, L. Immunological Drivers in Graves’ Disease: NK Cells as a Master Switcher. Front. Endocrinol. 2020, 11, 406. [Google Scholar] [CrossRef]
- Gallo, D.; De Vito, A.; Roncoroni, R.; Bruno, A.; Piantanida, E.; Bartalena, L.; Tanda, M.L.; Mortara, L.; Acquati, F. A potential role of human RNASET2 overexpression in the pathogenesis of Graves’ disease. Endocrine 2023, 79, 55–59. [Google Scholar] [CrossRef]
- Zhang, M.; Jiang, W.; Lu, G.; Wang, R.; Lv, Z.; Li, D. Insight Into Mouse Models of Hyperthyroidism. Front. Endocrinol. 2022, 13, 929750. [Google Scholar] [CrossRef]
- Shimojo, N.; Kohno, Y.; Yamaguchi, K.; Kikuoka, S.; Hoshioka, A.; Niimi, H.; Hirai, A.; Tamura, Y.; Saito, Y.; Kohn, L.D.; et al. Induction of Graves-like disease in mice by immunization with fibroblasts transfected with the thyrotropin receptor and a class II molecule. Proc. Natl. Acad. Sci. USA 1996, 93, 11074–11079. [Google Scholar] [CrossRef]
- Kaithamana, S.; Fan, J.; Osuga, Y.; Liang, S.G.; Prabhakar, B.S. Induction of experimental autoimmune Graves’ disease in BALB/c mice. J. Immunol. 1999, 163, 5157–5164. [Google Scholar] [CrossRef]
- Nagayama, Y. Graves’ animal models of Graves’ hyperthyroidism. Thyroid 2007, 17, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, O.; Nagayama, Y. Regulation of Graves’ Hyperthyroidism with Naturally Occurring CD4+CD25+ Regulatory T Cells in a Mouse Model. Endocrinology 2006, 147, 2417–2422. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, O.; Abiru, N.; Nakahara, M.; Nagayama, Y. CD8+CD122+ T cells, a newly identified regulatory T subset, negatively regulate Graves’ hyperthyroidism in a murine model. Endocrinology 2007, 148, 6040–6046. [Google Scholar] [CrossRef] [PubMed]
- Sempowski, G.D.; Cross, S.J.; Heinly, C.S.; Scearce, R.M.; Haynes, B.F. CD7 and CD28 are required for murine CD4+CD25+ regulatory T cell homeostasis and prevention of thyroiditis. J. Immunol. 2004, 172, 787–794. [Google Scholar] [CrossRef]
- Gangi, E.; Vasu, C.; Cheatem, D.; Prabhakar, B.S. IL-10-producing CD4+CD25+ regulatory T cells play a critical role in granulocyte-macrophage colony-stimulating factor-induced suppression of experimental autoimmune thyroiditis. J. Immunol. 2005, 174, 7006–7013. [Google Scholar] [CrossRef]
- Zhou, J.; Bi, M.; Fan, C.; Song, X.; Yang, R.; Zhao, S.; Li, L.; Li, Y.; Teng, W.; Shan, Z. Regulatory T cells but not T helper 17 cells are modulated in an animal model of Graves’ hyperthyroidism. Clin. Exp. Med. 2012, 12, 39–46. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhao, Y.; Zhu, X.; Liu, X. Low regulatory T cell and high IL-17 mRNA expression in a mouse Graves’ disease model. J. Endocrinol. Investig. 2017, 40, 397–407. [Google Scholar] [CrossRef]
- Brand, O.J.; Lowe, C.E.; Heward, J.M.; Franklyn, J.A.; Cooper, J.D.; Todd, J.; Gough, S.C. Association of the interleukin-2 receptor alpha (IL-2Ralpha)/CD25 gene region with Graves’ disease using a multilocus test and tag SNPs. Clin. Endocrinol. 2007, 66, 508–512. [Google Scholar] [CrossRef]
- Lee, H.J.; Li, C.W.; Hammerstad, S.S.; Stefan, M.; Tomer, Y. Immunogenetics of autoimmune thyroid diseases: A comprehensive review. J. Autoimmun. 2015, 64, 82–90. [Google Scholar] [CrossRef]
- Effraimidis, G.; Wiersinga, W.M. Mechanisms in endocrinology: Autoimmune thyroid disease: Old and new players. Eur. J. Endocrinol. 2014, 170, R241–R252. [Google Scholar] [CrossRef]
- Zhang, D.; Qiu, X.; Li, J.; Zheng, S.; Li, L.; Zhao, H. MiR-23a-3p-regulated abnormal acetylation of FOXP3 induces regulatory T cell function defect in Graves’ disease. Biol. Chem. 2019, 400, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Inoue, N.; Watanabe, M.; Morita, M.; Tomizawa, R.; Akamizu, T.; Tatsumi, K.; Hidaka, Y.; Iwatani, Y. Association of functional polymorphisms related to the transcriptional level of FOXP3 with prognosis of autoimmune thyroid diseases. Clin. Exp. Immunol. 2010, 162, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, S.; Tang, X.; Li, J.; Zou, P. Changes of regulatory T cells in Graves’ disease. J. Huazhong Univ. Sci. Technol. Med. Sci. 2006, 26, 545–547. [Google Scholar] [CrossRef]
- Pan, D.; Shin, Y.H.; Gopalakrishnan, G.; Hennessey, J.; De Groot, L.J. Regulatory T cells in Graves’ disease. Clin. Endocrinol. 2009, 71, 587–593. [Google Scholar] [CrossRef]
- Mao, C.; Wang, S.; Xiao, Y.; Xu, J.; Jiang, Q.; Jin, M.; Jiang, X.; Guo, H.; Ning, G.; Zhang, Y. Impairment of regulatory capacity of CD4+CD25+ regulatory T cells mediated by dendritic cell polarization and hyperthyroidism in Graves’ disease. J. Immunol. 2011, 186, 4734–4743. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Watanabe, M.; Iida, T.; Kuroda, S.; Matsuzuka, F.; Miyauchi, A.; Iwatani, Y. Apoptosis-induced decrease of intrathyroidal CD4+CD25+ regulatory T cells in autoimmune thyroid diseases. Thyroid 2007, 17, 25–31. [Google Scholar] [CrossRef]
- Kahaly, G.J.; Shimony, O.; Gellman, Y.N.; Lytton, S.D.; Eshkar-Sebban, L.; Rosenblum, N.; Refaeli, E.; Kassem, S.; Ilany, J.; Naor, D. Regulatory T-cells in Graves’ orbitopathy: Baseline findings and immunomodulation by anti-T lymphocyte globulin. J. Clin. Endocrinol. Metab. 2011, 96, 422–429. [Google Scholar] [CrossRef]
- Bossowski, A.; Moniuszko, M.; Dąbrowska, M.; Sawicka, B.; Rusak, M.; Jeznach, M.; Wójtowicz, J.; Bodzenta-Lukaszyk, A.; Bossowska, A. Lower proportions of CD4+CD25(high) and CD4+FoxP3, but not CD4+CD25+CD127(low) FoxP3+ T cell levels in children with autoimmune thyroid diseases. Autoimmunity 2013, 46, 222–230. [Google Scholar] [CrossRef]
- Liu, H.Y.; Shi, Z.Y.; Fan, D.; Zhang, S.X.; Wu, L.X.; Lu, K.Y.; Yang, S.Y.; Li, W.T.; Kang, J.F.; Li, C.H.; et al. Absolute reduction in peripheral regulatory T cells in patients with Graves’ disease and post-treatment recovery. Mol. Immunol. 2022, 144, 49–57. [Google Scholar] [CrossRef]
- Teniente-Serra, A.; Soldevila, B.; Quirant-Sánchez, B.; Fernández, M.A.; Ester Condins, A.; Puig-Domingo, M.; Pujol-Borrell, R.; Martínez-Cáceres, E.M. Distinct pattern of peripheral lymphocyte subsets in Graves’ disease with persistency of anti-TSHR autoantibodies. Autoimmunity 2019, 52, 220–227. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, Y.; Hu, S.; Zhang, M.; Shi, B.; Wang, Y. Decreased Treg Cell and TCR Expansion Are Involved in Long-Lasting Graves’ Disease. Front. Endocrinol. 2021, 12, 632492. [Google Scholar] [CrossRef] [PubMed]
- Klatka, M.; Grywalska, E.; Partyka, M.; Charytanowicz, M.; Kiszczak-Bochynska, E.; Rolinski, J. Th17 and Treg cells in adolescents with Graves’ disease. Impact of treatment with methimazole on these cell subsets. Autoimmunity 2014, 47, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muñoz, A.; Vitales-Noyola, M.; Ramos-Levi, A.; Serrano-Somavilla, A.; González-Amaro, R.; Marazuela, M. Levels of regulatory T cellsCD69+NKG2D+IL-10+ are increased in patients with autoimmune thyroid disorders. Endocrine 2016, 51, 478–489. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, A.; Martínez-Hernández, R.; Ramos-Leví, A.M.; Serrano-Somavilla, A.; González-Amaro, R.; Sánchez-Madrid, F.; de la Fuente, H.; Marazuela, M. Circulating Microvesicles Regulate Treg and Th17 Differentiation in Human Autoimmune Thyroid Disorders. J. Clin. Endocrinol. Metab. 2015, 100, E1531–E1539. [Google Scholar] [CrossRef]
- Marazuela, M.; García-López, M.A.; Figueroa-Vega, N.; de la Fuente, H.; Alvarado-Sánchez, B.; Monsiváis-Urenda, A.; Sánchez-Madrid, F.; González-Amaro, R. Regulatory T cells in human autoimmune thyroid disease. J. Clin. Endocrinol. Metab. 2006, 91, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Glick, A.B.; Wodzinski, A.; Fu, P.; Levine, A.D.; Wald, D.N. Impairment of regulatory T-cell function in autoimmune thyroid disease. Thyroid 2013, 23, 871–878. [Google Scholar] [CrossRef]
- Laurence, A.; Amarnath, S.; Mariotti, J.; Kim, Y.C.; Foley, J.; Eckhaus, M.; O’Shea, J.J.; Fowler, D.H. STAT3 transcription factor promotes instability of nTreg cells and limits generation of iTreg cells during acute murine graft-versus-host disease. Immunity 2012, 37, 209–222. [Google Scholar] [CrossRef]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef]
- Beriou, G.; Costantino, C.M.; Ashley, C.W.; Yang, L.; Kuchroo, V.K.; Baecher-Allan, C.; Hafler, D.A. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood 2009, 113, 4240–4249. [Google Scholar] [CrossRef]
- Voo, K.S.; Wang, Y.H.; Santori, F.R.; Boggiano, C.; Wang, Y.H.; Arima, K.; Bover, L.; Hanabuchi, S.; Khalili, J.; Marinova, E.; et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 4793–4798. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Y.; Ding, X.; Zhang, M.; He, M.; Zhao, Y.; Hu, S.; Zhao, F.; Wang, J.; Xie, B.; et al. The proportion of peripheral blood Tregs among the CD4+ T cells of autoimmune thyroid disease patients: A meta-analysis. Endocr. J. 2020, 67, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Huang, L.; Zhang, L. Helper T Cell 17 and Regulatory T Cell Levels in Peripheral Blood of Newly Diagnosed Patients with Autoimmune Thyroid Disease: A Meta-Analysis. Horm. Metab. Res. 2023, 55, 40–50. [Google Scholar] [CrossRef]
- Fountoulakis, S.; Vartholomatos, G.; Kolaitis, N.; Frillingos, S.; Philippou, G.; Tsatsoulis, A. HLA-DR expressing peripheral T regulatory cells in newly diagnosed patients with different forms of autoimmune thyroid disease. Thyroid 2008, 18, 1195–1200. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yuan, J.; Zhu, Y.F.; Yang, X.J.; Wang, Q.; Xu, J.; He, S.T.; Zhang, J.A. Imbalance of Th17/Treg in Different Subtypes of Autoimmune Thyroid Diseases. Cell. Physiol. Biochem. 2016, 40, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Zhou, J.; Fan, C.; Zhao, N.; Liu, Y.; Wang, S.; Cui, X.; Huang, M.; Guan, H.; Li, Y.; et al. Increased Circulating Th17 but Decreased CD4+Foxp3+ Treg and CD19+CD1dhiCD5+ Breg Subsets in New-Onset Graves’ Disease. Biomed. Res. Int. 2017, 2017, 8431838. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Vignali, D.A.; Rudensky, A.Y.; Niec, R.E.; Waldmann, H. The plasticity and stability of regulatory T cells. Nat. Rev. Immunol. 2013, 13, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S. Human Th1 dichotomy: Origin, phenotype and biologic activities. Immunology 2014, 144, 343–351. [Google Scholar] [CrossRef]
- Fan, Q.; Liu, Y.; Rao, J.; Zhang, Z.; Xiao, W.; Zhu, T.; Chai, X.; Ye, K.; Ning, N.; Yin, Z.; et al. Anti-Atherosclerosis Effect of Angong Niuhuang Pill via Regulating Th17/Treg Immune Balance and Inhibiting Chronic Inflammatory on ApoE−/− Mice Model of Early and Mid-Term Atherosclerosis. Front. Pharmacol. 2020, 10, 1584. [Google Scholar] [CrossRef]
- Su, X.; Yin, X.; Liu, Y.; Yan, X.; Zhang, S.; Wang, X.; Lin, Z.; Zhou, X.; Gao, J.; Wang, Z.; et al. Gut Dysbiosis Contributes to the Imbalance of Treg and Th17 Cells in Graves’ Disease Patients by Propionic Acid. J. Clin. Endocrinol. Metab. 2020, 105, dgaa511. [Google Scholar] [CrossRef]
- Tan, Y.; Chen, W.; Liu, C.; Zheng, X.; Guo, A.; Long, J. Effect of IL-21 on the Balance of Th17 Cells/Treg Cells in the Pathogenesis of Graves’ Disease. Endocr. Res. 2019, 44, 138–147. [Google Scholar] [CrossRef]
- Yao, G.; Qi, J.; Liang, J.; Shi, B.; Chen, W.; Li, W.; Tang, X.; Wang, D.; Lu, L.; Chen, W.; et al. Mesenchymal stem cell transplantation alleviates experimental Sjögren’s syndrome through IFN-β/IL-27 signaling axis. Theranostics 2019, 9, 8253–8265. [Google Scholar] [CrossRef]
- González-Amaro, R.; Marazuela, M. T regulatory (Treg) and T helper 17 (Th17) lymphocytes in thyroid autoimmunity. Endocrine 2016, 52, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Nanba, T.; Watanabe, M.; Inoue, N.; Iwatani, Y. Increases of the Th1/Th2 cell ratio in severe Hashimoto’s disease and in the proportion of Th17 cells in intractable Graves’ disease. Thyroid 2009, 19, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Yu, X.; Shen, L. Autoimmune thyroid diseases and Th17/Treg lymphocytes. Life Sci. 2018, 192, 160–165. [Google Scholar] [CrossRef]
- Vitales-Noyola, M.; Ramos-Levi, A.M.; Serrano-Somavilla, A.; Martínez-Hernández, R.; Sampedro-Nuñez, M.; Di Pasquale, C.; González-Amaro, R.; Marazuela, M. Expression and Function of the Costimulatory Receptor SLAMF1 Is Altered in Lymphocytes From Patients with Autoimmune Thyroiditis. J. Clin. Endocrinol. Metab. 2017, 102, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.T.; Hatton, R.D. Interplay between the TH17 and TReg cell lineages: A (co-)evolutionary perspective. Nat. Rev. Immunol. 2009, 9, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Crescioli, C.; Cosmi, L.; Borgogni, E.; Santarlasci, V.; Gelmini, S.; Sottili, M.; Sarchielli, E.; Mazzinghi, B.; Francalanci, M.; Pezzatini, A.; et al. Methimazole inhibits CXC chemokine ligand 10 secretion in human thyrocytes. J. Endocrinol. 2007, 195, 145–155. [Google Scholar] [CrossRef]
- Volpé, R. Evidence that the immunosuppressive effects of antithyroid drugs are mediated through actions on the thyroid cell, modulating thyrocyte-immunocyte signaling: A review. Thyroid 1994, 4, 217–223. [Google Scholar] [CrossRef]
- Lane, L.C.; Cheetham, T.D.; Perros, P.; Pearce, S.H.S. New Therapeutic Horizons for Graves’ Hyperthyroidism. Endocr. Rev. 2020, 41, 873–884. [Google Scholar] [CrossRef]
- McCallion, O.; Bilici, M.; Hester, J.; Issa, F. Regulatory T-cell therapy approaches. Clin. Exp. Immunol. 2023, 211, 96–107. [Google Scholar] [CrossRef]
- Wang, H.; Feng, X.; Yan, W.; Tian, D. Regulatory T Cells in Autoimmune Hepatitis: Unveiling Their Roles in Mouse Models and Patients. Front. Immunol. 2020, 11, 575572. [Google Scholar] [CrossRef] [PubMed]
- Danikowski, K.M.; Jayaraman, S.; Prabhakar, B.S. Regulatory T cells in multiple sclerosis and myasthenia gravis. J. Neuroinflamm. 2017, 14, 117. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Wang, W.; Li, Y.; Shan, Z.; Li, Y.; Teng, X.; Gao, Y.; Fan, C.; Teng, W. Selenium upregulates CD4+CD25+ regulatory T cells in iodine-induced autoimmune thyroiditis model of NOD.H-2(h4) mice. Endocr. J. 2010, 57, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Pantalena, L.C.; Liu, X.K.; Gaffen, S.L.; Liu, H.; Rohowsky-Kochan, C.; Ichiyama, K.; Yoshimura, A.; Steinman, L.; Christakos, S.; et al. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol. Cell. Biol. 2011, 31, 3653–3669. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kustrimovic, N.; Gallo, D.; Piantanida, E.; Bartalena, L.; Lai, A.; Zerbinati, N.; Tanda, M.L.; Mortara, L. Regulatory T Cells in the Pathogenesis of Graves’ Disease. Int. J. Mol. Sci. 2023, 24, 16432. https://doi.org/10.3390/ijms242216432
Kustrimovic N, Gallo D, Piantanida E, Bartalena L, Lai A, Zerbinati N, Tanda ML, Mortara L. Regulatory T Cells in the Pathogenesis of Graves’ Disease. International Journal of Molecular Sciences. 2023; 24(22):16432. https://doi.org/10.3390/ijms242216432
Chicago/Turabian StyleKustrimovic, Natasa, Daniela Gallo, Eliana Piantanida, Luigi Bartalena, Adriana Lai, Nicola Zerbinati, Maria Laura Tanda, and Lorenzo Mortara. 2023. "Regulatory T Cells in the Pathogenesis of Graves’ Disease" International Journal of Molecular Sciences 24, no. 22: 16432. https://doi.org/10.3390/ijms242216432
APA StyleKustrimovic, N., Gallo, D., Piantanida, E., Bartalena, L., Lai, A., Zerbinati, N., Tanda, M. L., & Mortara, L. (2023). Regulatory T Cells in the Pathogenesis of Graves’ Disease. International Journal of Molecular Sciences, 24(22), 16432. https://doi.org/10.3390/ijms242216432