
Identification of 13 Novel Loci in a Genome-Wide Association Study on Taiwanese with Hepatocellular Carcinoma

 , ,
, ,
Abstract
:1. Introduction
2. Results
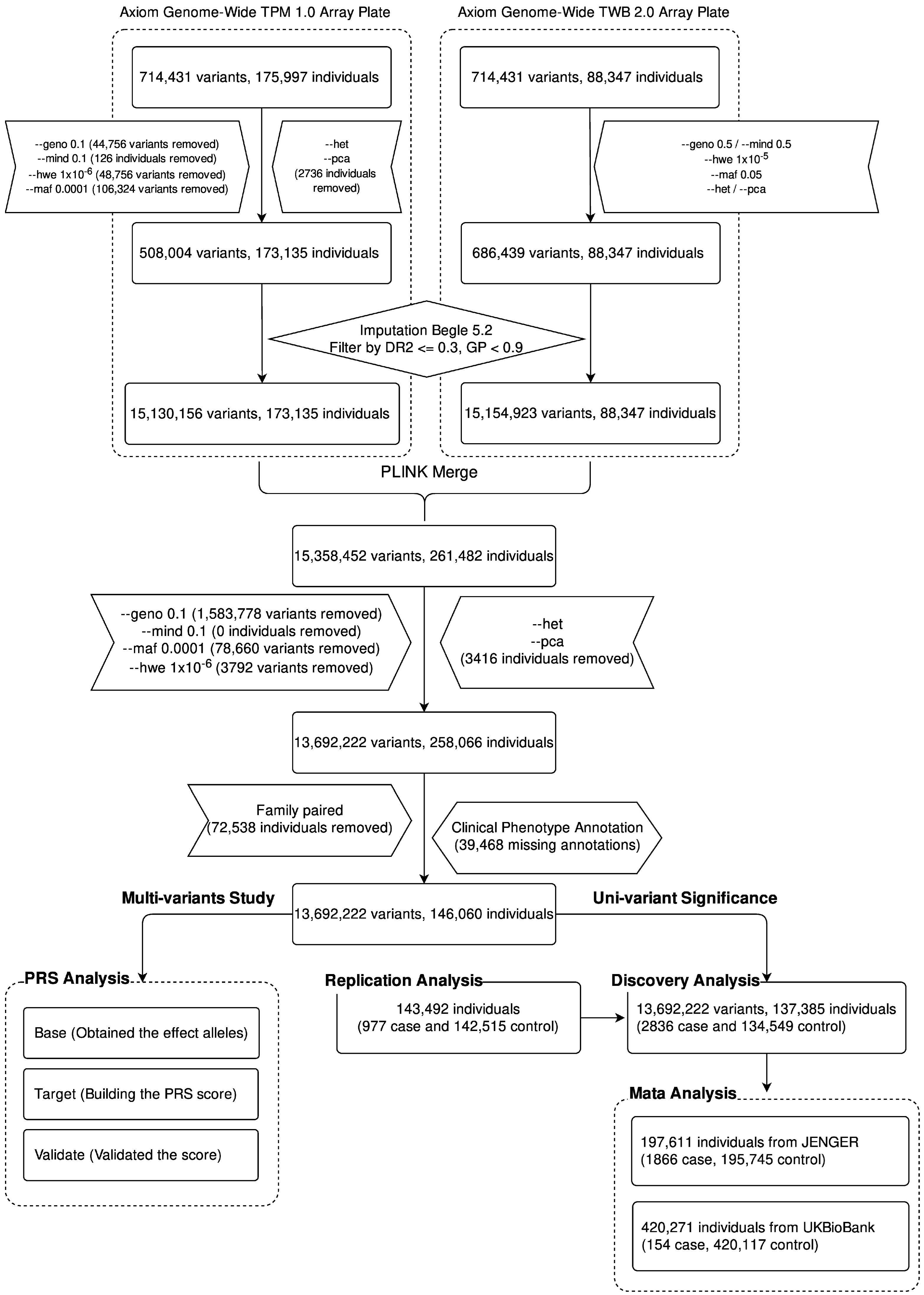
2.1. Study Flowchart
2.2. Demographic Characteristics of Patients
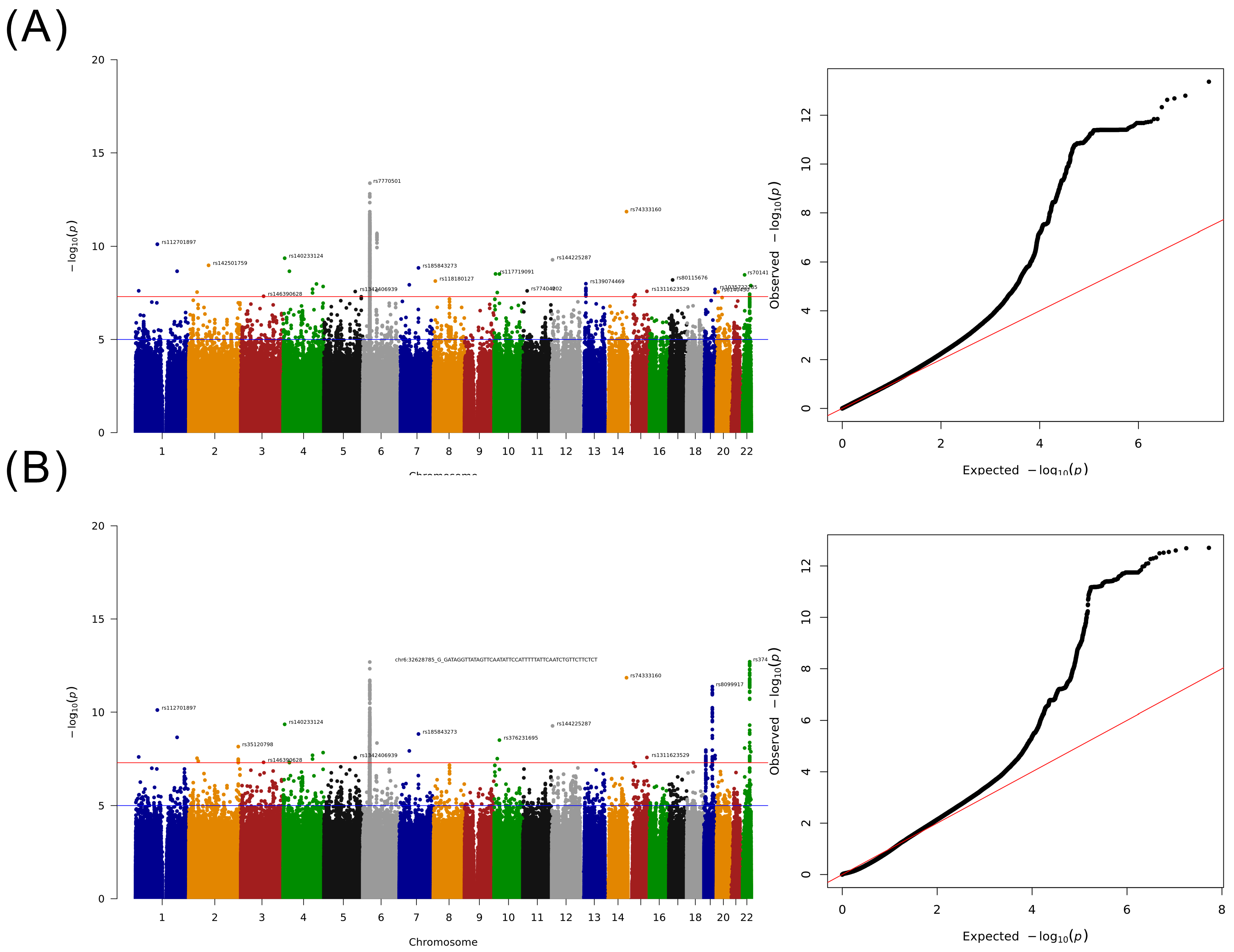
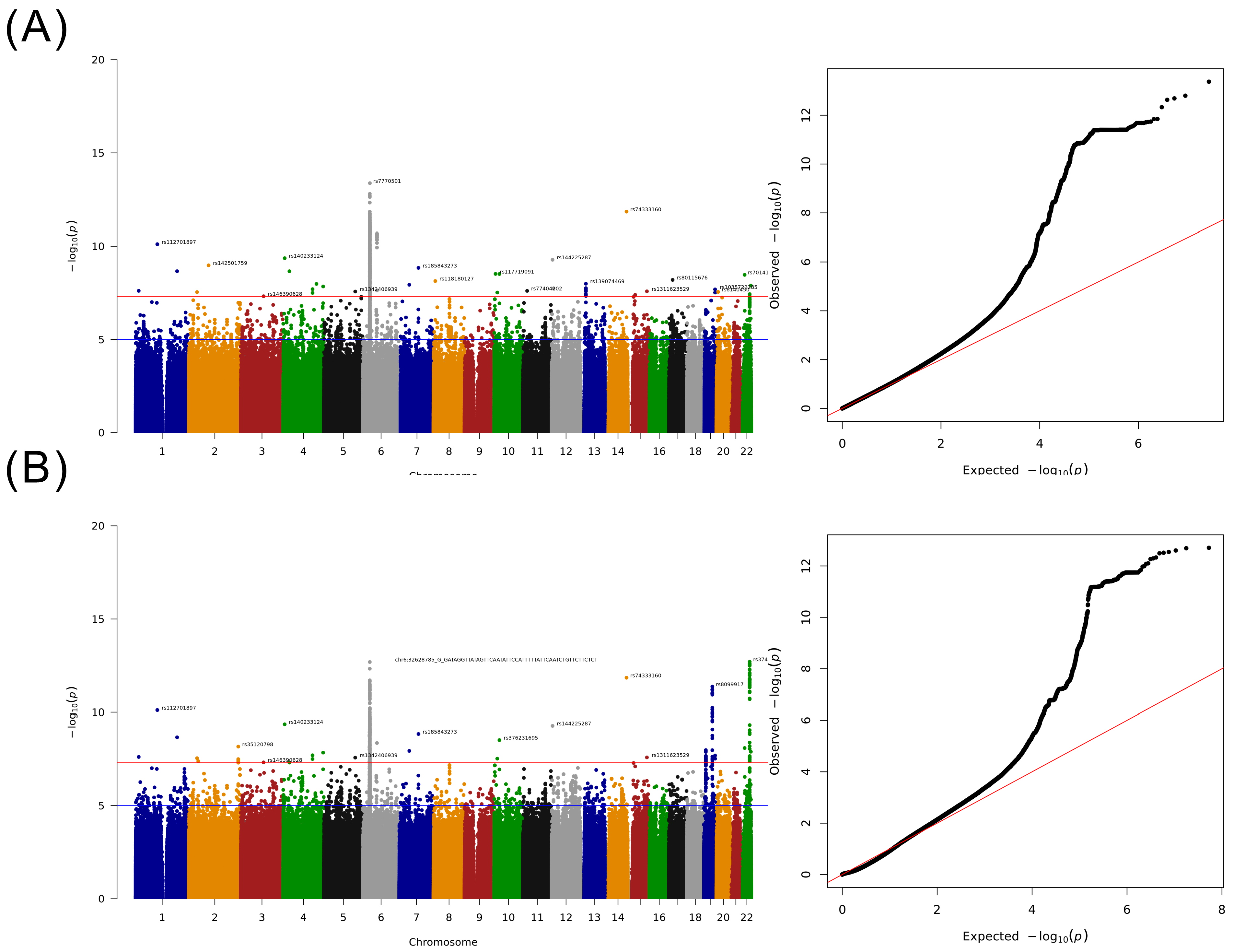
2.3. GWAS for Taiwanese Patients with HCC without a Specified Etiology
2.4. Detailed Analysis of HLA Loci
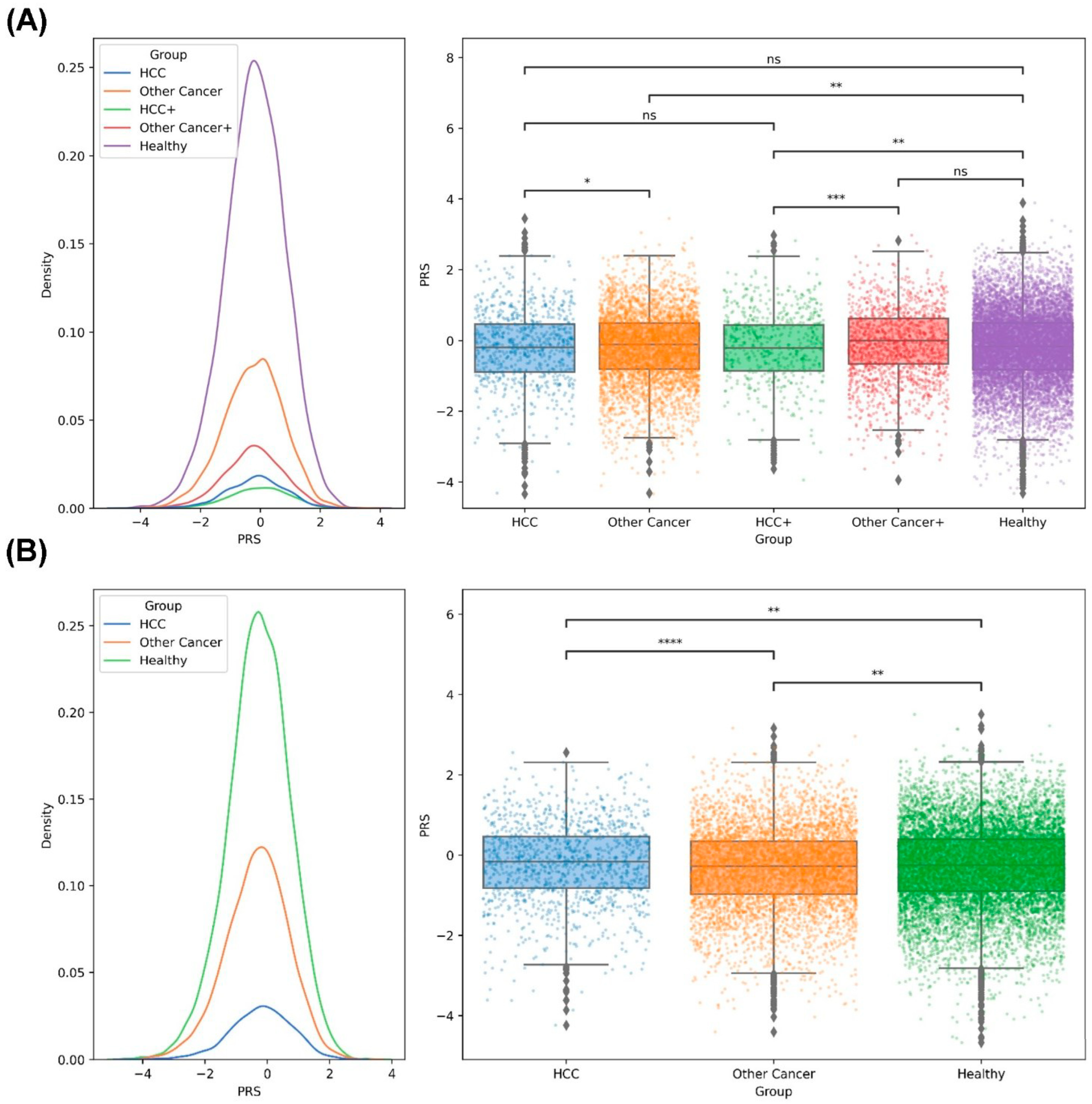
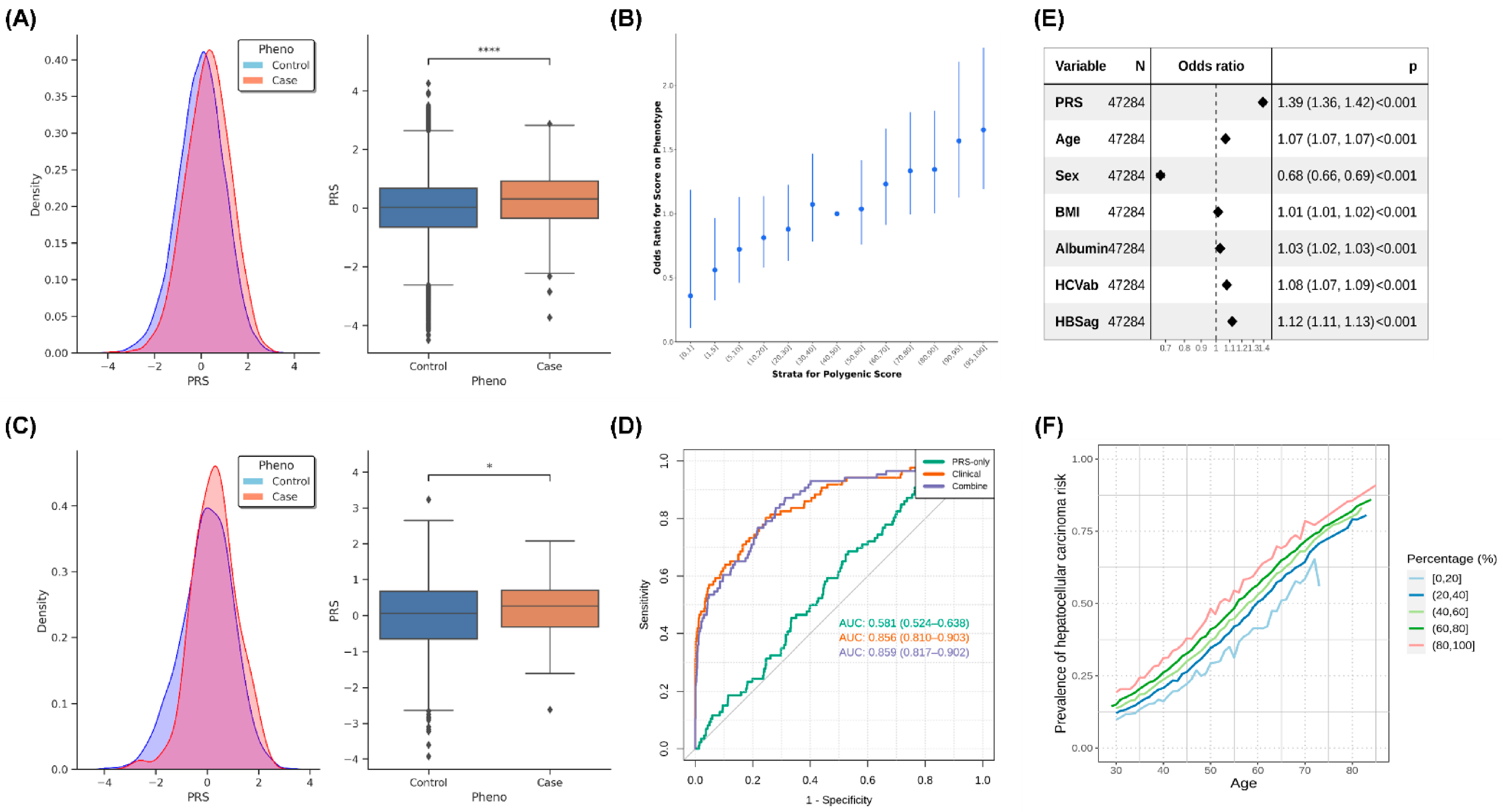
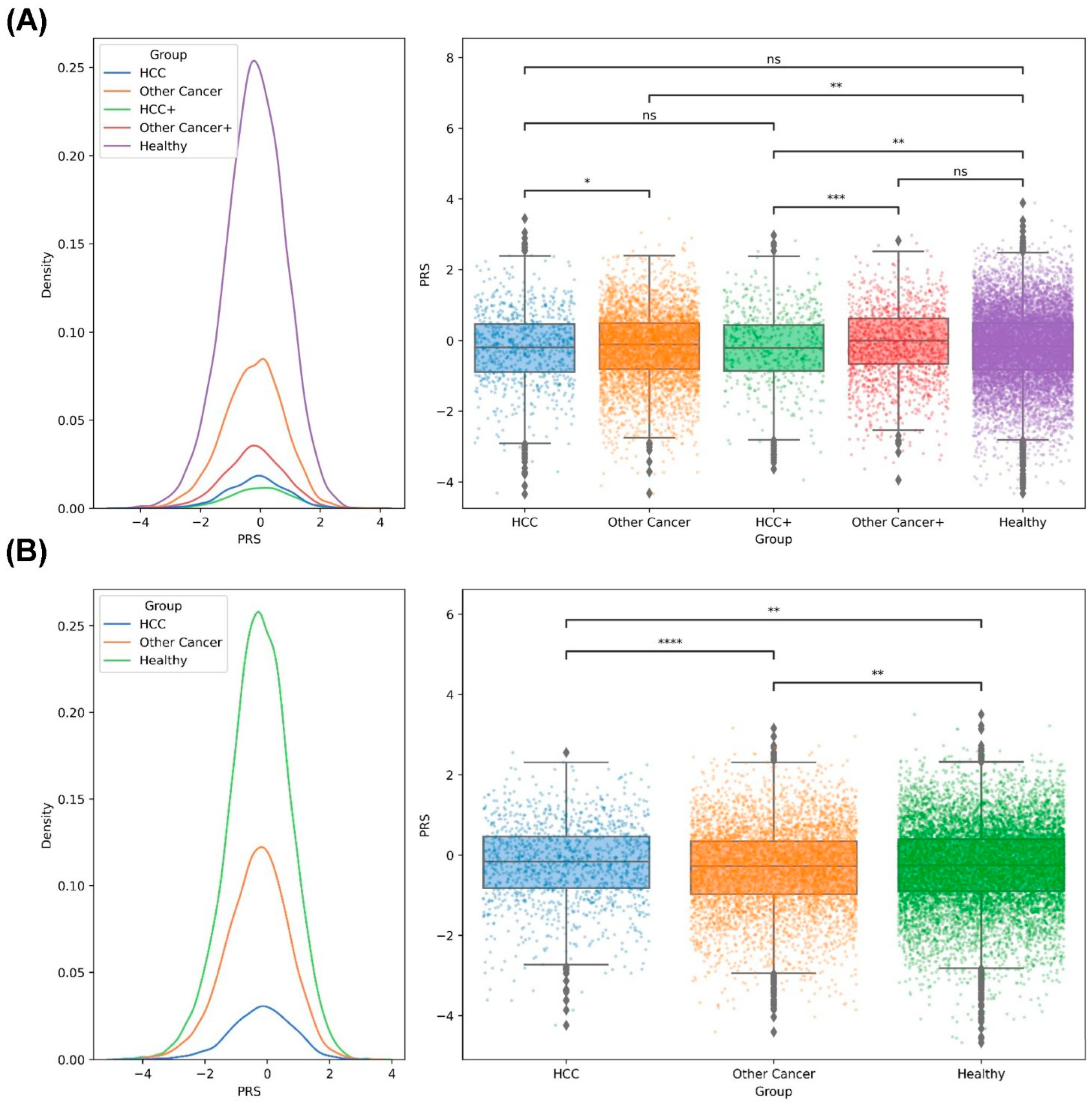
2.5. PRS Analysis of HCC in the Taiwanese Population
2.6. PRS Analysis of the Family Members of Taiwanese Patients with HCC
3. Methods and Materials
3.1. Participants and Cohorts
3.2. SNV Genotyping
3.3. Phasing and Imputation Workflow
3.4. MHC Class I and Class II Allele Imputation and Subtyping
3.5. GWAS
3.6. Phenome-Wide Association Study (PheWAS)
3.7. Meta-Analysis
3.8. Statistical Analysis
3.9. PRS Analysis
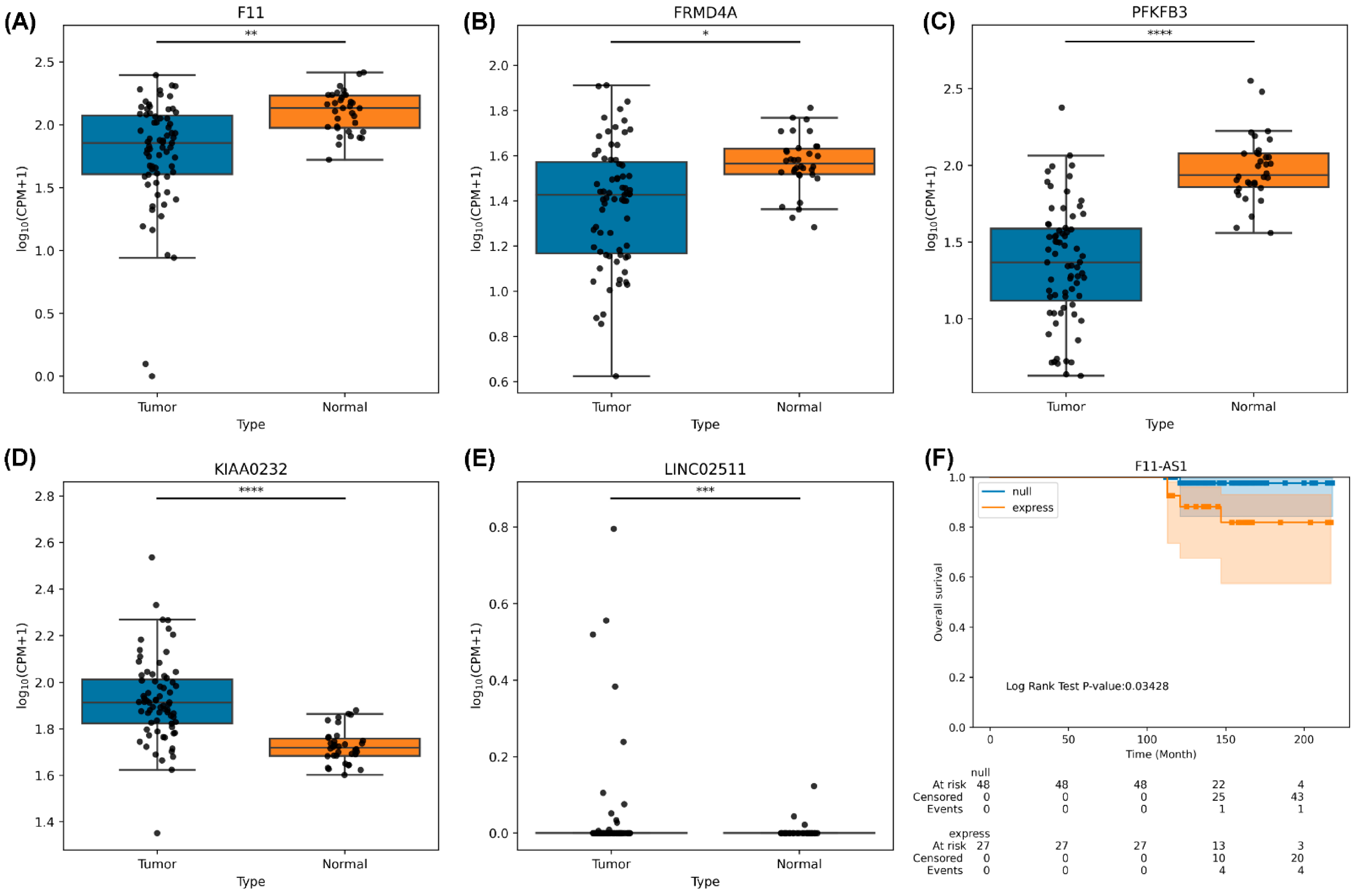
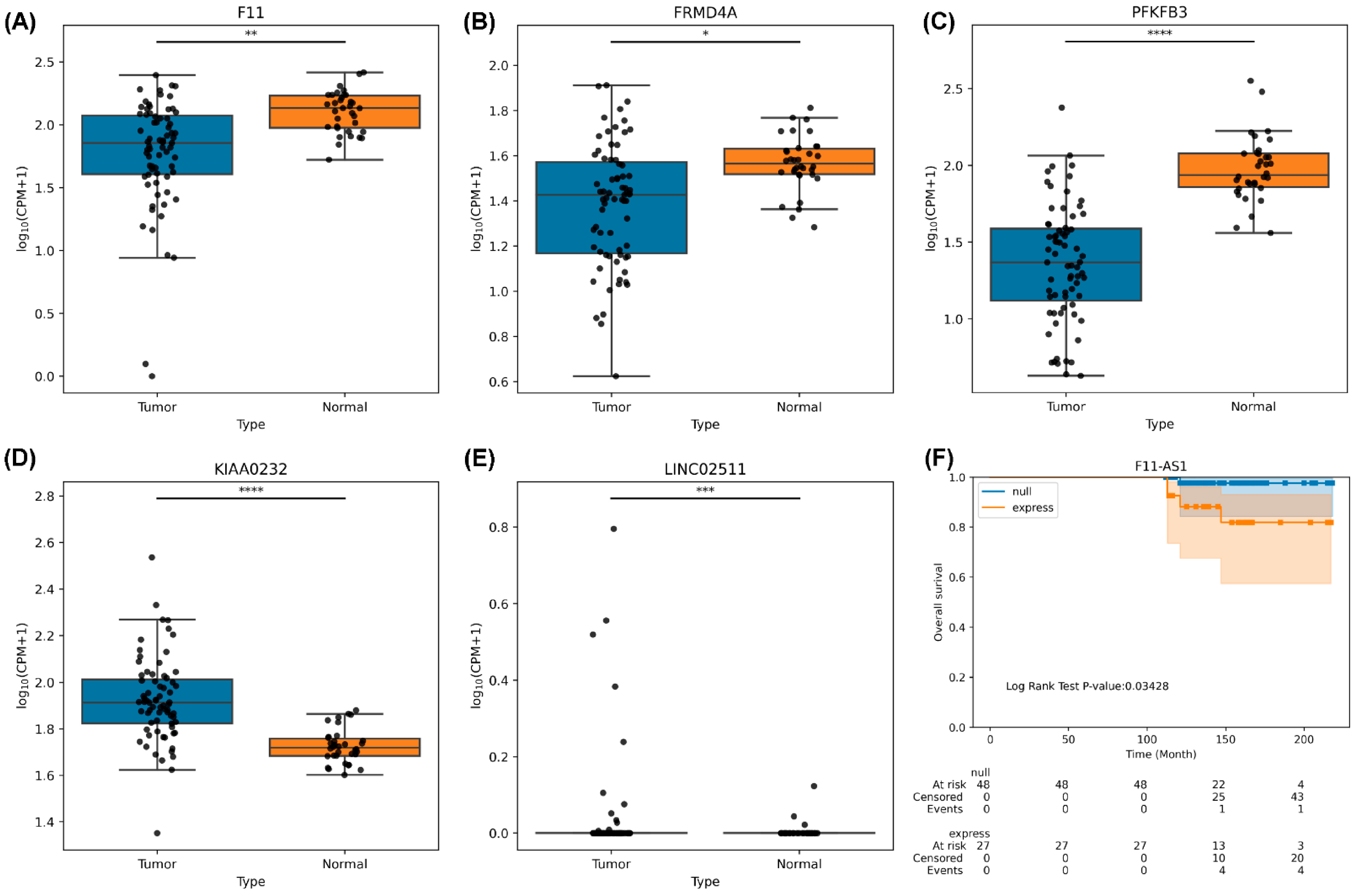
3.10. RNA Sequencing Analysis of HCC Tumor Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Area Under the Curve | (AUC) |
| Japan Biobank | (BBJ) |
| Body Mass Index | (BMI) |
| China Medical University Hospital | (CMUH) |
| Diabetes Mellitus | (DM) |
| Expression Quantitative Trait Loci | (eQTL) |
| Electronic Medical Record | (EMR) |
| Genome-wide association study | (GWAS) |
| Genome Research Consortium human build 38 | (hg38) |
| Hepatitis B Virus | (HBV) |
| Hepatitis C Virus | (HCV) |
| Hepatocellular Carcinoma | (HCC) |
| Human Leukocyte Antigen | (HLA) |
| Hardy–Weinberg equilibrium | (HWE) |
| Institutional Review Board | (IRB) |
| Linkage Disequilibrium | (LD) |
| Long Noncoding RNA | (lncRNA) |
| Minor Allele Frequency | (MAF) |
| Major Histocompatibility Complex | (MHC) |
| Nonalcoholic Fatty Liver Disease | (NAFLD) |
| Principal Component Analysis | (PCA) |
| Polygenic Risk Score | (PRS) |
| Single-Nucleotide Variant | (SNV) |
| Taiwan Biobank | (TWB) |
| Taiwan Precision Medicine Initiative | (TPMI) |
| Transcription Factor | (TF) |
| Whole Genome Sequence | (WGS) |
References
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. 1), 4–13. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Yu, M.W.; Lin, C.L.; Liu, C.J.; Yang, S.H.; Tseng, Y.L.; Wu, C.F. Influence of Metabolic Risk Factors on Risk of Hepatocellular Carcinoma and Liver-Related Death in Men With Chronic Hepatitis B: A Large Cohort Study. Gastroenterology 2017, 153, 1006–1017.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Yang, H.I. Natural history of chronic hepatitis B REVEALed. J. Gastroenterol. Hepatol. 2011, 26, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Nault, J.C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Letouze, E.; Shinde, J.; Renault, V.; Couchy, G.; Blanc, J.F.; Tubacher, E.; Bayard, Q.; Bacq, D.; Meyer, V.; Semhoun, J.; et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat. Commun. 2017, 8, 1315. [Google Scholar] [CrossRef]
- Villanueva, A.; Newell, P.; Hoshida, Y. Inherited hepatocellular carcinoma. Best. Pract. Res. Clin. Gastroenterol. 2010, 24, 725–734. [Google Scholar] [CrossRef]
- Turati, F.; Edefonti, V.; Talamini, R.; Ferraroni, M.; Malvezzi, M.; Bravi, F.; Franceschi, S.; Montella, M.; Polesel, J.; Zucchetto, A.; et al. Family history of liver cancer and hepatocellular carcinoma. Hepatology 2012, 55, 1416–1425. [Google Scholar] [CrossRef]
- Weledji, E.P. Familial hepatocellular carcinoma: ‘A model for studying preventive and therapeutic measures’. Ann. Med. Surg. 2018, 35, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Qian, J.; Yang, Y.; Zhao, W.; Dai, J.; Bei, J.X.; Foo, J.N.; McLaren, P.J.; Li, Z.; Yang, J.; et al. GWAS identifies novel susceptibility loci on 6p21.32 and 21q21.3 for hepatocellular carcinoma in chronic hepatitis B virus carriers. PLoS Genet. 2012, 8, e1002791. [Google Scholar] [CrossRef]
- Jiang, D.K.; Sun, J.; Cao, G.; Liu, Y.; Lin, D.; Gao, Y.Z.; Ren, W.H.; Long, X.D.; Zhang, H.; Ma, X.P.; et al. Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus-related hepatocellular carcinoma. Nat. Genet. 2013, 45, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhai, Y.; Song, Q.; Zhang, H.; Cao, P.; Ping, J.; Liu, X.; Guo, B.; Liu, G.; Song, J.; et al. Genome-Wide Association Study Identifies a New Locus at 7q21.13 Associated with Hepatitis B Virus-Related Hepatocellular Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.H.; Chen, C.J.; Lai, M.S.; Hsu, H.M.; Wu, T.C.; Kong, M.S.; Liang, D.C.; Shau, W.Y.; Chen, D.S. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N. Engl. J. Med. 1997, 336, 1855–1859. [Google Scholar] [CrossRef]
- Liao, S.H.; Chen, C.L.; Hsu, C.Y.; Chien, K.L.; Kao, J.H.; Chen, P.J.; Chen, T.H.; Chen, C.H. Long-term effectiveness of population-wide multifaceted interventions for hepatocellular carcinoma in Taiwan. J. Hepatol. 2021, 75, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Peng, S.F.; Huang, Y.P.; Tsai, C.H.; Tsai, F.J.; Huang, C.Y.; Tang, C.H.; Yang, J.S.; Hsu, Y.M.; Yin, M.C.; et al. Combinational treatment of all-trans retinoic acid (ATRA) and bisdemethoxycurcumin (BDMC)-induced apoptosis in liver cancer Hep3B cells. J. Food Biochem. 2020, 44, e13122. [Google Scholar] [CrossRef]
- Chang, W.S.; Tsai, C.W.; Yang, J.S.; Hsu, Y.M.; Shih, L.C.; Chiu, H.Y.; Bau, D.T.; Tsai, F.J. Resveratrol inhibited the metastatic behaviors of cisplatin-resistant human oral cancer cells via phosphorylation of ERK/p-38 and suppression of MMP-2/9. J. Food Biochem. 2021, 45, e13666. [Google Scholar] [CrossRef]
- Huang, S.F.; Chang, I.C.; Hong, C.C.; Yen, T.C.; Chen, C.L.; Wu, C.C.; Tsai, C.C.; Ho, M.C.; Lee, W.C.; Yu, H.C.; et al. Metabolic risk factors are associated with non-hepatitis B non-hepatitis C hepatocellular carcinoma in Taiwan, an endemic area of chronic hepatitis B. Hepatol. Commun. 2018, 2, 747–759. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Ishigaki, K.; Akiyama, M.; Kanai, M.; Takahashi, A.; Kawakami, E.; Sugishita, H.; Sakaue, S.; Matoba, N.; Low, S.-K.; Okada, Y.; et al. Large scale genome-wide association study in a Japanese population identified 45 novel susceptibility loci for 22 diseases. Nat. Genet. 2019, 52, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Chen, M.; Liu, J.; Hu, P.; Guan, H.; Jiao, X. LncRNA F11-AS1 suppresses liver hepatocellular carcinoma progression by competitively binding with miR-3146 to regulate PTEN expression. J. Cell Biochem. 2019, 120, 18457–18464. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wei, Z.; Huang, M.; Xu, G.; Wei, W.; Peng, B.; Nong, S.; Qin, H. Long non-coding RNA F11-AS1 inhibits HBV-related hepatocellular carcinoma progression by regulating NR1I3 via binding to microRNA-211-5p. J. Cell. Mol. Med. 2020, 24, 1848–1865. [Google Scholar] [CrossRef]
- Lei, G.L.; Li, Z.; Li, Y.Y.; Hong, Z.X.; Wang, S.; Bai, Z.F.; Sun, F.; Yan, J.; Yu, L.X.; Yang, P.H.; et al. Long noncoding RNA FAM66C promotes tumor progression and glycolysis in intrahepatic cholangiocarcinoma by regulating hsa-miR-23b-3p/KCND2 axis. Environ. Toxicol. 2021, 36, 2322–2332. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Cao, Y.; Yang, Z. C8B in Complement and Coagulation Cascades Signaling Pathway is a predictor for Survival in HBV-Related Hepatocellular Carcinoma Patients. Cancer Manag. Res. 2021, 13, 3503–3515. [Google Scholar] [CrossRef]
- Wang, R.; Li, M.; Wu, W.; Qiu, Y.; Hu, W.; Li, Z.; Wang, Z.; Yu, Y.; Liao, J.; Sun, W.; et al. NAV2 positively modulates inflammatory response of fibroblast-like synoviocytes through activating Wnt/β-catenin signaling pathway in rheumatoid arthritis. Clin. Transl. Med. 2021, 11, e376. [Google Scholar] [CrossRef]
- Goldie, S.J.; Mulder, K.W.; Tan, D.W.; Lyons, S.K.; Sims, A.H.; Watt, F.M. FRMD4A upregulation in human squamous cell carcinoma promotes tumor growth and metastasis and is associated with poor prognosis. Cancer Res. 2012, 72, 3424–3436. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.-C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.-A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef]
- Mejias, M.; Gallego, J.; Naranjo-Suarez, S.; Ramirez, M.; Pell, N.; Manzano, A.; Suñer, C.; Bartrons, R.; Mendez, R.; Fernandez, M. CPEB4 Increases Expression of PFKFB3 to Induce Glycolysis and Activate Mouse and Human Hepatic Stellate Cells, Promoting Liver Fibrosis. Gastroenterology 2020, 159, 273–288. [Google Scholar] [CrossRef]
- Oh, J.H.; Kim, Y.K.; Moon, S.; Kim, Y.J.; Kim, B.J. Genome-wide association study identifies candidate Loci associated with platelet count in koreans. Genom. Inf. 2014, 12, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsdottir, V.; Pedersen, H.K.; Allebrandt, K.V.; Brorsson, C.; van Leeuwen, N.; Banasik, K.; Mahajan, A.; Groves, C.J.; van de Bunt, M.; Dawed, A.Y.; et al. Integrative network analysis highlights biological processes underlying GLP-1 stimulated insulin secretion: A DIRECT study. PLoS ONE 2018, 13, e0189886. [Google Scholar] [CrossRef]
- Jeong, H.-C.; Park, S.-J.; Choi, J.-J.; Go, Y.-H.; Hong, S.-K.; Kwon, O.-S.; Shin, J.-G.; Kim, R.-K.; Lee, M.-O.; Lee, S.-J.; et al. PRMT8 Controls the Pluripotency and Mesodermal Fate of Human Embryonic Stem Cells By Enhancing the PI3K/AKT/SOX2 Axis. Stem Cells 2017, 35, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, S.; Dominko, T. Novel Protein Arginine Methyltransferase 8 Isoform Is Essential for Cell Proliferation. J. Cell Biochem. 2016, 117, 2056–2066. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qu, L.; Yang, Y.; Zhang, H.; Li, X.; Zhang, X. Single-cell Transcriptomic Architecture Unraveling the Complexity of Tumor Heterogeneity in Distal Cholangiocarcinoma. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1592–1609.e9. [Google Scholar] [CrossRef]
- Song, Y.; Qu, H.J. Identification and validation of a seven m6A-related lncRNAs signature predicting prognosis of ovarian cancer. BMC Cancer 2022, 22, 633. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Lian, Q.; Wang, S.; Zhang, G.; Wang, D.; Luo, G.; Tang, J.; Chen, L.; Gu, J. HCCDB: A Database of Hepatocellular Carcinoma Expression Atlas. Genom. Proteom. Bioinform. 2018, 16, 269–275. [Google Scholar] [CrossRef]
- Hong, Y.; Oh, S. Genome-wide association study of hepatitis in korean populations. Genom. Inf. 2014, 12, 203–207. [Google Scholar] [CrossRef]
- Liao, W.L.; Liu, T.Y.; Cheng, C.F.; Chou, Y.P.; Wang, T.Y.; Chang, Y.W.; Chen, S.Y.; Tsai, F.J. Analysis of HLA Variants and Graves’ Disease and Its Comorbidities Using a High Resolution Imputation System to Examine Electronic Medical Health Records. Front. Endocrinol. 2022, 13, 842673. [Google Scholar] [CrossRef]
- Lin, J.C.; Fan, C.T.; Liao, C.C.; Chen, Y.S. Taiwan Biobank: Making cross-database convergence possible in the Big Data era. Gigascience 2018, 7, gix110. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Delaneau, O.; Marchini, J.; Zagury, J.-F. A linear complexity phasing method for thousands of genomes. Nat. Methods 2012, 9, 179–181. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Liu, T.-Y.; Lin, C.-F.; Wu, H.-T.; Wu, Y.-L.; Chen, Y.-C.; Liao, C.-C.; Chou, Y.-P.; Chao, D.; Chang, Y.-S.; Lu, H.-F.; et al. Comparison of multiple imputation algorithms and verification using whole-genome sequencing in the CMUH genetic biobank. Biomedicine 2021, 11, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Shen, J.; Cox, C.; Wakefield, J.C.; Ehm, M.G.; Nelson, M.R.; Weir, B.S. HIBAG—HLA genotype imputation with attribute bagging. Pharmacogenomics J. 2014, 14, 192–200. [Google Scholar] [CrossRef]
- Lu, H.F.; Liu, T.Y.; Chou, Y.P.; Chang, S.S.; Hsieh, Y.W.; Chang, J.G.; Tsai, F.J. Comprehensive characterization of pharmacogenes in a Taiwanese Han population. Front. Genet. 2022, 13, 948616. [Google Scholar] [CrossRef] [PubMed]
- Boughton, A.P.; Welch, R.P.; Flickinger, M.; VandeHaar, P.; Taliun, D.; Abecasis, G.R.; Boehnke, M. LocusZoom.js: Interactive and embeddable visualization of genetic association study results. Bioinformatics 2021, 37, 3017–3018. [Google Scholar] [CrossRef]
- Carroll, R.J.; Bastarache, L.; Denny, J.C. R PheWAS: Data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 2014, 30, 2375–2376. [Google Scholar] [CrossRef]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef]
- Okada, Y.; Momozawa, Y.; Sakaue, S.; Kanai, M.; Ishigaki, K.; Akiyama, M.; Kishikawa, T.; Arai, Y.; Sasaki, T.; Kosaki, K.; et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 2018, 9, 1631. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Li, Y.; Abecasis, G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef]
- Liu, T.-Y.; Hsu, H.-Y.; You, Y.-S.; Hsieh, Y.-W.; Lin, T.-C.; Peng, C.-W.; Huang, H.-Y.; Chang, S.-S.; Tsai, F.-J. Efficacy of Warfarin Therapy Guided by Pharmacogenetics: A Real-World Investigation Among Han Taiwanese. Clin. Ther. 2023, 45, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; O’Reilly, P.F. PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience 2019, 8, giz082. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.Y.; Yu, M.W.; Lin, S.M.; Lee, S.D.; Chen, C.L.; Chen, D.S.; Chen, P.J. Genome-wide association analysis identifies a GLUL haplotype for familial hepatitis B virus-related hepatocellular carcinoma. Cancer 2017, 123, 3966–3976. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.C.; Chan, C.; Huang, Y.H.; Chen, Y.J.; Liao, S.F.; Lin, Y.J.; Huang, C.; Lu, S.N.; Jen, C.L.; Wang, L.Y. Genetic variants associated with serum alanine aminotransferase levels among patients with hepatitis C virus infection: A genome-wide association study. J. Viral Hepat. 2021, 28, 1265–1273. [Google Scholar] [CrossRef]
- Schwantes-An, T.H.; Darlay, R.; Mathurin, P.; Masson, S.; Liangpunsakul, S.; Mueller, S.; Aithal, G.P.; Eyer, F.; Gleeson, D.; Thompson, A.; et al. Genome-wide Association Study and Meta-analysis on Alcohol-Associated Liver Cirrhosis Identifies Genetic Risk Factors. Hepatology 2021, 73, 1920–1931. [Google Scholar] [CrossRef]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef]
- Bau, D.-T.; Liu, T.-Y.; Tsai, C.-W.; Chang, W.-S.; Gu, J.; Yang, J.-S.; Shih, L.-C.; Tsai, F.-J. A Genome-Wide Association Study Identified Novel Genetic Susceptibility Loci for Oral Cancer in Taiwan. Int. J. Mol. Sci. 2023, 24, 2789. [Google Scholar] [CrossRef]
- Hu, L.; Zhai, X.; Liu, J.; Chu, M.; Pan, S.; Jiang, J.; Zhang, Y.; Wang, H.; Chen, J.; Shen, H.; et al. Genetic variants in human leukocyte antigen/DP-DQ influence both hepatitis B virus clearance and hepatocellular carcinoma development. Hepatology 2012, 55, 1426–1431. [Google Scholar] [CrossRef]
- Zeng, Z.; Liu, H.; Xu, H.; Lu, H.; Yu, Y.; Xu, X.; Yu, M.; Zhang, T.; Tian, X.; Xi, H.; et al. Genome-wide association study identifies new loci associated with risk of HBV infection and disease progression. BMC Med. Genom. 2021, 14, 84. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Sawai, H.; Matsuura, K.; Sugiyama, M.; Ahn, S.H.; Park, J.Y.; Hige, S.; Kang, J.H.; Suzuki, K.; Kurosaki, M.; et al. Genome-wide association study confirming association of HLA-DP with protection against chronic hepatitis B and viral clearance in Japanese and Korean. PLoS ONE 2012, 7, e39175. [Google Scholar] [CrossRef]
- Sawai, H.; Nishida, N.; Khor, S.S.; Honda, M.; Sugiyama, M.; Baba, N.; Yamada, K.; Sawada, N.; Tsugane, S.; Koike, K.; et al. Genome-wide association study identified new susceptible genetic variants in HLA class I region for hepatitis B virus-related hepatocellular carcinoma. Sci. Rep. 2018, 8, 7958. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Huang, X.; Chen, J.; Cai, Y.; Xiong, L.; Mu, L.; Zhou, L. Host Genetic Variants in HLA Loci Influence Risk for Hepatitis B Virus Infection in Children. Hepat. Mon. 2016, 16, e37786. [Google Scholar] [CrossRef]
- Li, H.; Feng, B.; Miron, A.; Chen, X.; Beesley, J.; Bimeh, E.; Barrowdale, D.; John, E.M.; Daly, M.B.; Andrulis, I.L.; et al. Breast cancer risk prediction using a polygenic risk score in the familial setting: A prospective study from the Breast Cancer Family Registry and kConFab. Genet. Med. 2017, 19, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, E.; May, P.; Aldisi, R.; Spier, I.; Forstner, A.J.; Nöthen, M.M.; Aretz, S.; Krawitz, P.; Bobbili, D.R.; Maj, C. Breast and prostate cancer risk: The interplay of polygenic risk, rare pathogenic germline variants, and family history. Genet. Med. 2022, 24, 576–585. [Google Scholar] [CrossRef]
- Tarao, K.; Nozaki, A.; Ikeda, T.; Sato, A.; Komatsu, H.; Komatsu, T.; Taguri, M.; Tanaka, K. Real impact of liver cirrhosis on the development of hepatocellular carcinoma in various liver diseases-meta-analytic assessment. Cancer Med. 2019, 8, 1054–1065. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Case | Control | p-Value c | ||
|---|---|---|---|---|---|
| Demography | |||||
| Sex | male, n (%) | 1854 (65.37%) | 54,622 (40.60%) | 3.9 × 10−155 | |
| female, n (%) | 970 (34.20%) | 79234 (58.89%) | |||
| Unknown, n (%) | 12 (0.42%) | 693 (0.52%) | |||
| Age | year, mean (SD) | 65.5 ± 12.6 | 51.4 ± 17.8 b | 0.0 × 10+00 | |
| BMI | kg/m2, mean (SD) | 27.2 ± 5.4 | 25.0 ± 4.7 | 4.6 × 10−118 | |
| Unknown, n (%) | 160 (5.64%) | 19,104 (14.20%) | |||
| Liver Disease | |||||
| Cirrhosis a | n (%) | 1185 (41.78%) | 93 (0.07%) | 0.0 × 10+00 | |
| Unknown, n (%) | 1651 (58.22%) | 134,456 (99.93%) | |||
| Virus Infection | HBVsAg(+), n (%) | 1239 (43.69%) | 8503 (6.31%) | 0.0 × 10+00 | |
| HCV(+), n (%) | 707 (24.93%) | 2118 (1.57%) | 0.0 × 10+00 | ||
| Unknown, n (%) | 246 (8.67%) | 44,558 (33.12%) | |||
| HBVsAg(+) HCV(+), n (%) d | 118 (4.16%) | 211 (0.16%) | |||
| HBVsAg(−) HCV(−), n (%) | 761 (26.83%) | 75,528 (56.13%) | |||
| Metabolism | |||||
| Diabetes | Type II diabetes, n (%) | 979 (34.52%) | 12461 (9.26%) | 0.0 × 10+00 | |
| HBVsAg(+), n (%) | 139 (4.90%) | 285 (0.21%) | |||
| HCV(+), n (%) | 294 (10.37%) | 205 (0.15%) | |||
| HBVsAg(+) HCV(+), n (%) | 22 (0.78%) | 5 (0.00%) | |||
| diabetes (others), n (%) | 0 (0.00%) | 246 (0.18%) | |||
| Non-diabetes, n (%) | 1697 (59.84%) | 102,747 (76.36%) | |||
| Unknown, n (%) | 160 (5.64%) | 19,095 (14.19%) | |||
| Total | 2836 | 134,549 | |||
| Marker | Variant | MAF | Discovery | Nearest Gene | |||||
|---|---|---|---|---|---|---|---|---|---|
| Chr | Position | RA/EA | PAF a (%) | Case (AF, %) | Control (AF, %) | OR (95% CI) | p-Value b | ||
| rs187199523 | 1 | 194027489 | A/T | 2.48 | 5672 (4.60) | 283,510 (3.05) | 1.49 (1.30–1.69) | 2.17 × 10−9 | RP11-563D10.1 (ENSG00000227240) * |
| rs140233124 | 4 | 6834347 | A/- | 6.05 | 5664 (7.20) | 257,060 (4.91) | 1.40 (1.26–1.55) | 4.34×10−10 | KIAA0232 * |
| rs144285059 | 4 | 136895711 | -/A | 4.29 | 5672 (4.76) | 272,166 (3.19) | 1.44 (1.27–1.64) | 1.99 × 10−8 | LINC02511 |
| rs148610742 | 4 | 186288789 | C/T | 3.22 | 5668 (3.42) | 276,154 (2.12) | 1.54 (1.33–1.79) | 1.43 × 10−8 | F11 */F11-AS1 * |
| rs118180127 | 8 | 8513430 | T/A | 5.31 | 4894 (1.94) | 260,472 (3.90) | 0.55 (0.44–0.67) | 7.30 × 10−9 | CTD-3023L14.3 (ENSG00000253343) |
| rs117719091 | 10 | 6227313 | C/T | 4.05 | 4932 (0.85) | 264,072 (2.66) | 0.40 (0.29–0.54) | 3.02 × 10−9 | PFKFB3 * |
| rs17155112 | 10 | 14357172 | G/A | 2.77 | 5526 (2.01) | 273,888 (1.22) | 1.74 (1.43–2.12) | 3.02 × 10−8 | FRMD4A * |
| rs77404202 | 11 | 20117743 | C/T | 3.87 | 5144 (1.32) | 268,176 (2.91) | 0.50 (0.39–0.64) | 2.46 × 10−8 | NAV2 */NAV2-AS1 |
| rs144225287 | 12 | 3568611 | G/- | 4.01 | 5668 (5.51) | 269,502 (3.59) | 1.46 (1.29–1.64) | 5.27 × 10−10 | PRMT8 |
| rs150098717 | 12 | 8198462 | C/T | 3.43 | 5040 (0.83) | 266,258 (2.25) | 0.41 (0.30–0.56) | 1.81 × 10−8 | FAM66C/DEFB109F |
| rs74333160 | 14 | 101184577 | T/G | 5.21 | 5664 (6.94) | 257,812 (4.51) | 1.47 (1.32–1.64) | 1.42 × 10−12 | AL355836.4 (ENSG00000288245) |
| rs80115676 | 17 | 17375355 | A/G | 6.25 | 5014 (1.76) | 264,032 (3.61) | 0.53 (0.43–0.66) | 6.34 × 10−9 | RPL13P12 * |
| rs6140450 | 20 | 7873320 | T/C | 5.11 | 5016 (1.42) | 264,980 (3.14) | 0.51 (0.40–0.65) | 2.79 × 10−8 | RP1-209B9.2 (ENSG00000277315) |
| Trait | Risk | Protect | ||||
|---|---|---|---|---|---|---|
| HLA-Type | OR (95% CI) | p-Value a | HLA-Type | OR (95% CI) | p-Value a | |
| HCC | DQA1*04:01 | 1.71 (1.22–2.46) | 3.30 × 10−3 | B*54:01 | 0.72 (0.61–0.85) | 4.19 × 10−3 |
| DQB1*04:02 | 1.69 (1.21–2.45) | 4.33 × 10−3 | DRB1*14:54 | 0.72 (0.62–0.84) | 2.22 × 10−4 | |
| DRB1*06:09 | 1.53 (1.21–1.96) | 5.75 × 10−4 | DQA1*06:01 | 0.74 (0.68–0.80) | 1.16 × 10−10 | |
| DRB1*13:02 | 1.52 (1.21–1.94) | 8.28 × 10−4 | DRB1*12:01 | 0.75 (0.65–0.87) | 9.88 × 10−4 | |
| DPB1*04:02 | 1.41 (1.05–1.92) | 9.03 × 10−2 | DRB1*12:02 | 0.78 (0.71–0.85) | 7.90 × 10−7 | |
| DQB1*06:02 | 1.38 (1.17–1.63) | 4.08 × 10−4 | B*38:02 | 0.81 (0.70–0.94) | 8.79 × 10−2 | |
| DQB1*03:02 | 1.35 (1.21–1.51) | 4.43 × 10−7 | DQB1*03:01 | 0.81 (0.76–0.86) | 4.34 × 10−10 | |
| DQA1*03:01 | 1.29 (1.14–1.47) | 2.66 × 10−4 | DQA1*01:04 | 0.85 (0.77–0.95) | 8.45 × 10−3 | |
| DRB1*15:01 | 1.24 (1.10–1.40) | 9.61 × 10−4 | DPB1*05:01 | 0.87 (0.82–0.92) | 5.21 × 10−5 | |
| DQA1*01:02 | 1.17 (1.08–1.26) | 3.35 × 10−4 | ||||
| HBV infection | DRB1*13:01 | 5.45 (3.14–10.35) | 2.21 × 10−14 | DPB1*05:01 | 0.69 (0.67–0.72) | 1.56 × 10−102 |
| DQB1*06:03 | 4.60 (2.53–9.34) | 9.95 × 10−10 | DRB1*14:54 | 0.70 (0.64–0.76) | 1.48 × 10−16 | |
| DRB1*13:02 | 3.48 (2.93–4.17) | 1.12 × 10−63 | DQA1*06:01 | 0.73 (0.69–0.76) | 5.18 × 10−38 | |
| DQB1*06:09 | 3.45 (2.90–4.14) | 1.30 × 10−62 | DPA1*02:02 | 0.73 (0.71–0.76) | 8.94 × 10−88 | |
| B*44:03 | 2.33 (1.46–3.95) | 6.49 × 10−4 | DPB1*19:01 | 0.76 (0.67–0.86) | 2.09 × 10−05 | |
| DPB1*09:01 | 2.09 (1.72–2.56) | 8.39 × 10−16 | DQB1*03:01 | 0.76 (0.74–0.79) | 2.22 × 10−54 | |
| DPB1*17:01 | 1.90 (1.54–2.37) | 1.45 × 10−10 | DPB1*13:01 | 0.77 (0.72–0.82) | 3.61 × 10−15 | |
| DQB1*03:02 | 1.89 (1.77–2.03) | 6.72 × 10−85 | DRB1*12:02 | 0.78 (0.74–0.82) | 5.66 × 10−23 | |
| DRB1*01:01 | 1.84 (1.37–2.52) | 3.11 × 10−5 | DQA1*01:04 | 0.81 (0.76–0.85) | 6.36 × 10−14 | |
| DQA1*03:01 | 1.80 (1.67–1.95) | 2.22 × 10−57 | DQB1*03:03 | 0.82 (0.79–0.86) | 7.67 × 10−22 | |
| HCV infection | C*07:04 | 1.72 (1.12–2.78) | 5.90 × 10−2 | DPB1*104:01 | 0.48 (0.27–0.91) | 8.26 × 10−2 |
| C*08:01 | 1.14 (1.03–1.26) | 7.51 × 10−2 | DRB1*07:01 | 0.81 (0.70–0.95) | 7.87 × 10−2 | |
| DPB1*13:01 | 1.13 (1.02–1.25) | 7.41 × 10−2 | DRB1*13:02 | 0.82 (0.71–0.95) | 5.97 × 10−2 | |
| DQB1*03:01 | 1.10 (1.04–1.16) | 2.12 × 10−3 | DQB1*06:09 | 0.83 (0.72–0.97) | 7.61 × 10−2 | |
| DPB1*05:01 | 1.06 (1.01–1.12) | 7.73 × 10−2 | DRB1*03:01 | 0.86 (0.79–0.93) | 4.01 × 10−3 | |
| DPA1*02:02 | 1.04 (1.00–1.09) | 9.04 × 10−2 | DQB1*02:01 | 0.87 (0.81–0.94) | 4.22 × 10−3 | |
| C*03:02 | 0.87 (0.82–0.93) | 1.77 × 10−3 | ||||
| DQA1*05:01 | 0.88 (0.82–0.95) | 2.32 × 10−2 | ||||
| B*58:01 | 0.89 (0.82–0.96) | 4.78 × 10−2 | ||||
| DPB1*02:02 | 0.90 (0.82–0.99) | 9.99 × 10−2 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.-Y.; Liao, C.-C.; Chang, Y.-S.; Chen, Y.-C.; Chen, H.-D.; Lai, I.-L.; Peng, C.-Y.; Chung, C.-C.; Chou, Y.-P.; Tsai, F.-J.; et al. Identification of 13 Novel Loci in a Genome-Wide Association Study on Taiwanese with Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 16417. https://doi.org/10.3390/ijms242216417
Liu T-Y, Liao C-C, Chang Y-S, Chen Y-C, Chen H-D, Lai I-L, Peng C-Y, Chung C-C, Chou Y-P, Tsai F-J, et al. Identification of 13 Novel Loci in a Genome-Wide Association Study on Taiwanese with Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2023; 24(22):16417. https://doi.org/10.3390/ijms242216417
Chicago/Turabian StyleLiu, Ting-Yuan, Chi-Chou Liao, Ya-Sian Chang, Yu-Chia Chen, Hong-Da Chen, I-Lu Lai, Cheng-Yuan Peng, Chin-Chun Chung, Yu-Pao Chou, Fuu-Jen Tsai, and et al. 2023. "Identification of 13 Novel Loci in a Genome-Wide Association Study on Taiwanese with Hepatocellular Carcinoma" International Journal of Molecular Sciences 24, no. 22: 16417. https://doi.org/10.3390/ijms242216417
APA StyleLiu, T.-Y., Liao, C.-C., Chang, Y.-S., Chen, Y.-C., Chen, H.-D., Lai, I.-L., Peng, C.-Y., Chung, C.-C., Chou, Y.-P., Tsai, F.-J., Jeng, L.-B., & Chang, J.-G. (2023). Identification of 13 Novel Loci in a Genome-Wide Association Study on Taiwanese with Hepatocellular Carcinoma. International Journal of Molecular Sciences, 24(22), 16417. https://doi.org/10.3390/ijms242216417