
PROTAC-Based Protein Degradation as a Promising Strategy for Targeted Therapy in Sarcomas




Abstract
:1. Introduction
2. Fundamentals of Targeted Protein Degradation
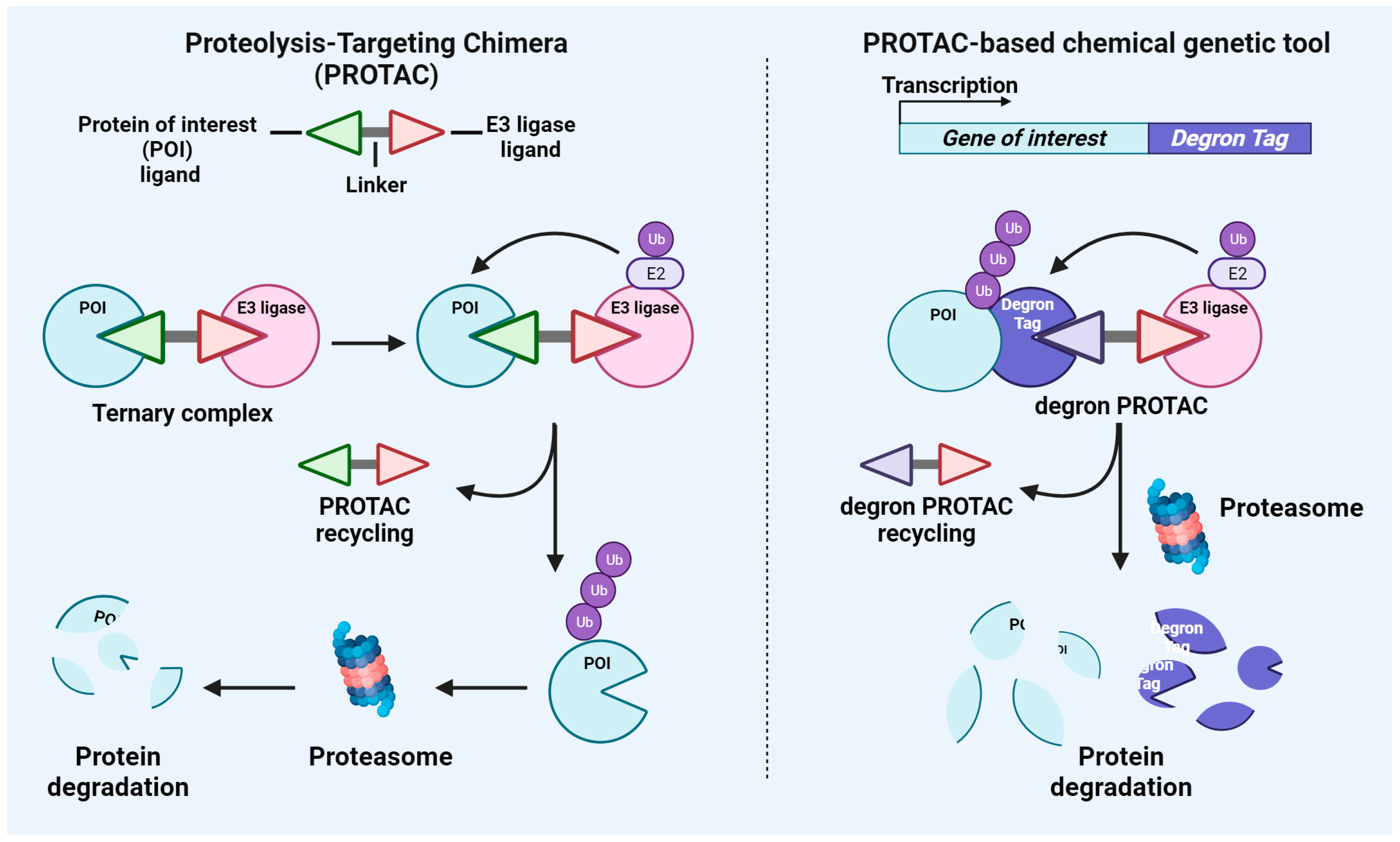
2.1. PROTAC
2.2. PROTAC-Based Chemical Genetic Tool
3. Can Targeted Protein Degradation Overcome Major Challenges in Cancer Treatment?
3.1. Targeted Protein Degradation and Undruggable Cancer Targets
3.2. Targeted Protein Degradation and Drug Resistance
3.3. Targeted Protein Degradation and Target Specificity
4. Perspectives for the Use of Targeted Protein Degradation-Based Strategies in Sarcoma Treatment
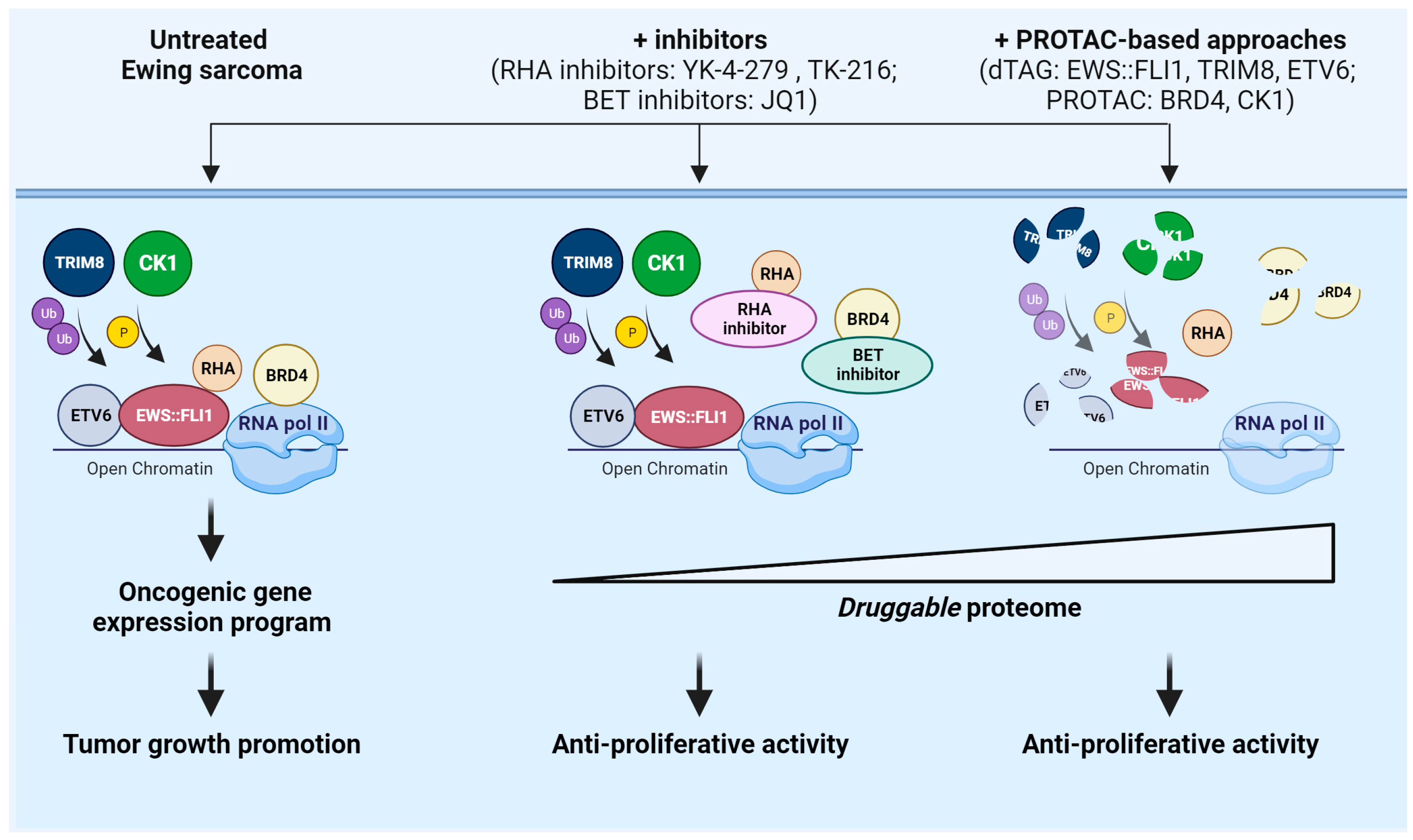
4.1. Targeting the Undruggable EWS::FLI1 in Ewing Sarcoma
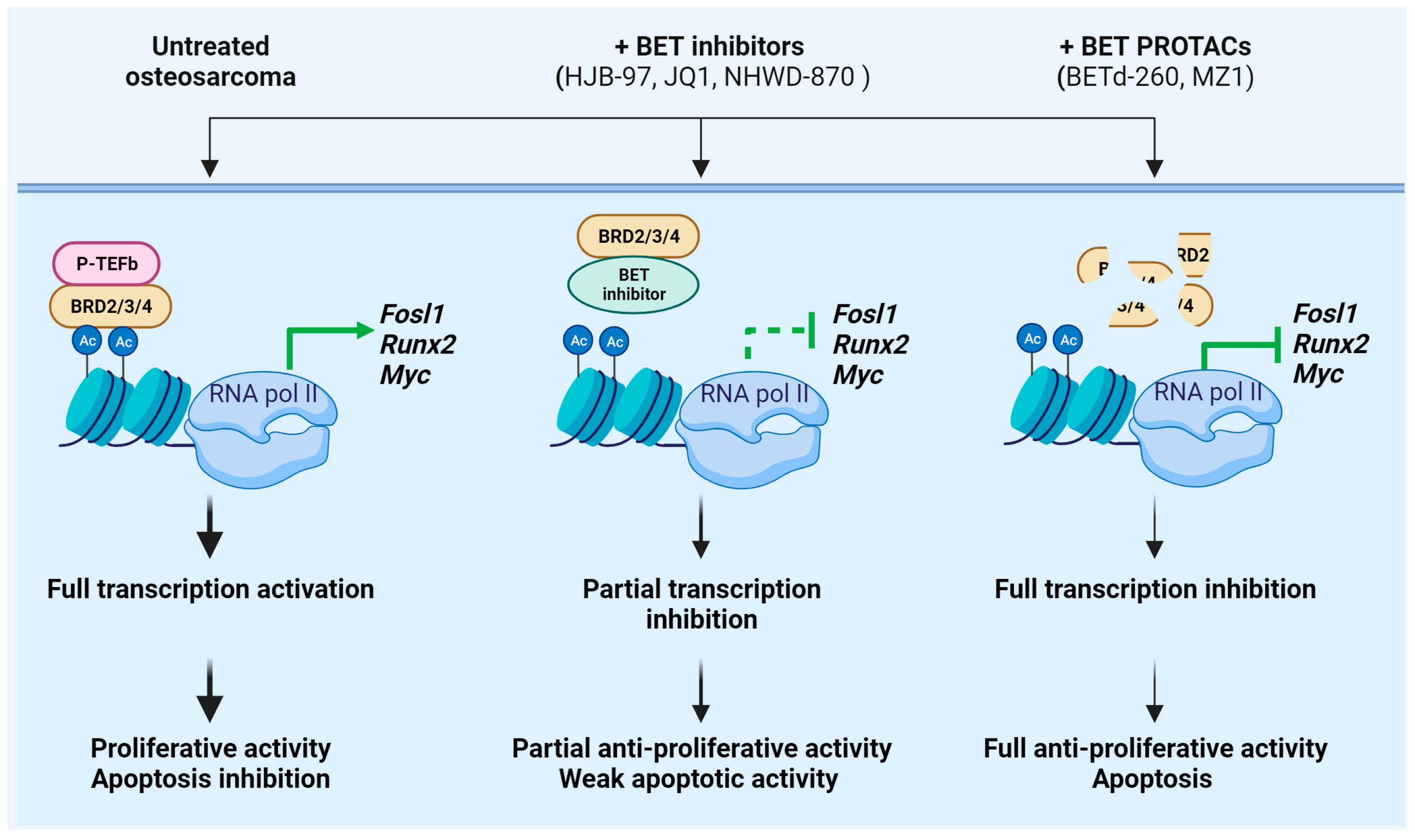
4.2. Overcoming the Resistance to BET Protein Inhibition in Osteosarcoma
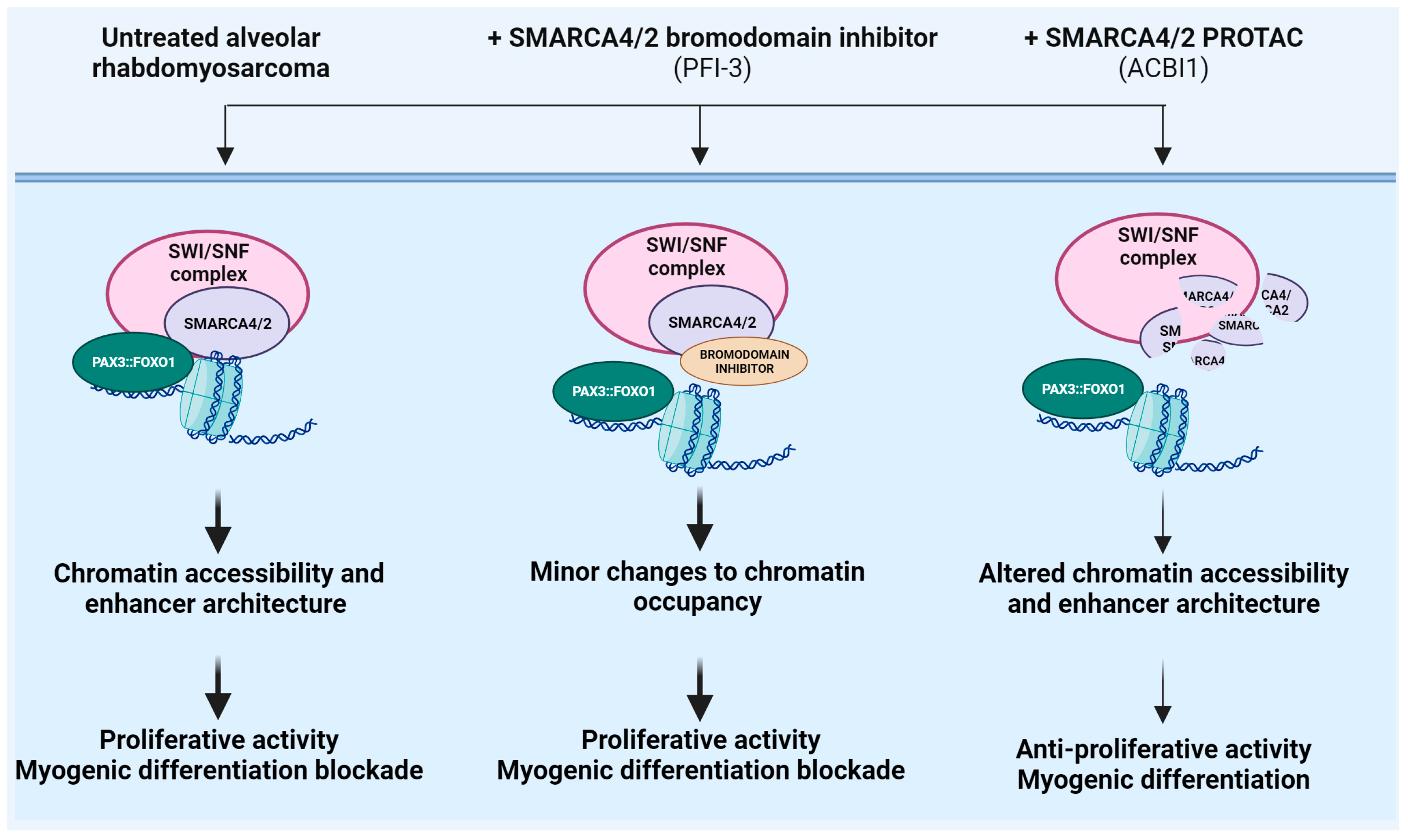
4.3. Enhancing Target Selectivity to SMARCA4/A2 in Rhabdomyosarcoma
4.4. Maximizing BRD9 Blockade in Synovial Sarcoma
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Choi, J.H.; Ro, J.Y. The 2020 WHO Classification of Tumors of Bone: An Updated Review. Adv. Anat. Pathol. 2021, 28, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Ro, J.Y. The 2020 WHO Classification of Tumors of Soft Tissue: Selected Changes and New Entities. Adv. Anat. Pathol. 2021, 28, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.Y.; Hindi, N.; Bollard, J.; Aguiar, S., Jr.; Angel, M.; Araya, B.; Badilla, R.; Bernabeu, D.; Campos, F.; Caro-Sanchez, C.H.S.; et al. SELNET clinical practice guidelines for soft tissue sarcoma and GIST. Cancer Treat. Rev. 2022, 102, 102312. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; Di Pompo, G.; Distel, M.; Dorado-Garcia, H.; Garcia-Castro, J.; et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol. Med. 2020, 12, e11131. [Google Scholar] [CrossRef] [PubMed]
- Wallander, K.; Ofverholm, I.; Boye, K.; Tsagkozis, P.; Papakonstantinou, A.; Lin, Y.; Haglund de Flon, F. Sarcoma care in the era of precision medicine. J. Intern. Med. 2023, 294, 690–707. [Google Scholar] [CrossRef]
- Gronchi, A.; Palmerini, E.; Quagliuolo, V.; Martin Broto, J.; Lopez Pousa, A.; Grignani, G.; Brunello, A.; Blay, J.Y.; Tendero, O.; Diaz Beveridge, R.; et al. Neoadjuvant Chemotherapy in High-Risk Soft Tissue Sarcomas: Final Results of a Randomized Trial From Italian (ISG), Spanish (GEIS), French (FSG), and Polish (PSG) Sarcoma Groups. J. Clin. Oncol. 2020, 38, 2178–2186. [Google Scholar] [CrossRef]
- Tang, F.; Tie, Y.; Wei, Y.Q.; Tu, C.Q.; Wei, X.W. Targeted and immuno-based therapies in sarcoma: Mechanisms and advances in clinical trials. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188606. [Google Scholar] [CrossRef]
- Sourrouille, I.; Macovei, R.; Faron, M.; Le Pechoux, C.; Mir, O.; Adam, J.; Dumont, S.; Terrier, P.; Le Cesne, A.; Honore, C. Long-Term Outcome After Surgery for a Localized Retroperitoneal Soft Tissue Sarcoma in Elderly Patients: Results from a Retrospective, Single-Center Study. Ann. Surg. Oncol. 2018, 25, 2201–2208. [Google Scholar] [CrossRef]
- Dufresne, A.; Brahmi, M.; Karanian, M.; Blay, J.Y. Using biology to guide the treatment of sarcomas and aggressive connective-tissue tumours. Nat. Rev. Clin. Oncol. 2018, 15, 443–458. [Google Scholar] [CrossRef]
- Bekes, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Kelm, J.M.; Pandey, D.S.; Malin, E.; Kansou, H.; Arora, S.; Kumar, R.; Gavande, N.S. PROTAC’ing oncoproteins: Targeted protein degradation for cancer therapy. Mol. Cancer 2023, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mullin-Bernstein, Z.; Oliver, S.; Skipper, T.A.; Atack, T.C.; Bick, N.; Ching, M.; Guirguis, A.A.; Kwon, J.; Langan, C.; et al. Systematic profiling of conditional degron tag technologies for target validation studies. Nat. Commun. 2022, 13, 5495. [Google Scholar] [CrossRef] [PubMed]
- Critchley, W.R.; Pellet-Many, C.; Ringham-Terry, B.; Harrison, M.A.; Zachary, I.C.; Ponnambalam, S. Receptor Tyrosine Kinase Ubiquitination and De-Ubiquitination in Signal Transduction and Receptor Trafficking. Cells 2018, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Henneberg, L.T.; Schulman, B.A. Decoding the messaging of the ubiquitin system using chemical and protein probes. Cell Chem. Biol. 2021, 28, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Suzuki, T.; Chiba, T. The ligation systems for ubiquitin and ubiquitin-like proteins. Mol. Cells 1998, 8, 503–512. [Google Scholar]
- Dang, F.; Nie, L.; Wei, W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021, 28, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, C.; Chang, L.; Zhang, Z.; Yang, J.; Maslen, S.; Skehel, M.; Barford, D. Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 2016, 536, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Werner, A.; Iwasaki, S.; McGourty, C.A.; Medina-Ruiz, S.; Teerikorpi, N.; Fedrigo, I.; Ingolia, N.T.; Rape, M. Cell-fate determination by ubiquitin-dependent regulation of translation. Nature 2015, 525, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Davidson, K.; Pickering, A.M. The proteasome: A key modulator of nervous system function, brain aging, and neurodegenerative disease. Front. Cell Dev. Biol. 2023, 11, 1124907. [Google Scholar] [CrossRef]
- Gu, J.; Pang, L.; Yan, D.; Wang, C.; Song, Y.; Jin, Z.; Xu, Z.; Mao, Y.; Liu, S.; Chen, S. Ubiquitin-proteasome system-mediated ubiquitination modification patterns and characterization of tumor microenvironment infiltration, stemness and cellular senescence in low-grade glioma. Aging 2023, 15, 2970–2998. [Google Scholar] [CrossRef]
- Jiao, J.; Curley, M.; Graca, F.A.; Robles-Murguia, M.; Shirinifard, A.; Finkelstein, D.; Xu, B.; Fan, Y.; Demontis, F. Modulation of protease expression by the transcription factor Ptx1/PITX regulates protein quality control during aging. Cell Rep. 2023, 42, 111970. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.J.; Rahimi, N.; Tadi, P. Biochemistry, Ubiquitination. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Dewey, J.A.; Delalande, C.; Azizi, S.A.; Lu, V.; Antonopoulos, D.; Babnigg, G. Molecular Glue Discovery: Current and Future Approaches. J. Med. Chem. 2023, 66, 9278–9296. [Google Scholar] [CrossRef] [PubMed]
- Ruffilli, C.; Roth, S.; Rodrigo, M.; Boyd, H.; Zelcer, N.; Moreau, K. Proteolysis Targeting Chimeras (PROTACs): A Perspective on Integral Membrane Protein Degradation. ACS Pharmacol. Transl. Sci. 2022, 5, 849–858. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Weng, G.; Cai, X.; Cao, D.; Du, H.; Shen, C.; Deng, Y.; He, Q.; Yang, B.; Li, D.; Hou, T. PROTAC-DB 2.0: An updated database of PROTACs. Nucleic Acids Res. 2023, 51, D1367–D1372. [Google Scholar] [CrossRef]
- Weng, G.; Shen, C.; Cao, D.; Gao, J.; Dong, X.; He, Q.; Yang, B.; Li, D.; Wu, J.; Hou, T. PROTAC-DB: An online database of PROTACs. Nucleic Acids Res. 2021, 49, D1381–D1387. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.; Jin, M.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K.; et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Yamamoto, J.; Suwa, T.; Murase, Y.; Tateno, S.; Mizutome, H.; Asatsuma-Okumura, T.; Shimizu, N.; Kishi, T.; Momose, S.; Kizaki, M.; et al. ARID2 is a pomalidomide-dependent CRL4(CRBN) substrate in multiple myeloma cells. Nat. Chem. Biol. 2020, 16, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Flaxman, H.A.; Xu, W.; Vallavoju, N.; Lloyd, H.C.; Wang, B.; Shen, D.; Pratt, M.R.; Woo, C.M. The E3 ligase adapter cereblon targets the C-terminal cyclic imide degron. Nature 2022, 610, 775–782. [Google Scholar] [CrossRef]
- Cardote, T.A.F.; Gadd, M.S.; Ciulli, A. Crystal Structure of the Cul2-Rbx1-EloBC-VHL Ubiquitin Ligase Complex. Structure 2017, 25, 901–911 e903. [Google Scholar] [CrossRef]
- Hon, W.C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002, 417, 975–978. [Google Scholar] [CrossRef]
- Min, J.H.; Yang, H.; Ivan, M.; Gertler, F.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of an HIF-1alpha -pVHL complex: Hydroxyproline recognition in signaling. Science 2002, 296, 1886–1889. [Google Scholar] [CrossRef]
- Tovell, H.; Testa, A.; Zhou, H.; Shpiro, N.; Crafter, C.; Ciulli, A.; Alessi, D.R. Design and Characterization of SGK3-PROTAC1, an Isoform Specific SGK3 Kinase PROTAC Degrader. ACS Chem. Biol. 2019, 14, 2024–2034. [Google Scholar] [CrossRef]
- Girardini, M.; Maniaci, C.; Hughes, S.J.; Testa, A.; Ciulli, A. Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem. 2019, 27, 2466–2479. [Google Scholar] [CrossRef]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef]
- Zografou-Barredo, N.A.; Hallatt, A.J.; Goujon-Ricci, J.; Cano, C. A beginner’s guide to current synthetic linker strategies towards VHL-recruiting PROTACs. Bioorg. Med. Chem. 2023, 88–89, 117334. [Google Scholar] [CrossRef]
- Li, F.; Hu, Q.; Zhang, X.; Sun, R.; Liu, Z.; Wu, S.; Tian, S.; Ma, X.; Dai, Z.; Yang, X.; et al. DeepPROTACs is a deep learning-based targeted degradation predictor for PROTACs. Nat. Commun. 2022, 13, 7133. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.; Ferguson, F.M.; Seong, B.K.A.; Kuljanin, M.; Leggett, A.L.; Mohardt, M.L.; Robichaud, A.; Conway, A.S.; Buckley, D.L.; Mancias, J.D.; et al. Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat. Commun. 2020, 11, 4687. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Yang, W.; Rozamus, L.W.; Hatada, M.; Amara, J.F.; Rollins, C.T.; Stevenson, L.F.; Magari, S.R.; Wood, S.A.; Courage, N.L.; et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. USA 1998, 95, 10437–10442. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.M.; Glennie, L.; Brewer, A.; Zhao, J.F.; Crooks, J.; Shpiro, N.; Sapkota, G.P. Target protein localization and its impact on PROTAC-mediated degradation. Cell Chem. Biol. 2022, 29, 1482–1504 e1487. [Google Scholar] [CrossRef]
- Yenerall, P.; Sung, T.; Palyada, K.; Qian, J.; Arat, S.; Kumpf, S.W.; Wang, S.W.; Biddle, K.; Esparza, C.; Chang, S.; et al. Use of the dTAG system in vivo to degrade CDK2 and CDK5 in adult mice and explore potential safety liabilities. Toxicol. Sci. 2023, 194, 53–69. [Google Scholar] [CrossRef]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef]
- Tovell, H.; Testa, A.; Maniaci, C.; Zhou, H.; Prescott, A.R.; Macartney, T.; Ciulli, A.; Alessi, D.R. Rapid and Reversible Knockdown of Endogenously Tagged Endosomal Proteins via an Optimized HaloPROTAC Degrader. ACS Chem. Biol. 2019, 14, 882–892. [Google Scholar] [CrossRef]
- BasuRay, S.; Wang, Y.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9521–9526. [Google Scholar] [CrossRef]
- Caine, E.A.; Mahan, S.D.; Johnson, R.L.; Nieman, A.N.; Lam, N.; Warren, C.R.; Riching, K.M.; Urh, M.; Daniels, D.L. Targeted Protein Degradation Phenotypic Studies Using HaloTag CRISPR/Cas9 Endogenous Tagging Coupled with HaloPROTAC3. Curr. Protoc. Pharmacol. 2020, 91, e81. [Google Scholar] [CrossRef] [PubMed]
- Alabi, S.; Jaime-Figueroa, S.; Yao, Z.; Gao, Y.; Hines, J.; Samarasinghe, K.T.G.; Vogt, L.; Rosen, N.; Crews, C.M. Mutant-selective degradation by BRAF-targeting PROTACs. Nat. Commun. 2021, 12, 920. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kalogeropulou, A.F.; Domingos, S.; Makukhin, N.; Nirujogi, R.S.; Singh, F.; Shpiro, N.; Saalfrank, A.; Sammler, E.; Ganley, I.G.; et al. Discovery of XL01126: A Potent, Fast, Cooperative, Selective, Orally Bioavailable, and Blood-Brain Barrier Penetrant PROTAC Degrader of Leucine-Rich Repeat Kinase 2. J. Am. Chem. Soc. 2022, 144, 16930–16952. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, Q.; Jiang, T.; Li, S.; Ye, J.; Zheng, J.; Wang, X.; Liu, Y.; Deng, M.; Ke, D.; et al. A novel small-molecule PROTAC selectively promotes tau clearance to improve cognitive functions in Alzheimer-like models. Theranostics 2021, 11, 5279–5295. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, S.D.; Yang, B.; Fallan, C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: Recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564. [Google Scholar] [CrossRef]
- Liu, J.R.; Yu, C.W.; Hung, P.Y.; Hsin, L.W.; Chern, J.W. High-selective HDAC6 inhibitor promotes HDAC6 degradation following autophagy modulation and enhanced antitumor immunity in glioblastoma. Biochem. Pharmacol. 2019, 163, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Guardigni, M.; Pruccoli, L.; Santini, A.; Simone, A.; Bersani, M.; Spyrakis, F.; Frabetti, F.; Uliassi, E.; Andrisano, V.; Pagliarani, B.; et al. PROTAC-Induced Glycogen Synthase Kinase 3beta Degradation as a Potential Therapeutic Strategy for Alzheimer’s Disease. ACS Chem. Neurosci. 2023, 14, 1963–1970. [Google Scholar] [CrossRef]
- Li, J.; Chen, X.; Lu, A.; Liang, C. Targeted protein degradation in cancers: Orthodox PROTACs and beyond. Innovation 2023, 4, 100413. [Google Scholar] [CrossRef]
- Chirnomas, D.; Hornberger, K.R.; Crews, C.M. Protein degraders enter the clinic—A new approach to cancer therapy. Nat. Rev. Clin. Oncol. 2023, 20, 265–278. [Google Scholar] [CrossRef]
- Lee, G.T.; Nagaya, N.; Desantis, J.; Madura, K.; Sabaawy, H.E.; Kim, W.J.; Vaz, R.J.; Cruciani, G.; Kim, I.Y. Effects of MTX-23, a Novel PROTAC of Androgen Receptor Splice Variant-7 and Androgen Receptor, on CRPC Resistant to Second-Line Antiandrogen Therapy. Mol. Cancer Ther. 2021, 20, 490–499. [Google Scholar] [CrossRef]
- Grinshpun, A. Clinician’s guide to targeted estrogen receptor degradation using PROTAC in patients with estrogen receptor-positive metastatic breast cancer. Curr. Opin. Oncol. 2023, 35, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Manda, S.; Lee, N.K.; Oh, D.C.; Lee, J. Design, Synthesis, and Biological Evaluation of Proteolysis Targeting Chimeras (PROTACs) for the Dual Degradation of IGF-1R and Src. Molecules 2020, 25, 1948. [Google Scholar] [CrossRef] [PubMed]
- Maneiro, M.A.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Gao, F.; Pontigon, D.; Gnawali, G.; Xu, H.; Wang, W. Bioorthogonal PROTAC Prodrugs Enabled by On-Target Activation. J. Am. Chem. Soc. 2023, 145, 14155–14163. [Google Scholar] [CrossRef]
- Yu, D.; Fan, H.; Zhou, Z.; Zhang, Y.; Sun, J.; Wang, L.; Jia, Y.; Tian, J.; Campbell, A.; Mi, W.; et al. Hydrogen Peroxide-Inducible PROTACs for Targeted Protein Degradation in Cancer Cells. Chembiochem 2023, 24, e202300422. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Ma, L.; He, Z.; Wang, D.; Liu, Y.; Lin, Q.; Zhang, T.; Gray, N.; Kaniskan, H.U.; et al. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020, 6, eaay5154. [Google Scholar] [CrossRef]
- Shi, C.; Zhang, H.; Wang, P.; Wang, K.; Xu, D.; Wang, H.; Yin, L.; Zhang, S.; Zhang, Y. PROTAC induced-BET protein degradation exhibits potent anti-osteosarcoma activity by triggering apoptosis. Cell Death Dis. 2019, 10, 815. [Google Scholar] [CrossRef]
- Seong, B.K.A.; Dharia, N.V.; Lin, S.; Donovan, K.A.; Chong, S.; Robichaud, A.; Conway, A.; Hamze, A.; Ross, L.; Alexe, G.; et al. TRIM8 modulates the EWS/FLI oncoprotein to promote survival in Ewing sarcoma. Cancer Cell 2021, 39, 1262–1278 e1267. [Google Scholar] [CrossRef]
- Lu, D.Y.; Ellegast, J.M.; Ross, K.N.; Malone, C.F.; Lin, S.; Mabe, N.W.; Dharia, N.V.; Meyer, A.; Conway, A.; Su, A.H.; et al. The ETS transcription factor ETV6 constrains the transcriptional activity of EWS-FLI to promote Ewing sarcoma. Nat. Cell Biol. 2023, 25, 285–297. [Google Scholar] [CrossRef]
- Gollavilli, P.N.; Pawar, A.; Wilder-Romans, K.; Natesan, R.; Engelke, C.G.; Dommeti, V.L.; Krishnamurthy, P.M.; Nallasivam, A.; Apel, I.J.; Xu, T.; et al. EWS/ETS-Driven Ewing Sarcoma Requires BET Bromodomain Proteins. Cancer Res. 2018, 78, 4760–4773. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Jiang, Y.; Wang, Y.; Vital, T.; Zhang, J.; Laggner, C.; Nguyen, K.T.; Zhu, Z.; Prevatte, A.W.; et al. SPOP and OTUD7A Control EWS-FLI1 Protein Stability to Govern Ewing Sarcoma Growth. Adv. Sci. 2021, 8, e2004846. [Google Scholar] [CrossRef] [PubMed]
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019, 15, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, J.; Liu, Q.; McDonald, W.H.; Bomber, M.L.; Layden, H.M.; Ellis, J.; Borinstein, S.C.; Hiebert, S.W.; Stengel, K.R. PAX3-FOXO1 coordinates enhancer architecture, eRNA transcription, and RNA polymerase pause release at select gene targets. Mol. Cell 2022, 82, 4428–4442 e4427. [Google Scholar] [CrossRef] [PubMed]
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; Van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife 2018, 7, e41305. [Google Scholar] [CrossRef]
- Fuchs, J.R.; Schulte, B.C.; Fuchs, J.W.; Agulnik, M. Emerging targeted and cellular therapies in the treatment of advanced and metastatic synovial sarcoma. Front. Oncol. 2023, 13, 1123464. [Google Scholar] [CrossRef]
- Lim, Y.S.; Yoo, S.M.; Patil, V.; Kim, H.W.; Kim, H.H.; Suh, B.; Park, J.Y.; Jeong, N.R.; Park, C.H.; Ryu, J.H.; et al. Orally bioavailable BTK PROTAC active against wild-type and C481 mutant BTKs in human lymphoma CDX mouse models. Blood Adv. 2023, 7, 92–105. [Google Scholar] [CrossRef]
- Zhang, X.; Tu, L.; Chai, H.; Li, Z.; Fu, Y.; Zheng, X.; Zeng, S.; Cheng, L. The Activity of Novel BCR-ABL Small-Molecule Degraders Containing Pyrimidine Rings and Their Role in Overcoming Drug Resistance. J. Oncol. 2022, 2022, 4056398. [Google Scholar] [CrossRef]
- Burslem, G.M.; Schultz, A.R.; Bondeson, D.P.; Eide, C.A.; Savage Stevens, S.L.; Druker, B.J.; Crews, C.M. Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res. 2019, 79, 4744–4753. [Google Scholar] [CrossRef]
- Ghidini, A.; Clery, A.; Halloy, F.; Allain, F.H.T.; Hall, J. RNA-PROTACs: Degraders of RNA-Binding Proteins. Angew. Chem. Int. Ed. 2021, 60, 3163–3169. [Google Scholar] [CrossRef]
- Kaneshige, A.; Bai, L.; Wang, M.; McEachern, D.; Meagher, J.L.; Xu, R.; Wang, Y.; Jiang, W.; Metwally, H.; Kirchhoff, P.D.; et al. A selective small-molecule STAT5 PROTAC degrader capable of achieving tumor regression in vivo. Nat. Chem. Biol. 2023, 19, 703–711. [Google Scholar] [CrossRef]
- Li, D.; Yu, X.; Kottur, J.; Gong, W.; Zhang, Z.; Storey, A.J.; Tsai, Y.H.; Uryu, H.; Shen, Y.; Byrum, S.D.; et al. Discovery of a dual WDR5 and Ikaros PROTAC degrader as an anti-cancer therapeutic. Oncogene 2022, 41, 3328–3340. [Google Scholar] [CrossRef] [PubMed]
- Yesbolatova, A.; Tominari, Y.; Kanemaki, M.T. Ligand-induced genetic degradation as a tool for target validation. Drug Discov. Today Technol. 2019, 31, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Radoux, C.J.; Hercules, A.; Ochoa, D.; Dunham, I.; Zalmas, L.P.; Hessler, G.; Ruf, S.; Shanmugasundaram, V.; Hann, M.M.; et al. The PROTACtable genome. Nat. Rev. Drug Discov. 2021, 20, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Pizzagalli, M.D.; Kartnig, F.; Dvorak, V.; Essletzbichler, P.; Winter, G.E.; Superti-Furga, G. Targeted Degradation of SLC Transporters Reveals Amenability of Multi-Pass Transmembrane Proteins to Ligand-Induced Proteolysis. Cell Chem. Biol. 2020, 27, 728–739 e729. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, K.T.G.; Jaime-Figueroa, S.; Burgess, M.; Nalawansha, D.A.; Dai, K.; Hu, Z.; Bebenek, A.; Holley, S.A.; Crews, C.M. Targeted degradation of transcription factors by TRAFTACs: TRAnscription Factor TArgeting Chimeras. Cell Chem. Biol. 2021, 28, 648–661 e645. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Kaniskan, H.U.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. [Google Scholar] [CrossRef] [PubMed]
- Worner, K.; Liu, Q.; Maschhoff, K.; Hu, W. Identification of RNA-binding proteins’ direct effects on gene expression via the degradation tag system. RNA 2023, 29, 1453–1457. [Google Scholar] [CrossRef]
- Jia, C.; Tang, H.; Yang, Y.; Yuan, S.; Han, T.; Fang, M.; Huang, S.; Hu, R.; Li, C.; Geng, W. Ubiquitination of IGF2BP3 by E3 ligase MKRN2 regulates the proliferation and migration of human neuroblastoma SHSY5Y cells. Biochem. Biophys. Res. Commun. 2020, 529, 43–50. [Google Scholar] [CrossRef]
- Buhimschi, A.D.; Armstrong, H.A.; Toure, M.; Jaime-Figueroa, S.; Chen, T.L.; Lehman, A.M.; Woyach, J.A.; Johnson, A.J.; Byrd, J.C.; Crews, C.M. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57, 3564–3575. [Google Scholar] [CrossRef]
- Cieslak, M.; Slowianek, M. Cereblon-Recruiting PROTACs: Will New Drugs Have to Face Old Challenges? Pharmaceutics 2023, 15, 812. [Google Scholar] [CrossRef]
- Kurimchak, A.M.; Herrera-Montavez, C.; Montserrat-Sangra, S.; Araiza-Olivera, D.; Hu, J.; Neumann-Domer, R.; Kuruvilla, M.; Bellacosa, A.; Testa, J.R.; Jin, J.; et al. The drug efflux pump MDR1 promotes intrinsic and acquired resistance to PROTACs in cancer cells. Sci. Signal. 2022, 15, eabn2707. [Google Scholar] [CrossRef] [PubMed]
- Shirasaki, R.; Matthews, G.M.; Gandolfi, S.; de Matos Simoes, R.; Buckley, D.L.; Raja Vora, J.; Sievers, Q.L.; Bruggenthies, J.B.; Dashevsky, O.; Poarch, H.; et al. Functional Genomics Identify Distinct and Overlapping Genes Mediating Resistance to Different Classes of Heterobifunctional Degraders of Oncoproteins. Cell Rep. 2021, 34, 108532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Riley-Gillis, B.; Vijay, P.; Shen, Y. Acquired Resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes. Mol. Cancer Ther. 2019, 18, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Song, S.; Liu, L.; Yu, Y.; Zhang, R.; Li, Y.; Cao, W.; Xiao, Y.; Fang, G.; Li, Z.; Wang, X.; et al. Inhibition of BRD4 attenuates transverse aortic constriction- and TGF-beta-induced endothelial-mesenchymal transition and cardiac fibrosis. J. Mol. Cell. Cardiol. 2019, 127, 83–96. [Google Scholar] [CrossRef]
- Nowak, R.P.; DeAngelo, S.L.; Buckley, D.; He, Z.; Donovan, K.A.; An, J.; Safaee, N.; Jedrychowski, M.P.; Ponthier, C.M.; Ishoey, M.; et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 2018, 14, 706–714. [Google Scholar] [CrossRef]
- Yan, Z.; Lyu, X.; Lin, D.; Wu, G.; Gong, Y.; Ren, X.; Xiao, J.; Lou, J.; Huang, H.; Chen, Y.; et al. Selective degradation of cellular BRD3 and BRD4-L promoted by PROTAC molecules in six cancer cell lines. Eur. J. Med. Chem. 2023, 254, 115381. [Google Scholar] [CrossRef]
- Li, Y.J.; Zhang, C.; Martincuks, A.; Herrmann, A.; Yu, H. STAT proteins in cancer: Orchestration of metabolism. Nat. Rev. Cancer 2023, 23, 115–134. [Google Scholar] [CrossRef]
- Liu, J.; Peng, Y.; Wei, W. Light-Controllable PROTACs for Temporospatial Control of Protein Degradation. Front. Cell Dev. Biol. 2021, 9, 678077. [Google Scholar] [CrossRef]
- Bratovic, M. Click, release, destroy. Nat. Chem. Biol. 2023, 19, 921. [Google Scholar] [CrossRef]
- Mullen, D.; Vajpeyi, R.; Capo-Chichi, J.M.; Nowak, K.; Wong, N.; Chetty, R. Gastrointestinal Stromal Tumours (GISTs) with KRAS Mutation: A Rare but Important Subset of GISTs. Case Rep. Gastrointest. Med. 2023, 2023, 4248128. [Google Scholar] [CrossRef]
- Olmos, D.; Postel-Vinay, S.; Molife, L.R.; Okuno, S.H.; Schuetze, S.M.; Paccagnella, M.L.; Batzel, G.N.; Yin, D.; Pritchard-Jones, K.; Judson, I.; et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: A phase 1 expansion cohort study. Lancet Oncol. 2010, 11, 129–135. [Google Scholar] [CrossRef]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Flores, G.; Grohar, P.J. One oncogene, several vulnerabilities: EWS/FLI targeted therapies for Ewing sarcoma. J. Bone Oncol. 2021, 31, 100404. [Google Scholar] [CrossRef]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef]
- Selvanathan, S.P.; Graham, G.T.; Grego, A.R.; Baker, T.M.; Hogg, J.R.; Simpson, M.; Batish, M.; Crompton, B.; Stegmaier, K.; Tomazou, E.M.; et al. EWS-FLI1 modulated alternative splicing of ARID1A reveals novel oncogenic function through the BAF complex. Nucleic Acids Res. 2019, 47, 9619–9636. [Google Scholar] [CrossRef]
- Conn, E.; Hour, S.; Allegakoen, D.; Graham, G.; Petro, J.; Kouassi-Brou, M.; Hong, S.H.; Selvanathan, S.; Celik, H.; Toretsky, J.; et al. Development of an Ewing sarcoma cell line with resistance to EWS-FLI1 inhibitor YK-4-279. Mol. Med. Rep. 2020, 21, 1667–1675. [Google Scholar] [CrossRef]
- Gong, H.; Xue, B.; Ru, J.; Pei, G.; Li, Y. Targeted Therapy for EWS-FLI1 in Ewing Sarcoma. Cancers 2023, 15, 4035. [Google Scholar] [CrossRef]
- Gierisch, M.E.; Pfistner, F.; Lopez-Garcia, L.A.; Harder, L.; Schafer, B.W.; Niggli, F.K. Proteasomal Degradation of the EWS-FLI1 Fusion Protein Is Regulated by a Single Lysine Residue. J. Biol. Chem. 2016, 291, 26922–26933. [Google Scholar] [CrossRef]
- Gierisch, M.E.; Pedot, G.; Walser, F.; Lopez-Garcia, L.A.; Jaaks, P.; Niggli, F.K.; Schafer, B.W. USP19 deubiquitinates EWS-FLI1 to regulate Ewing sarcoma growth. Sci. Rep. 2019, 9, 951. [Google Scholar] [CrossRef]
- Mancarella, C.; Morrione, A.; Scotlandi, K. Unraveling the IGF System Interactome in Sarcomas Exploits Novel Therapeutic Options. Cells 2021, 10, 2075. [Google Scholar] [CrossRef]
- Winter, G.E.; Mayer, A.; Buckley, D.L.; Erb, M.A.; Roderick, J.E.; Vittori, S.; Reyes, J.M.; di Iulio, J.; Souza, A.; Ott, C.J.; et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol. Cell 2017, 67, 5–18 e19. [Google Scholar] [CrossRef]
- Lilienthal, I.; Herold, N. Targeting Molecular Mechanisms Underlying Treatment Efficacy and Resistance in Osteosarcoma: A Review of Current and Future Strategies. Int. J. Mol. Sci. 2020, 21, 6885. [Google Scholar] [CrossRef]
- Sayles, L.C.; Breese, M.R.; Koehne, A.L.; Leung, S.G.; Lee, A.G.; Liu, H.Y.; Spillinger, A.; Shah, A.T.; Tanasa, B.; Straessler, K.; et al. Genome-Informed Targeted Therapy for Osteosarcoma. Cancer Discov. 2019, 9, 46–63. [Google Scholar] [CrossRef]
- Benini, S.; Baldini, N.; Manara, M.C.; Chano, T.; Serra, M.; Rizzi, S.; Lollini, P.L.; Picci, P.; Scotlandi, K. Redundancy of autocrine loops in human osteosarcoma cells. Int. J. Cancer 1999, 80, 581–588. [Google Scholar] [CrossRef]
- Anderson, P.M.; Bielack, S.S.; Gorlick, R.G.; Skubitz, K.; Daw, N.C.; Herzog, C.E.; Monge, O.R.; Lassaletta, A.; Boldrini, E.; Papai, Z.; et al. A phase II study of clinical activity of SCH 717454 (robatumumab) in patients with relapsed osteosarcoma and Ewing sarcoma. Pediatr. Blood Cancer 2016, 63, 1761–1770. [Google Scholar] [CrossRef]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: A non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2019, 20, 120–133. [Google Scholar] [CrossRef]
- Schott, C.; Shah, A.T.; Sweet-Cordero, E.A. Genomic Complexity of Osteosarcoma and Its Implication for Preclinical and Clinical Targeted Therapies. Adv. Exp. Med. Biol. 2020, 1258, 1–19. [Google Scholar] [CrossRef]
- Lamoureux, F.; Baud’huin, M.; Rodriguez Calleja, L.; Jacques, C.; Berreur, M.; Redini, F.; Lecanda, F.; Bradner, J.E.; Heymann, D.; Ory, B. Selective inhibition of BET bromodomain epigenetic signalling interferes with the bone-associated tumour vicious cycle. Nat. Commun. 2014, 5, 3511. [Google Scholar] [CrossRef]
- Lee, D.H.; Qi, J.; Bradner, J.E.; Said, J.W.; Doan, N.B.; Forscher, C.; Yang, H.; Koeffler, H.P. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. Int. J. Cancer 2015, 136, 2055–2064. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, G.; Mu, H.; Ma, X.; Wang, Z.; Lv, Y.; Zhang, T.; Xu, J.; Wang, J.; Li, Y.; et al. Bromodomain Inhibition Attenuates the Progression and Sensitizes the Chemosensitivity of Osteosarcoma by Repressing GP130/STAT3 Signaling. Front. Oncol. 2021, 11, 642134. [Google Scholar] [CrossRef]
- Zhou, B.; Hu, J.; Xu, F.; Chen, Z.; Bai, L.; Fernandez-Salas, E.; Lin, M.; Liu, L.; Yang, C.Y.; Zhao, Y.; et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem. 2018, 61, 462–481. [Google Scholar] [CrossRef]
- Baker, E.K.; Taylor, S.; Gupte, A.; Sharp, P.P.; Walia, M.; Walsh, N.C.; Zannettino, A.C.; Chalk, A.M.; Burns, C.J.; Walkley, C.R. BET inhibitors induce apoptosis through a MYC independent mechanism and synergise with CDK inhibitors to kill osteosarcoma cells. Sci. Rep. 2015, 5, 10120. [Google Scholar] [CrossRef]
- Abraham, J.; Nunez-Alvarez, Y.; Hettmer, S.; Carrio, E.; Chen, H.I.; Nishijo, K.; Huang, E.T.; Prajapati, S.I.; Walker, R.L.; Davis, S.; et al. Lineage of origin in rhabdomyosarcoma informs pharmacological response. Genes Dev. 2014, 28, 1578–1591. [Google Scholar] [CrossRef]
- Ramadan, F.; Fahs, A.; Ghayad, S.E.; Saab, R. Signaling pathways in Rhabdomyosarcoma invasion and metastasis. Cancer Metastasis Rev. 2020, 39, 287–301. [Google Scholar] [CrossRef]
- Manceau, L.; Richard Albert, J.; Lollini, P.L.; Greenberg, M.V.C.; Gilardi-Hebenstreit, P.; Ribes, V. Divergent transcriptional and transforming properties of PAX3-FOXO1 and PAX7-FOXO1 paralogs. PLoS Genet. 2022, 18, e1009782. [Google Scholar] [CrossRef]
- Cao, L.; Yu, Y.; Bilke, S.; Walker, R.L.; Mayeenuddin, L.H.; Azorsa, D.O.; Yang, F.; Pineda, M.; Helman, L.J.; Meltzer, P.S. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010, 70, 6497–6508. [Google Scholar] [CrossRef]
- Laubscher, D.; Gryder, B.E.; Sunkel, B.D.; Andresson, T.; Wachtel, M.; Das, S.; Roschitzki, B.; Wolski, W.; Wu, X.S.; Chou, H.C.; et al. BAF complexes drive proliferation and block myogenic differentiation in fusion-positive rhabdomyosarcoma. Nat. Commun. 2021, 12, 6924. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Barr, F.G. Therapeutic Approaches Targeting PAX3-FOXO1 and Its Regulatory and Transcriptional Pathways in Rhabdomyosarcoma. Molecules 2018, 23, 2798. [Google Scholar] [CrossRef]
- Bharathy, N.; Cleary, M.M.; Kim, J.A.; Nagamori, K.; Crawford, K.A.; Wang, E.; Saha, D.; Settelmeyer, T.P.; Purohit, R.; Skopelitis, D.; et al. SMARCA4 biology in alveolar rhabdomyosarcoma. Oncogene 2022, 41, 1647–1656. [Google Scholar] [CrossRef]
- Shah, A.M.; Guo, L.; Morales, M.G.; Jaichander, P.; Chen, K.; Huang, H.; Cano Hernandez, K.; Xu, L.; Bassel-Duby, R.; Olson, E.N.; et al. TWIST2-mediated chromatin remodeling promotes fusion-negative rhabdomyosarcoma. Sci. Adv. 2023, 9, eade8184. [Google Scholar] [CrossRef] [PubMed]
- Wanior, M.; Kramer, A.; Knapp, S.; Joerger, A.C. Exploiting vulnerabilities of SWI/SNF chromatin remodelling complexes for cancer therapy. Oncogene 2021, 40, 3637–3654. [Google Scholar] [CrossRef] [PubMed]
- Papillon, J.P.N.; Nakajima, K.; Adair, C.D.; Hempel, J.; Jouk, A.O.; Karki, R.G.; Mathieu, S.; Mobitz, H.; Ntaganda, R.; Smith, T.; et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J. Med. Chem. 2018, 61, 10155–10172. [Google Scholar] [CrossRef] [PubMed]
- Vangamudi, B.; Paul, T.A.; Shah, P.K.; Kost-Alimova, M.; Nottebaum, L.; Shi, X.; Zhan, Y.; Leo, E.; Mahadeshwar, H.S.; Protopopov, A.; et al. The SMARCA2/4 ATPase Domain Surpasses the Bromodomain as a Drug Target in SWI/SNF-Mutant Cancers: Insights from cDNA Rescue and PFI-3 Inhibitor Studies. Cancer Res. 2015, 75, 3865–3878. [Google Scholar] [CrossRef]
- Ladanyi, M. Fusions of the SYT and SSX genes in synovial sarcoma. Oncogene 2001, 20, 5755–5762. [Google Scholar] [CrossRef]
- Li, J.; Mulvihill, T.S.; Li, L.; Barrott, J.J.; Nelson, M.L.; Wagner, L.; Lock, I.C.; Pozner, A.; Lambert, S.L.; Ozenberger, B.B.; et al. A Role for SMARCB1 in Synovial Sarcomagenesis Reveals That SS18-SSX Induces Canonical BAF Destruction. Cancer Discov. 2021, 11, 2620–2637. [Google Scholar] [CrossRef]
- Haldar, M.; Hancock, J.D.; Coffin, C.M.; Lessnick, S.L.; Capecchi, M.R. A conditional mouse model of synovial sarcoma: Insights into a myogenic origin. Cancer Cell 2007, 11, 375–388. [Google Scholar] [CrossRef]
- Isfort, I.; Cyra, M.; Elges, S.; Kailayangiri, S.; Altvater, B.; Rossig, C.; Steinestel, K.; Grunewald, I.; Huss, S.; Esseling, E.; et al. SS18-SSX-Dependent YAP/TAZ Signaling in Synovial Sarcoma. Clin. Cancer Res. 2019, 25, 3718–3731. [Google Scholar] [CrossRef]
- McBride, M.J.; Pulice, J.L.; Beird, H.C.; Ingram, D.R.; D’Avino, A.R.; Shern, J.F.; Charville, G.W.; Hornick, J.L.; Nakayama, R.T.; Garcia-Rivera, E.M.; et al. The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 2018, 33, 1128–1141 e1127. [Google Scholar] [CrossRef]
- Landuzzi, L.; Manara, M.C.; Pazzaglia, L.; Lollini, P.L.; Scotlandi, K. Innovative Breakthroughs for the Treatment of Advanced and Metastatic Synovial Sarcoma. Cancers 2023, 15, 3887. [Google Scholar] [CrossRef]
- Mu, J.; Sun, X.; Zhao, Z.; Sun, H.; Sun, P. BRD9 inhibition promotes PUMA-dependent apoptosis and augments the effect of imatinib in gastrointestinal stromal tumors. Cell Death Dis. 2021, 12, 962. [Google Scholar] [CrossRef] [PubMed]
- Cooley, C.; Su, L. HDAC2 links ubiquitination to tumor suppression in synovial sarcoma. Mol. Cell. Oncol. 2021, 8, 1914291. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Wang, J.; Shiozawa, K.; Jones, K.B.; Zhang, Y.; Prokop, J.W.; Davenport, G.G.; Nihira, N.T.; Hao, Z.; Wong, D.; et al. HDAC2 Regulates Site-Specific Acetylation of MDM2 and Its Ubiquitination Signaling in Tumor Suppression. iScience 2019, 13, 43–54. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Types | Target Protein | TPD Approach | References |
|---|---|---|---|
| Solid tumors | |||
| Prostate cancer | AR | ARCC-4 PROTAC | [30] |
| AR Splice Variant-7 | MTX-23 PROTAC | [61] | |
| Breast cancer | Estrogen receptor | ARV-471 PROTAC | [62] |
| AC682 PROTAC | |||
| DT2216 PROTAC | |||
| SGK3 | SGK3-PROTAC1, HaloTag tool | [38,50] | |
| IGF-1R/Src | CPR3/CPR4 PROTACs | [63] | |
| BRD4 | Ab-PROTAC 3 | [64] | |
| BRD4 | Bioorthogonal PROTAC | [65] | |
| Lung cancer | IGF-1R/Src | CPR3/CPR4 PROTACs | [63] |
| BRD4 | H2O2-inducible PROTAC | [66] | |
| EML4-ALK fusion | dALK opto-PROTAC | [67] | |
| Osteosarcoma | BET proteins | MZ1 PROTAC | [29] |
| BETd-260 PROTAC | [68] | ||
| Ewing sarcoma | EWS::FLI1 | dTAG tool | [43] |
| TRIM8 | dTAG tool | [69] | |
| ETV6 | dTAG tool | [70] | |
| BRD4 | BETd PROTAC | [71] | |
| CK1 | CK1α PROTAC | [72] | |
| Glioblastoma | HDAC6 | J22352 PROTAC | [57] |
| Rhabdomyosarcoma | SMARCA4/A2 | ACBI-1 PROTAC | [73] |
| PAX3::FOXO1 | dTAG tool | [74] | |
| Melanoma | Mutant BRAF | SJF-0661 PROTAC | [53] |
| Synovial sarcoma | BRD9 | dBRD9-A PROTAC | [75] |
| CFT8634 | [76] | ||
| FHD-609 | [76] | ||
| Cervical carcinoma | BET proteins | MZ1 PROTAC | [29] |
| Hematological tumors | |||
| Diffuse large B-cell lymphoma | BTK | UBX-382 PROTAC | [77] |
| Chronic myeloid leukemia | BCR-ABL1 | DMP11 PROTAC | [78] |
| GMB-475 PROTAC | [79] | ||
| LIN28 | RNA-PROTAC | [80] | |
| STAT5 | AK-2292 PROTAC | [81] | |
| MLL-rearranged leukemia | IKZF | MS40 PROTAC | [82] |
| Acute myeloid leukemia | CDK2/CDK5 | dTAG tool | [43] |
| Anaplastic large cell lymphoma | NPM-ALK fusion | dALK opto-PROTAC | [67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancarella, C.; Morrione, A.; Scotlandi, K. PROTAC-Based Protein Degradation as a Promising Strategy for Targeted Therapy in Sarcomas. Int. J. Mol. Sci. 2023, 24, 16346. https://doi.org/10.3390/ijms242216346
Mancarella C, Morrione A, Scotlandi K. PROTAC-Based Protein Degradation as a Promising Strategy for Targeted Therapy in Sarcomas. International Journal of Molecular Sciences. 2023; 24(22):16346. https://doi.org/10.3390/ijms242216346
Chicago/Turabian StyleMancarella, Caterina, Andrea Morrione, and Katia Scotlandi. 2023. "PROTAC-Based Protein Degradation as a Promising Strategy for Targeted Therapy in Sarcomas" International Journal of Molecular Sciences 24, no. 22: 16346. https://doi.org/10.3390/ijms242216346
APA StyleMancarella, C., Morrione, A., & Scotlandi, K. (2023). PROTAC-Based Protein Degradation as a Promising Strategy for Targeted Therapy in Sarcomas. International Journal of Molecular Sciences, 24(22), 16346. https://doi.org/10.3390/ijms242216346