
FTO Sensitizes Oral Squamous Cell Carcinoma to Ferroptosis via Suppressing ACSL3 and GPX4


Abstract
:1. Introduction
2. Results
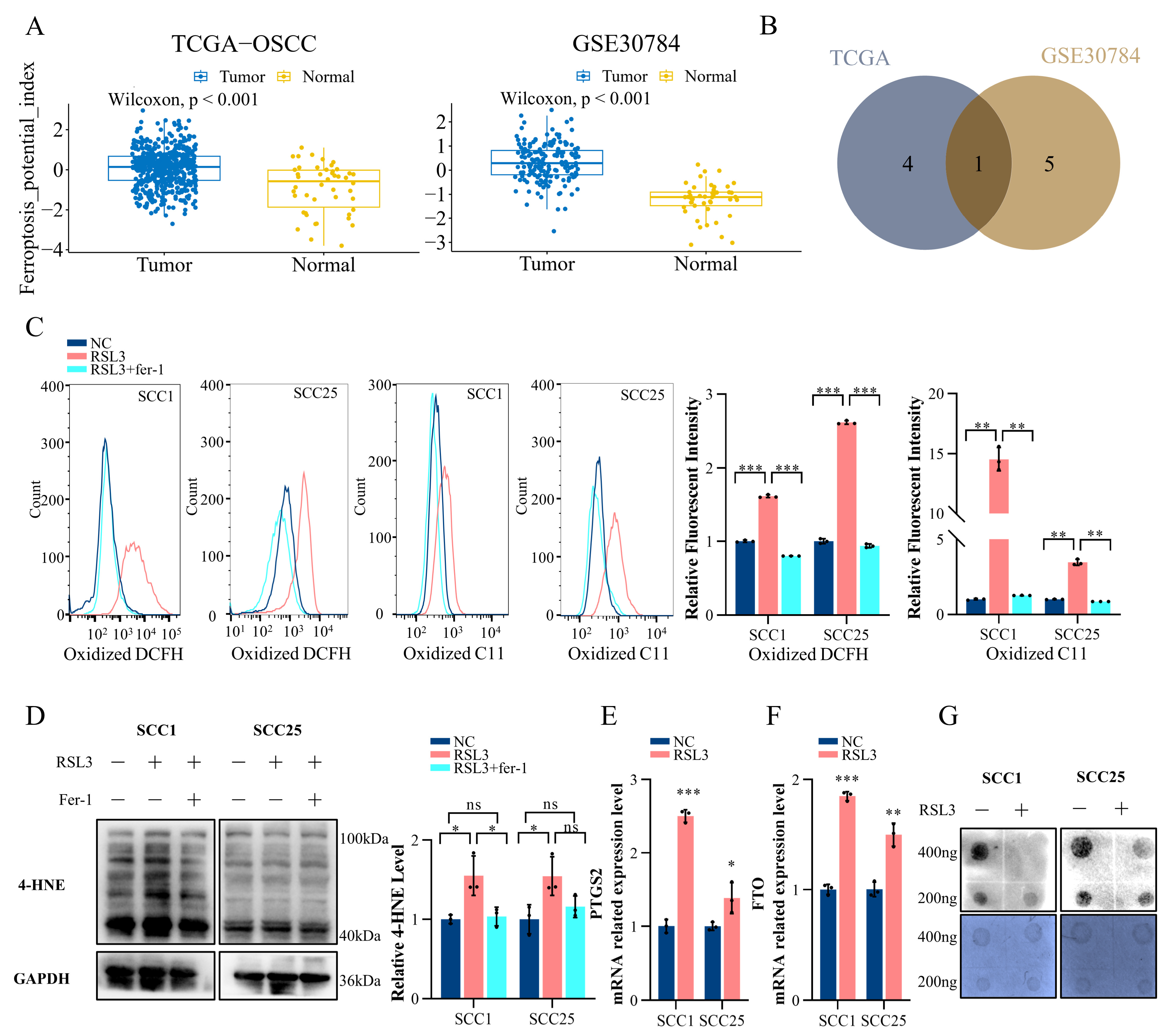
2.1. Ferroptosis in OSCC Is Correlated with the m6A Modification
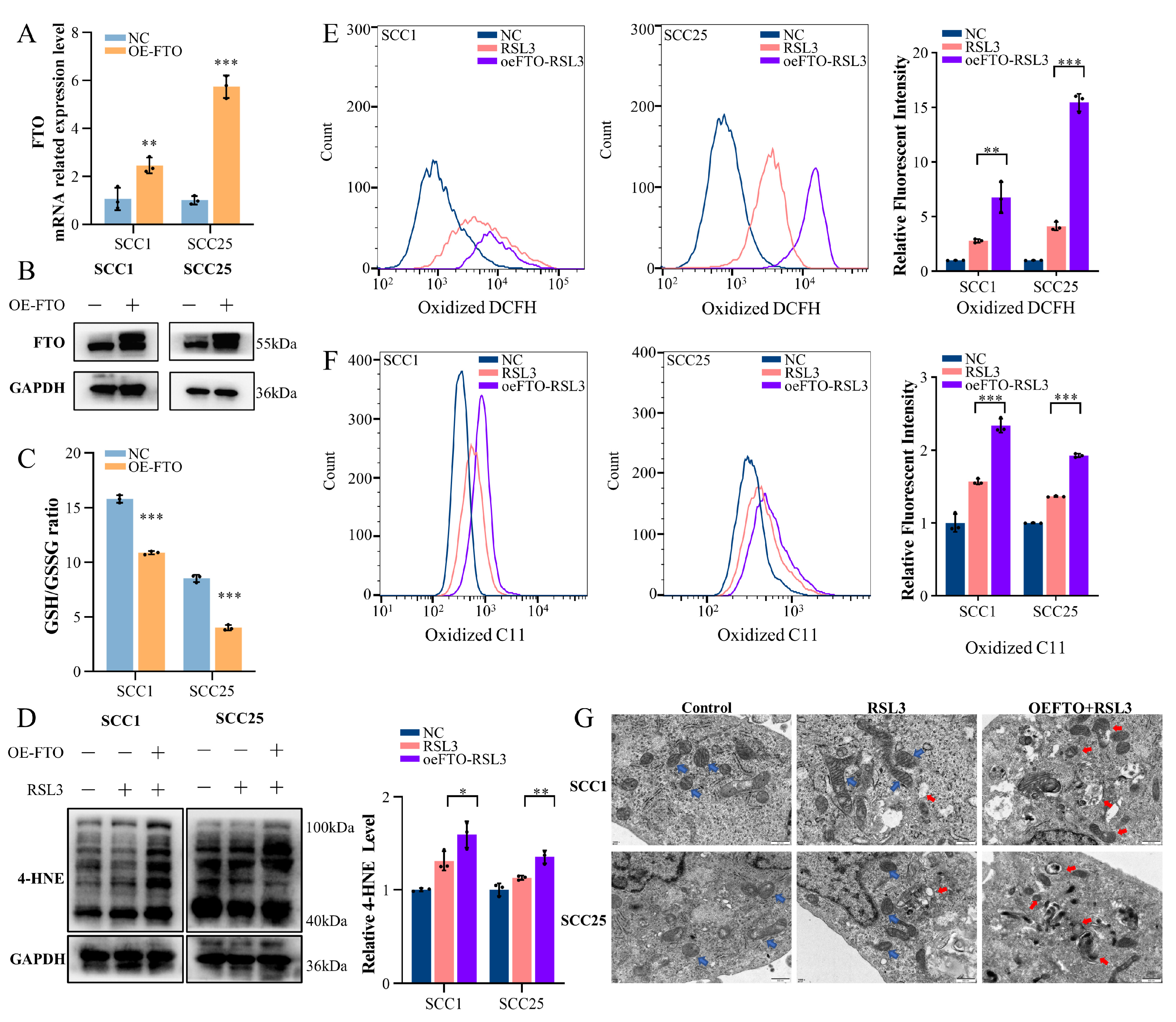
2.2. Upregulation of FTO Expression Enhances Ferroptosis in OSCC Cells
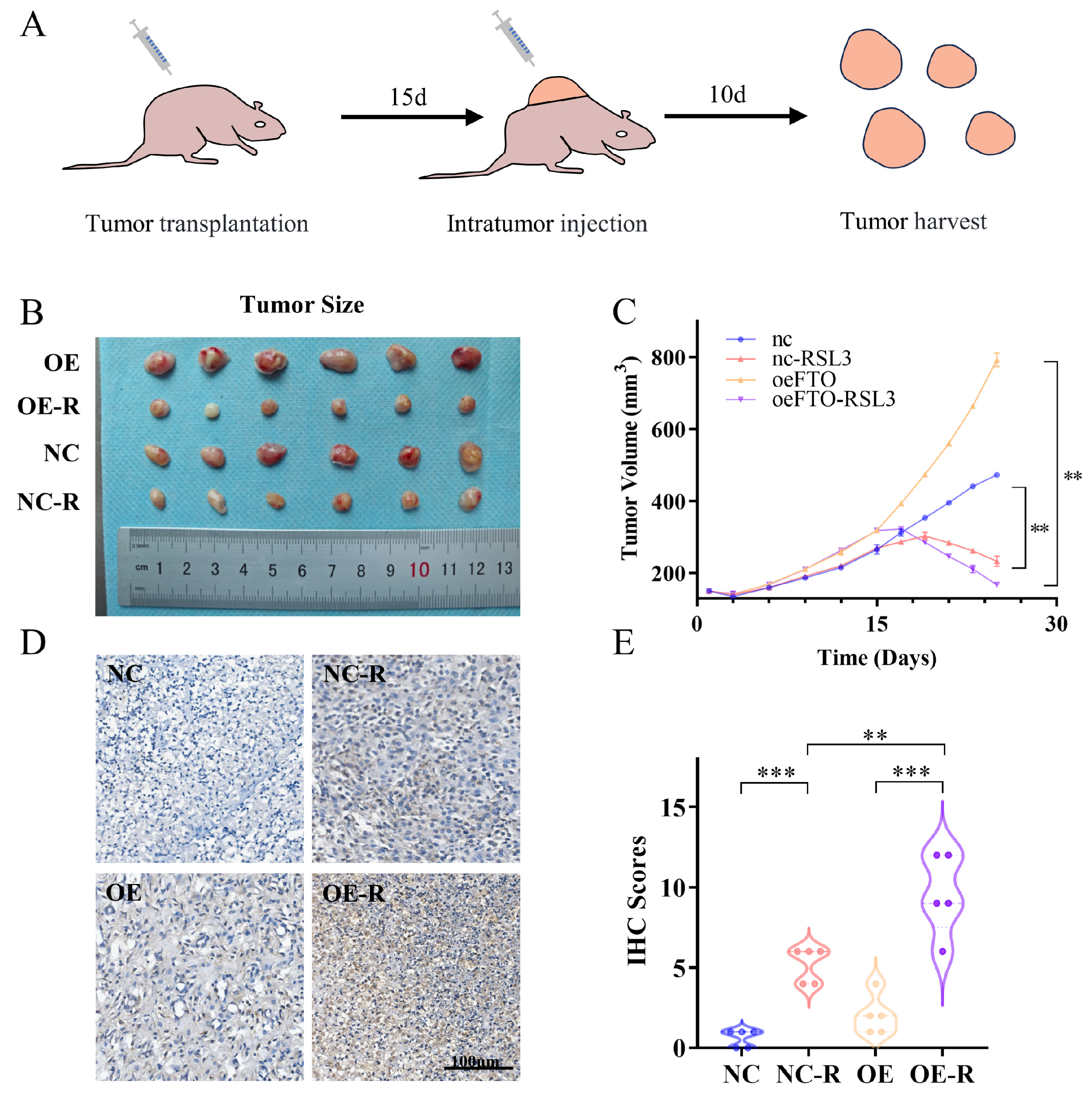
2.3. FTO Enhances OSCC Cell Ferroptosis In Vivo
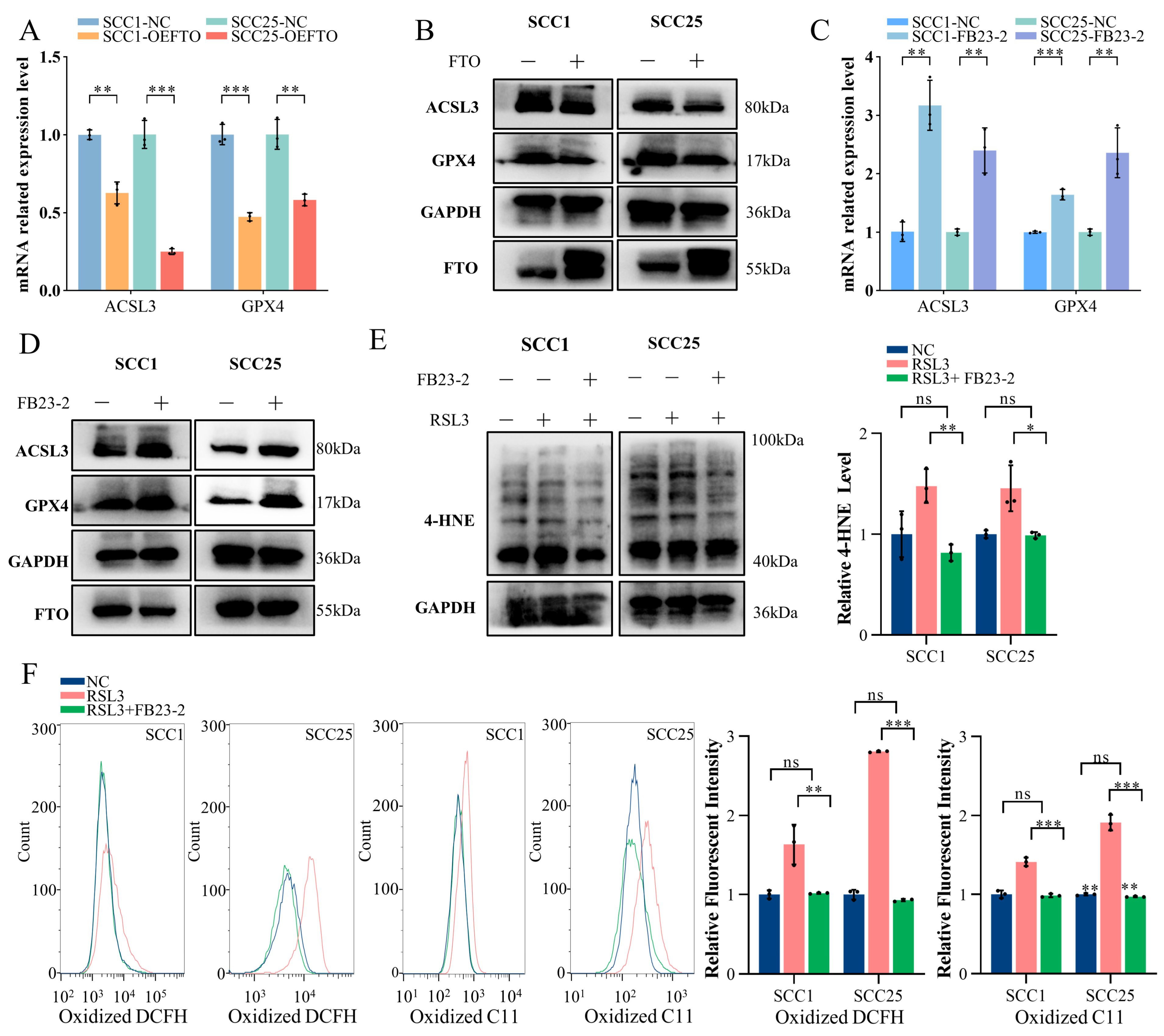
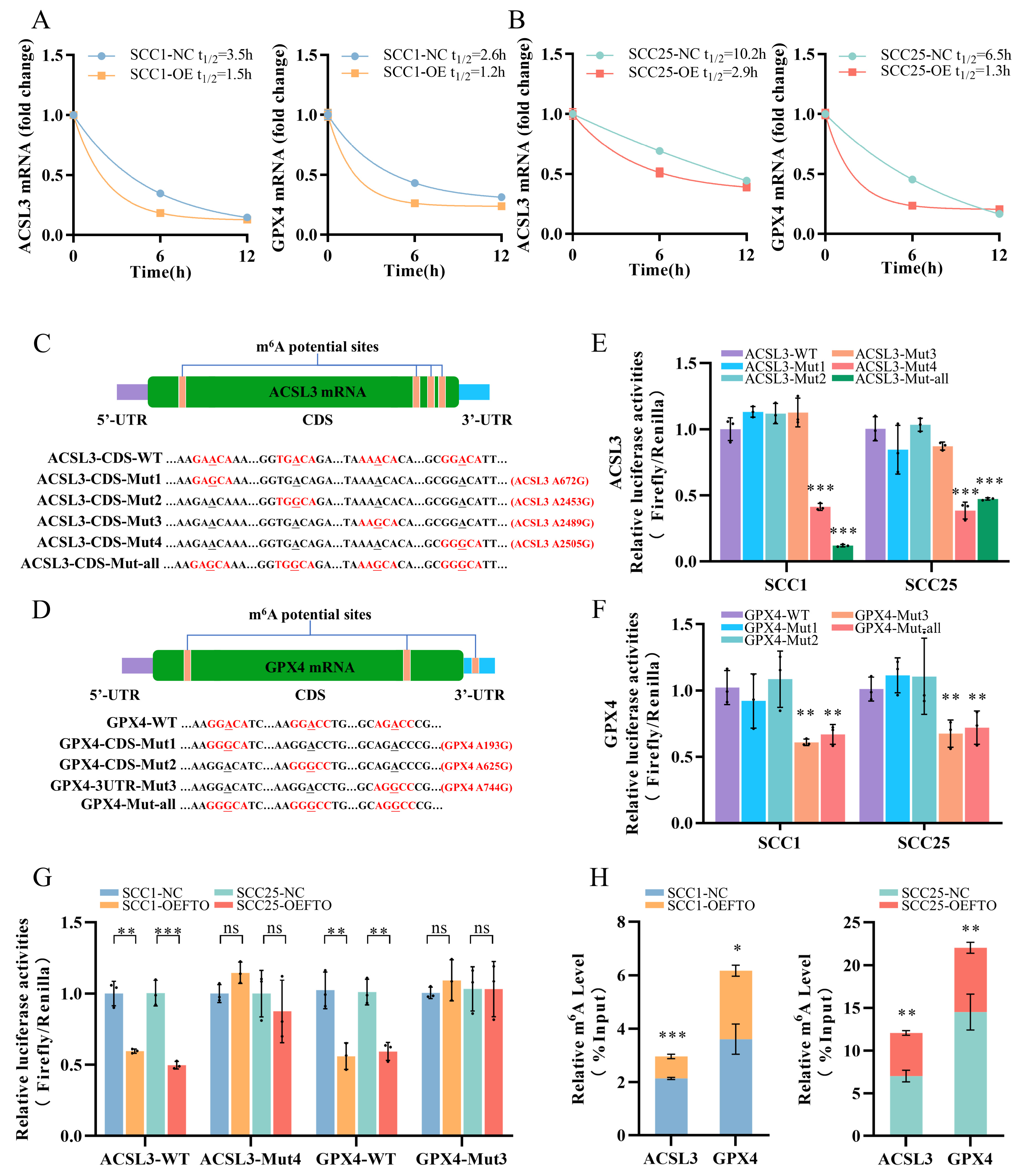
2.4. FTO Promotes Ferroptosis in OSCC Cells by Downgrading ACSL3 and GPX4 Expression
2.5. FTO Demethylates the m6A Modification on ACSL3 and GPX4 mRNA
3. Discussion
4. Materials and Methods
4.1. Data Collection and Analysis, and Ferroptosis Potential Index Calculation
4.2. Cell Line Culture
4.3. Construction of Stable Gene Expression Cell Lines
4.4. Real-Time Quantity-PCR
4.5. Western Blot
4.6. m6A Dot Blot Assay
4.7. Transmission Electron Microscopy (TEM)
4.8. ROS and Lipid Peroxidation Detection
4.9. Tumor Xenograft and Immunohistochemistry
4.10. GSH/GSSG Assay
4.11. Small Interfering RNA (siRNA) Transfection
4.12. RNA Stability Assays
4.13. Dual-Luciferase Reporter Assay
4.14. Methylated RNA Immunoprecipitation
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Oral squamous cell carcinoma | OSCC |
| N6-methyladenosine | m6A |
| Reactive oxygen species | ROS |
| Ferroptosis potential index | FPI |
| The Cancer Genome Atlas | TCGA |
| 4-hydroxynonenal | 4-HNE |
| Real-time quantity-PCR | RT-qPCR |
| Glutathione | GSH |
| Immunohistochemistry | IHC |
| N6-methylated RNA immunoprecipitation | MeRIP |
| Lung adenocarcinoma | LUAD |
| Polyunsaturated fatty acids | PUFAs |
| Monounsaturated fatty acids | MUFAs |
| Small interfering RNA | siRNA |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bagan, J.; Sarrion, G.; Jimenez, Y. Oral cancer: Clinical features. Oral Oncol. 2010, 46, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Kamangar, F.; Dores, G.M.; Anderson, W.F. Patterns of Cancer Incidence, Mortality, and Prevalence Across Five Continents: Defining Priorities to Reduce Cancer Disparities in Different Geographic Regions of the World. J. Clin. Oncol. 2006, 24, 2137–2150. [Google Scholar] [CrossRef] [PubMed]
- Shield, K.D.; Ferlay, J.; Jemal, A.; Sankaranarayanan, R.; Chaturvedi, A.K.; Bray, F.; Soerjomataram, I. The global incidence of lip, oral cavity, and pharyngeal cancers by subsite in 2012. CA Cancer J. Clin. 2017, 67, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.P.; Tsai, M.K.; Chung, W.S.; Hsu, H.L.; Chang, Y.C.; Chan, H.T.; Chiang, P.H.; Cheng, T.Y.; Tsai, S.P. Cancer risks from betel quid chewing beyond oral cancer: A multiple-site carcinogen when acting with smoking. Cancer Causes Control 2010, 21, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Bagan, J.V.; Scully, C. Recent advances in Oral Oncology 2007: Epidemiology, aetiopathogenesis, diagnosis and prognostication. Oral Oncol. 2008, 44, 103–108. [Google Scholar] [CrossRef]
- Peng, S.Y.; Tang, J.Y.; Li, R.N.; Huang, H.W.; Wu, C.Y.; Chiu, C.C.; Chang, F.R.; Zhang, H.W.; Lee, Y.J.; Sheu, J.H.; et al. Oxidative Stress-Dependent Synergistic Antiproliferation, Apoptosis, and DNA Damage of Ultraviolet-C and Coral-Derived Sinularin Combined Treatment for Oral Cancer Cells. Cancers 2021, 13, 2450. [Google Scholar] [CrossRef]
- Olmos, M.; Lutz, R.; Büntemeyer, T.-O.; Glajzer, J.; Nobis, C.-P.; Ries, J.; Möst, T.; Eckstein, M.; Hecht, M.; Gostian, A.-O.; et al. Case report: Patient specific combination of surgery and immunotherapy in advanced squamous cell carcinoma of the head and neck—A case series and review of literature. Front. Immunol. 2022, 13, 970823. [Google Scholar] [CrossRef]
- Wang, T.; Sun, S.; Zeng, X.; Li, J. ICI-based therapies: A new strategy for oral potentially malignant disorders. Oral. Oncol. 2023, 140, 106388. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Zandberg, D.P.; Algazi, A.P.; Jimeno, A.; Good, J.S.; Fayette, J.; Bouganim, N.; Ready, N.E.; Clement, P.M.; Even, C.; Jang, R.W.; et al. Durvalumab for recurrent or metastatic head and neck squamous cell carcinoma: Results from a single-arm, phase II study in patients with ≥25% tumour cell PD-L1 expression who have progressed on platinum-based chemotherapy. Eur. J. Cancer 2019, 107, 142–152. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The role of m6A modification in the biological functions and diseases. Signal Transduct. Target. Ther. 2021, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Mei, W.; Qu, C.; Lu, J.; Shang, L.; Cao, F.; Li, F. Role of m6A writers, erasers and readers in cancer. Exp. Hematol. Oncol. 2022, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wu, R.; Ming, L. The role of m6A RNA methylation in cancer. Biomed. Pharmacother. 2019, 112, 108613. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Li, Y.; Su, R.; Deng, X.; Chen, Y.; Chen, J. FTO in cancer: Functions, molecular mechanisms, and therapeutic implications. Trends Cancer 2022, 8, 598–614. [Google Scholar] [CrossRef]
- Li, X.; Chen, W.; Gao, Y.; Song, J.; Gu, Y.; Zhang, J.; Cheng, X.; Ai, Y. Fat mass and obesity-associated protein regulates arecoline-exposed oral cancer immune response through programmed cell death-ligand 1. Cancer Sci. 2022, 113, 2962–2973. [Google Scholar] [CrossRef]
- Wang, F.; Liao, Y.; Zhang, M.; Zhu, Y.; Wang, W.; Cai, H.; Liang, J.; Song, F.; Hou, C.; Huang, S.; et al. N6-methyladenosine demethyltransferase FTO-mediated autophagy in malignant development of oral squamous cell carcinoma. Oncogene 2021, 40, 3885–3898. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, M.; Bi, R.; Su, Y.; Quan, F.; Lin, Y.; Yue, C.; Cui, X.; Zhao, Q.; Liu, S.; et al. ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury. Redox Biol. 2022, 51, 102262. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, Y.; Mao, C.; Liu, S.; Xiao, D.; Huang, J.; Tao, Y. Emerging mechanisms and targeted therapy of ferroptosis in cancer. Mol. Ther. 2021, 29, 2185–2208. [Google Scholar] [CrossRef]
- Liang, W.; Ferrara, N. Iron Metabolism in the Tumor Microenvironment: Contributions of Innate Immune Cells. Front. Immunol. 2021, 11, 626812. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Shen, C.; Liu, J.; Liu, H.; Li, G.; Wang, H.; Tian, H.; Mao, Y.; Hua, D. Timosaponin AIII induces lipid peroxidation and ferroptosis by enhancing Rab7-mediated lipophagy in colorectal cancer cells. Phytomedicine 2023, 122, 155079. [Google Scholar] [CrossRef]
- Tu, J.; Meng, X.; Wang, J.; Han, Z.; Yu, Z.; Sun, H. 3β-Hydroxy-12-oleanen-27-oic Acid Exerts an Antiproliferative Effect on Human Colon Carcinoma HCT116 Cells via Targeting FDFT1. Int. J. Mol. Sci. 2023, 24, 15020. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Li, X.; Hu, S.; Cheng, J.; Cai, R. Regulation of Ferroptosis in Lung Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 14614. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, H.; Hu, D.; Wang, Y.; Shao, W.; Zhong, J.; Yang, S.; Liu, J.; Zhang, J. Insights into N6-methyladenosine and programmed cell death in cancer. Mol. Cancer 2022, 21, 32. [Google Scholar] [CrossRef]
- Liu, L.; He, J.; Sun, G.; Huang, N.; Bian, Z.; Xu, C.; Zhang, Y.; Cui, Z.; Xu, W.; Sun, F.; et al. The N6-methyladenosine modification enhances ferroptosis resistance through inhibiting SLC7A11 mRNA deadenylation in hepatoblastoma. Clin. Transl. Med. 2022, 12, e778. [Google Scholar] [CrossRef] [PubMed]
- Ji, F.H.; Fu, X.H.; Li, G.Q.; He, Q.; Qiu, X.G. FTO Prevents Thyroid Cancer Progression by SLC7A11 m6A Methylation in a Ferroptosis-Dependent Manner. Front. Endocrinol. 2022, 13, 857765. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Guo, M.; Li, Y.; Wang, Y.; Qiu, Y.; Shao, J.; Zhang, F.; Xu, X.; Yin, G.; Wang, S.; et al. m(6)A methylation is required for dihydroartemisinin to alleviate liver fibrosis by inducing ferroptosis in hepatic stellate cells. Free Radic. Biol. Med. 2022, 182, 246–259. [Google Scholar] [CrossRef]
- Shen, M.; Li, Y.; Wang, Y.; Shao, J.; Zhang, F.; Yin, G.; Chen, A.; Zhang, Z.; Zheng, S. N(6)-methyladenosine modification regulates ferroptosis through autophagy signaling pathway in hepatic stellate cells. Redox Biol. 2021, 47, 102151. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Liu, Z.-X.; Li, L.-M.; Sun, H.-L.; Liu, S.-M. Link between m6A Modification and Cancers. Front. Bioeng. Biotechnol. 2018, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piątkowski, P.; Bagiński, B.; Wirecki, T.K.; de Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Sun, P.; Zheng, J.; Wu, M.; Yang, C.; Cheng, M.; Yin, M.; Cui, C.; Wang, G.; Yuan, L.; et al. JNK Signaling Promotes Bladder Cancer Immune Escape by Regulating METTL3-Mediated m6A Modification of PD-L1 mRNA. Cancer Res. 2022, 82, 1789–1802. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Zhang, L.; Liu, J.; Wan, Y.; Jiang, Y.; Yang, J.; Sun, R.; Ma, X.; Sun, G.; Meng, H.; et al. ALKBH5-HOXA10 loop-mediated JAK2 m6A demethylation and cisplatin resistance in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2021, 40, 284. [Google Scholar] [CrossRef]
- Yang, Y.; Cai, J.; Yang, X.; Wang, K.; Sun, K.; Yang, Z.; Zhang, L.; Yang, L.; Gu, C.; Huang, X.; et al. Dysregulated m6A modification promotes lipogenesis and development of non-alcoholic fatty liver disease and hepatocellular carcinoma. Mol. Ther. 2022, 30, 2342–2353. [Google Scholar] [CrossRef]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677–691.e610. [Google Scholar] [CrossRef]
- Wang, J.; Yu, H.; Dong, W.; Zhang, C.; Hu, M.; Ma, W.; Jiang, X.; Li, H.; Yang, P.; Xiang, D. N6-Methyladenosine–Mediated Up-Regulation of FZD10 Regulates Liver Cancer Stem Cells’ Properties and Lenvatinib Resistance through WNT/β-Catenin and Hippo Signaling Pathways. Gastroenterology 2023, 164, 990–1005. [Google Scholar] [CrossRef]
- Jin, D.; Guo, J.; Wu, Y.; Du, J.; Yang, L.; Wang, X.; Di, W.; Hu, B.; An, J.; Kong, L.; et al. RETRACTED ARTICLE: m6A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J. Hematol. Oncol. 2019, 12, 135. [Google Scholar] [CrossRef]
- Zhao, Y.; Wen, S.; Li, H.; Pan, C.-W.; Wei, Y.; Huang, T.; Li, Z.; Yang, Y.; Fan, S.; Zhang, Y. Enhancer RNA promotes resistance to radiotherapy in bone-metastatic prostate cancer by m6A modification. Theranostics 2023, 13, 596–610. [Google Scholar] [CrossRef]
- Shriwas, O.; Mohapatra, P.; Mohanty, S.; Dash, R. The Impact of m6A RNA Modification in Therapy Resistance of Cancer: Implication in Chemotherapy, Radiotherapy, and Immunotherapy. Front. Oncol. 2021, 10, 612337. [Google Scholar] [CrossRef]
- Chen, J.; Ye, M.; Bai, J.; Hu, C.; Lu, F.; Gu, D.; Yu, P.; Tang, Q. Novel insights into the interplay between m6A modification and programmed cell death in cancer. Int. J. Biol. Sci. 2023, 19, 1748–1763. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lv, D.; Yan, C.; Su, H.; Zhang, X.; Shi, Y.; Ying, K. METTL3 promotes lung adenocarcinoma tumor growth and inhibits ferroptosis by stabilizing SLC7A11 m6A modification. Cancer Cell Int. 2022, 22, 11. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Gao, T.; Pang, B.; Su, X.; Guo, C.; Zhang, R.; Pang, Q. RNA binding protein NKAP protects glioblastoma cells from ferroptosis by promoting SLC7A11 mRNA splicing in an m6A-dependent manner. Cell Death Dis. 2022, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, G.; Wei, J.; Cao, H.; Jiang, G. ALKBH5 inhibits thyroid cancer progression by promoting ferroptosis through TIAM1–Nrf2/HO-1 axis. Mol. Cell. Biochem. 2023, 478, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, T.; Zhang, X.; Miao, Y.; Tian, X.; Yu, K.; Xu, X.; Niu, Y.; Guo, S.; Zhang, C.; et al. The m6A reader YTHDC2 inhibits lung adenocarcinoma tumorigenesis by suppressing SLC7A11-dependent antioxidant function. Redox Biol. 2021, 38, 101801. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, X.; Yu, K.; Xu, X.; Chen, T.; Shi, Y.; Wang, Y.; Qiu, S.; Guo, S.; Cui, J.; et al. Targeting SLC3A2 subunit of system XC− is essential for m6A reader YTHDC2 to be an endogenous ferroptosis inducer in lung adenocarcinoma. Free Radic. Biol. Med. 2021, 168, 25–43. [Google Scholar] [CrossRef]
- Xu, X.; Cui, J.; Wang, H.; Ma, L.; Zhang, X.; Guo, W.; Xue, X.; Wang, Y.; Qiu, S.; Tian, X.; et al. IGF2BP3 is an essential N6-methyladenosine biotarget for suppressing ferroptosis in lung adenocarcinoma cells. Mater. Today Bio 2022, 17, 100503. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Azzam, S.K.; Alsafar, H.; Sajini, A.A. FTO m6A Demethylase in Obesity and Cancer: Implications and Underlying Molecular Mechanisms. Int. J. Mol. Sci. 2022, 23, 3800. [Google Scholar] [CrossRef]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef]
- Shimura, T.; Kandimalla, R.; Okugawa, Y.; Ohi, M.; Toiyama, Y.; He, C.; Goel, A. Novel evidence for m6A methylation regulators as prognostic biomarkers and FTO as a potential therapeutic target in gastric cancer. Br. J. Cancer 2022, 126, 228–237. [Google Scholar] [CrossRef]
- Bi, J.; Yang, S.; Li, L.; Dai, Q.; Borcherding, N.; Wagner, B.A.; Buettner, G.R.; Spitz, D.R.; Leslie, K.K.; Zhang, J.; et al. Metadherin enhances vulnerability of cancer cells to ferroptosis. Cell Death Dis. 2019, 10, 682. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, T.; Wang, X.; Xiong, F.; Hu, Z.; Qiao, X.; Yuan, X.; Wang, D. ACSL3 and ACSL4, Distinct Roles in Ferroptosis and Cancers. Cancers 2022, 14, 5896. [Google Scholar] [CrossRef] [PubMed]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, Q.; Zuo, Z.-X.; Yuan, S.-Q.; Yu, K.; Zhang, Q.; Zhang, X.; Sheng, H.; Ju, H.-Q.; Cheng, H.; et al. Systematic Analysis of the Aberrances and Functional Implications of Ferroptosis in Cancer. iScience 2020, 23, 101302. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef]
- Weng, J.; Wang, C.; Wang, Y.; Tang, H.; Liang, J.; Liu, X.; Huang, H.; Hou, J. Beclin1 inhibits proliferation, migration and invasion in tongue squamous cell carcinoma cell lines. Oral. Oncol. 2014, 50, 983–990. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence |
|---|---|
| FTO | Forward: 5′-ACTTGGCTCCCTTATCTGACC-3′ |
| Reverse: 5′-TGTGCAGTGTGAGAAAGGCTT-3′ | |
| ACSL3 | Forward: 5′-GCCGAGTGGATGATAGCTGC-3′ |
| Reverse: 5′-ATGGCTGGACCTCCTAGAGTG-3′ | |
| GPX4 | Forward: 5′-GAGGCAAGACCGAAGTAAACTAC-3′ |
| Reverse: 5′-CCGAACTGGTTACACGGGAA-3′ | |
| PTGS2 | Forward: 5′-CTGGCGCTCAGCCATACAG-3′ |
| Reverse: 5′-CGCACTTATACTGGTCAAATCCC-3′ | |
| β-ACTIN | Forward: 5′-CTACCTCATGAAGATCCTCACCGA-3′ |
| Reverse: 5′-TTCTCCTTAATGTCACGCACGATT-3′ | |
| ACSL3 m6A | Forward: 5′-GACCCCTGAAACTGGTCTGG-3′ |
| Reverse: 5′-CCTTGCAGCTTGAGACATGC-3′ | |
| GPX4 m6A | Forward: 5′-TGGGGCAGACCCGAAAATC-3′ |
| Reverse: 5′-TATTCCCACAAGGTAGCCAGG-3′ |
| Antibody | Dilution | Manufacturer |
|---|---|---|
| anti-ACSL3 primary antibody | 1:2500 | Abcam, UK |
| anti-FTO primary antibody | 1:5000 | Abcam, UK |
| anti-GPX4 primary antibody | 1:3000 | Abcam, UK |
| anti-4-HNE primary antibody | 1:2500 | Abcam, UK |
| anti-GAPDH primary antibody | 1:1000 | Servicebio, China |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Li, H.; Cai, H.; Liang, J.; Jiang, Y.; Song, F.; Hou, C.; Hou, J. FTO Sensitizes Oral Squamous Cell Carcinoma to Ferroptosis via Suppressing ACSL3 and GPX4. Int. J. Mol. Sci. 2023, 24, 16339. https://doi.org/10.3390/ijms242216339
Wang Z, Li H, Cai H, Liang J, Jiang Y, Song F, Hou C, Hou J. FTO Sensitizes Oral Squamous Cell Carcinoma to Ferroptosis via Suppressing ACSL3 and GPX4. International Journal of Molecular Sciences. 2023; 24(22):16339. https://doi.org/10.3390/ijms242216339
Chicago/Turabian StyleWang, Ziyi, Hongyu Li, Hongshi Cai, Jianfeng Liang, Yaoqi Jiang, Fan Song, Chen Hou, and Jinsong Hou. 2023. "FTO Sensitizes Oral Squamous Cell Carcinoma to Ferroptosis via Suppressing ACSL3 and GPX4" International Journal of Molecular Sciences 24, no. 22: 16339. https://doi.org/10.3390/ijms242216339
APA StyleWang, Z., Li, H., Cai, H., Liang, J., Jiang, Y., Song, F., Hou, C., & Hou, J. (2023). FTO Sensitizes Oral Squamous Cell Carcinoma to Ferroptosis via Suppressing ACSL3 and GPX4. International Journal of Molecular Sciences, 24(22), 16339. https://doi.org/10.3390/ijms242216339