

CRISPR/Cas9-Mediated Genome Editing in Cancer Therapy
Abstract
:1. Introduction
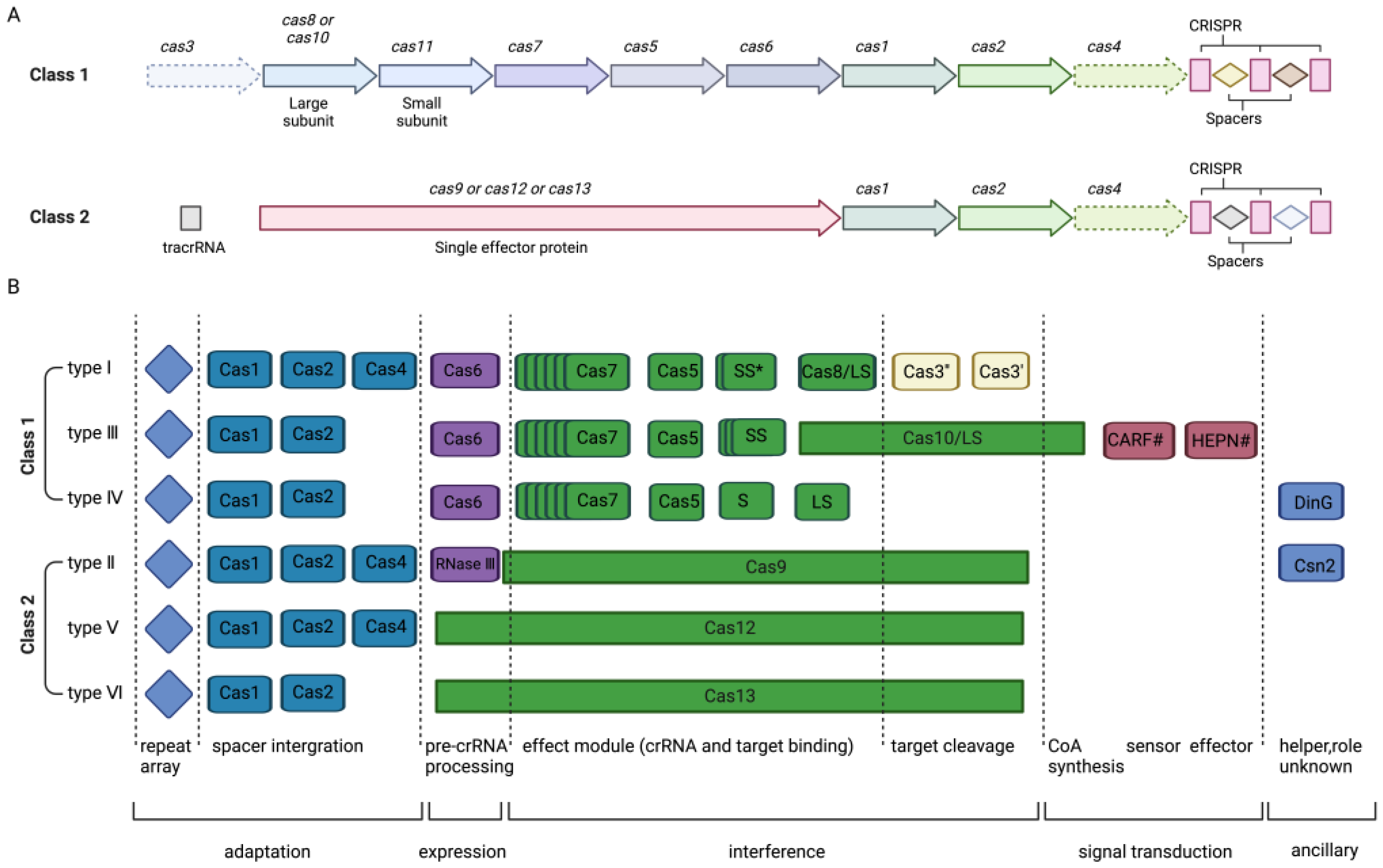
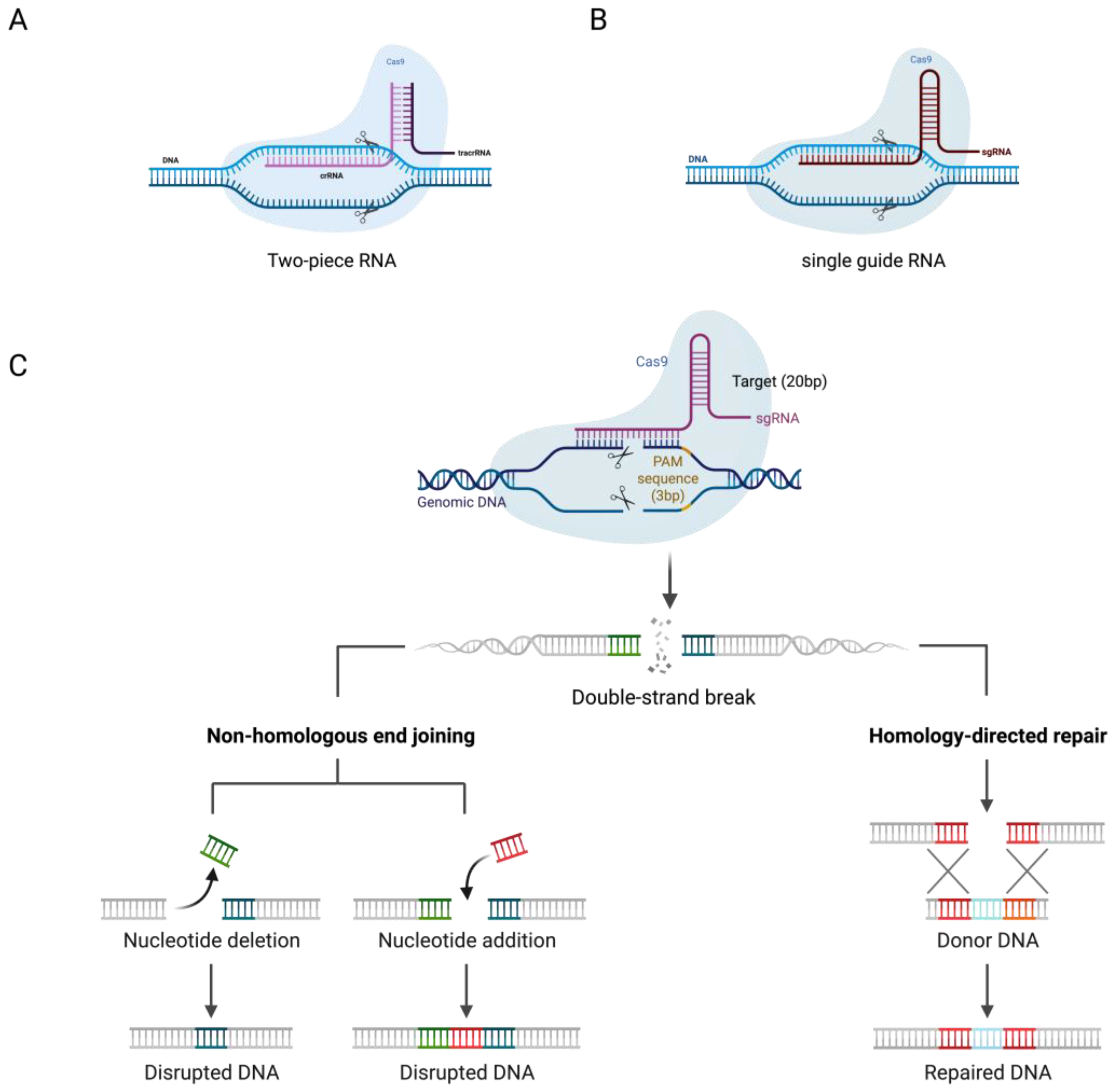
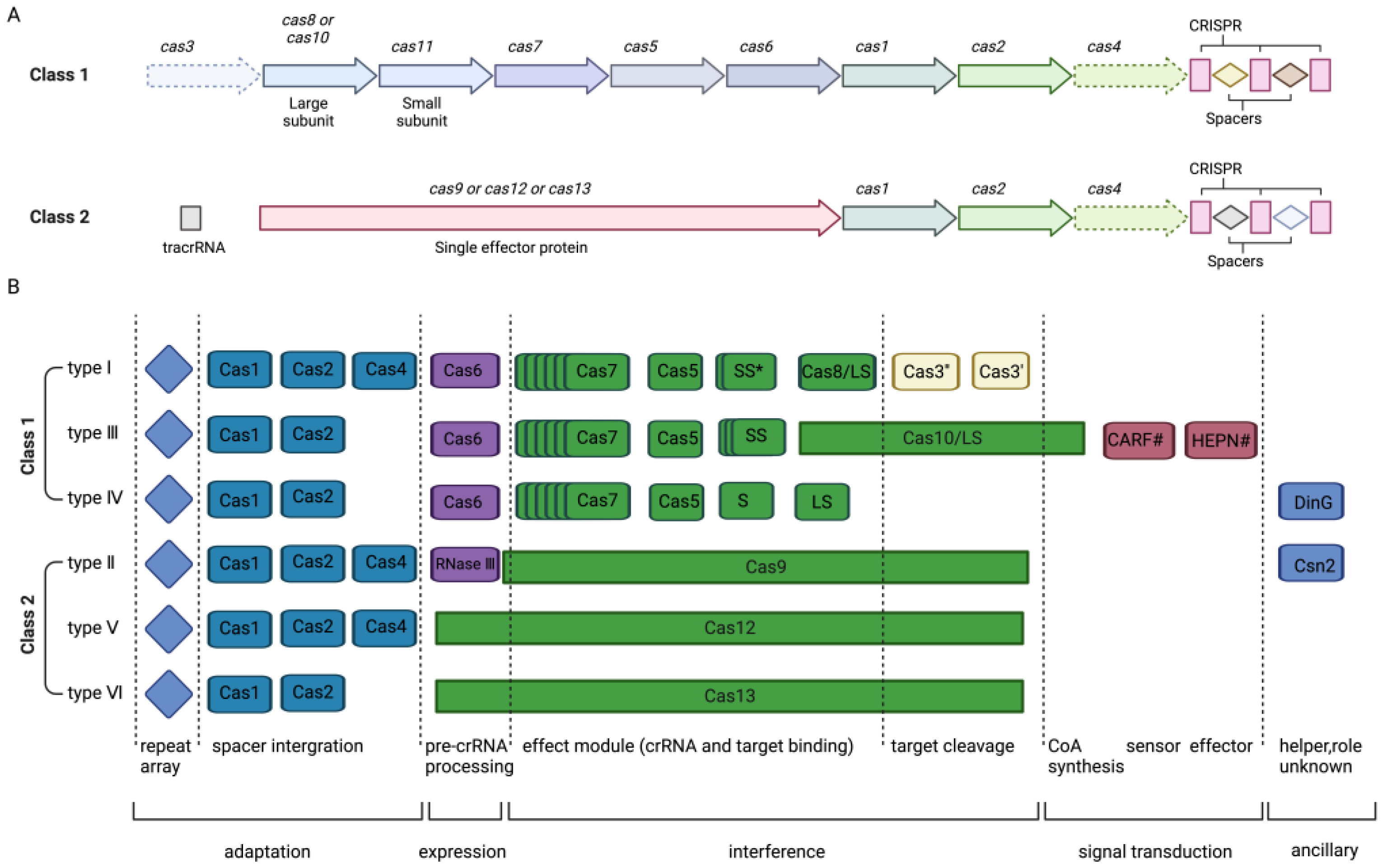
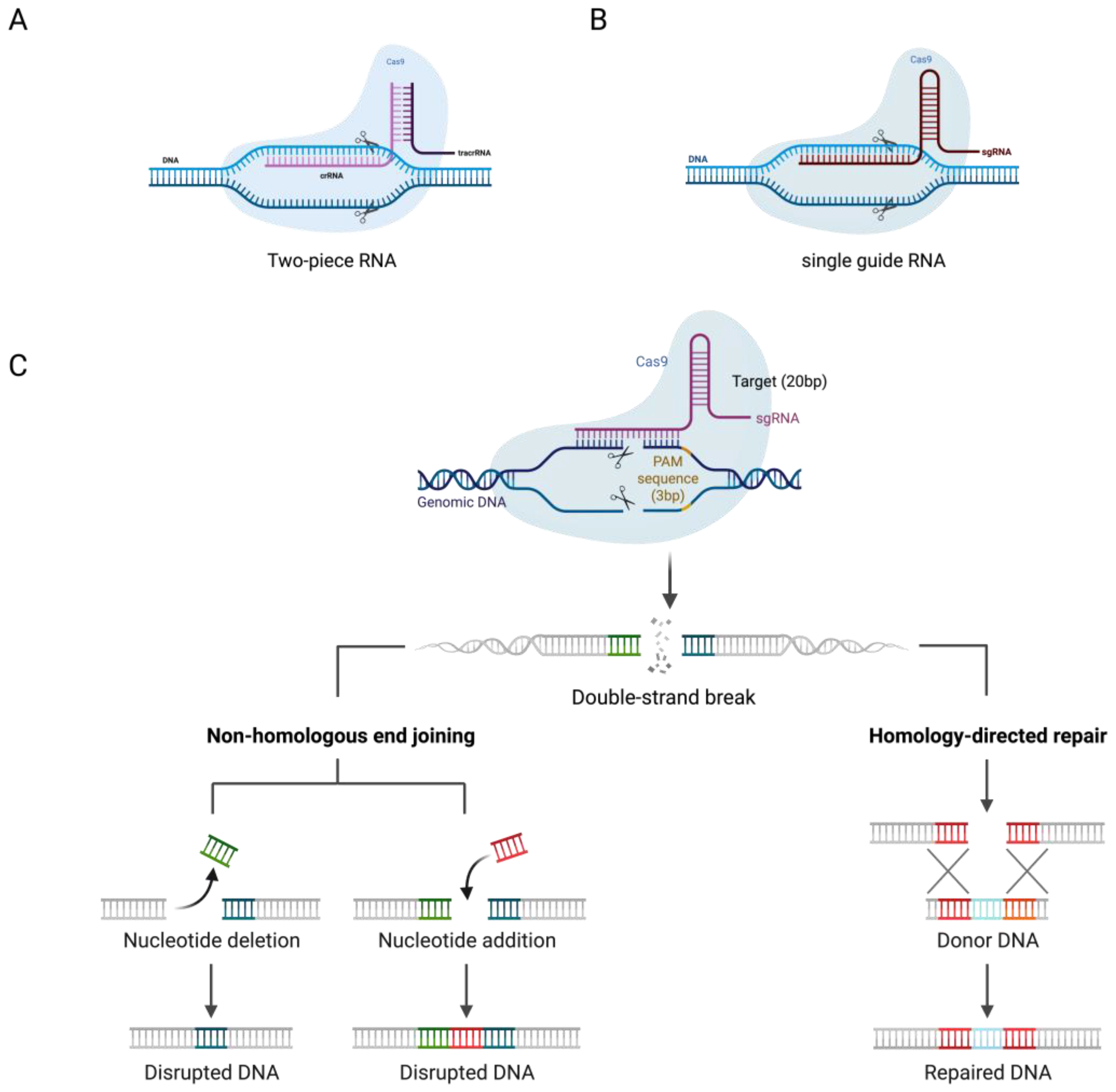
2. CRISPR/Cas System Mechanism
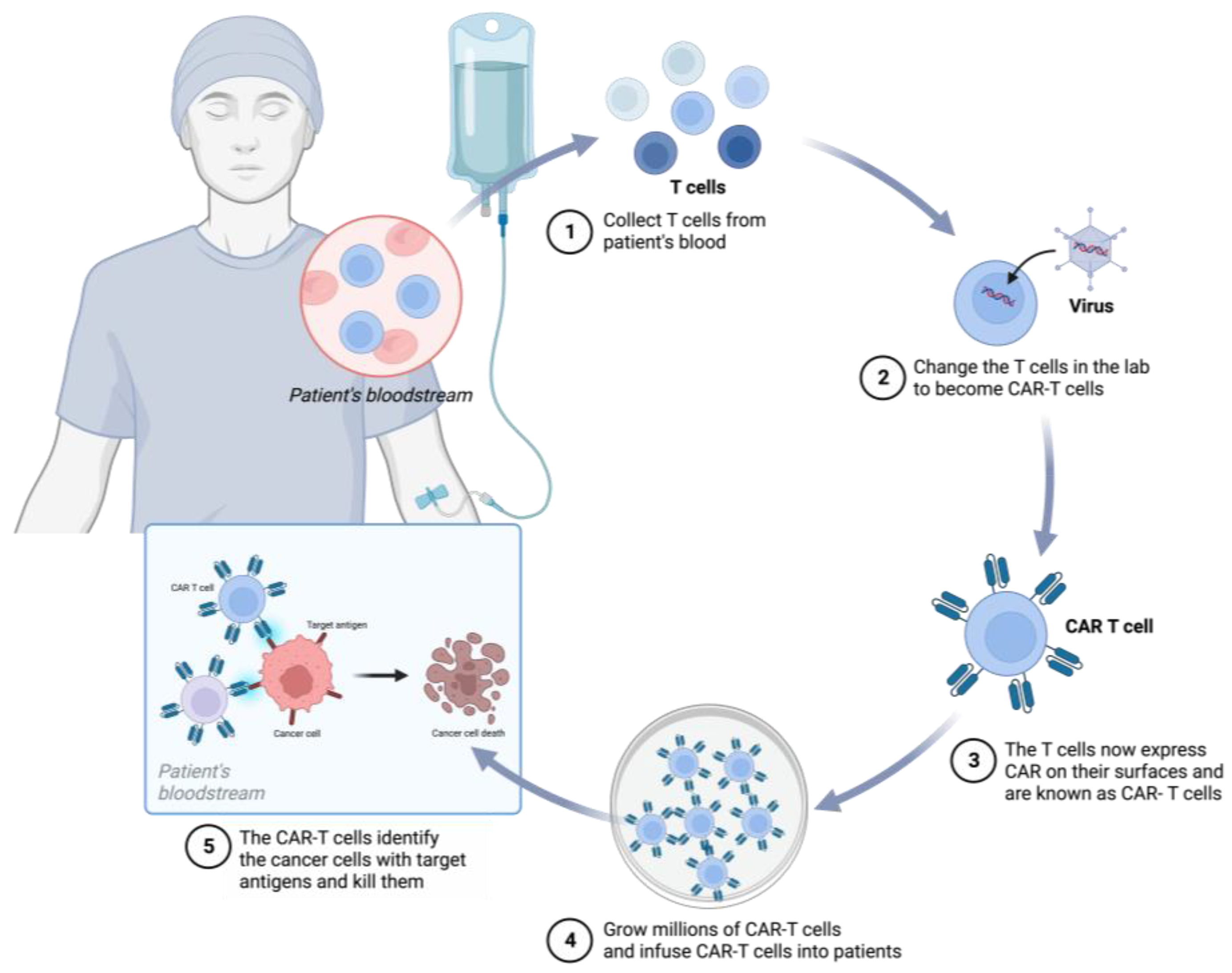
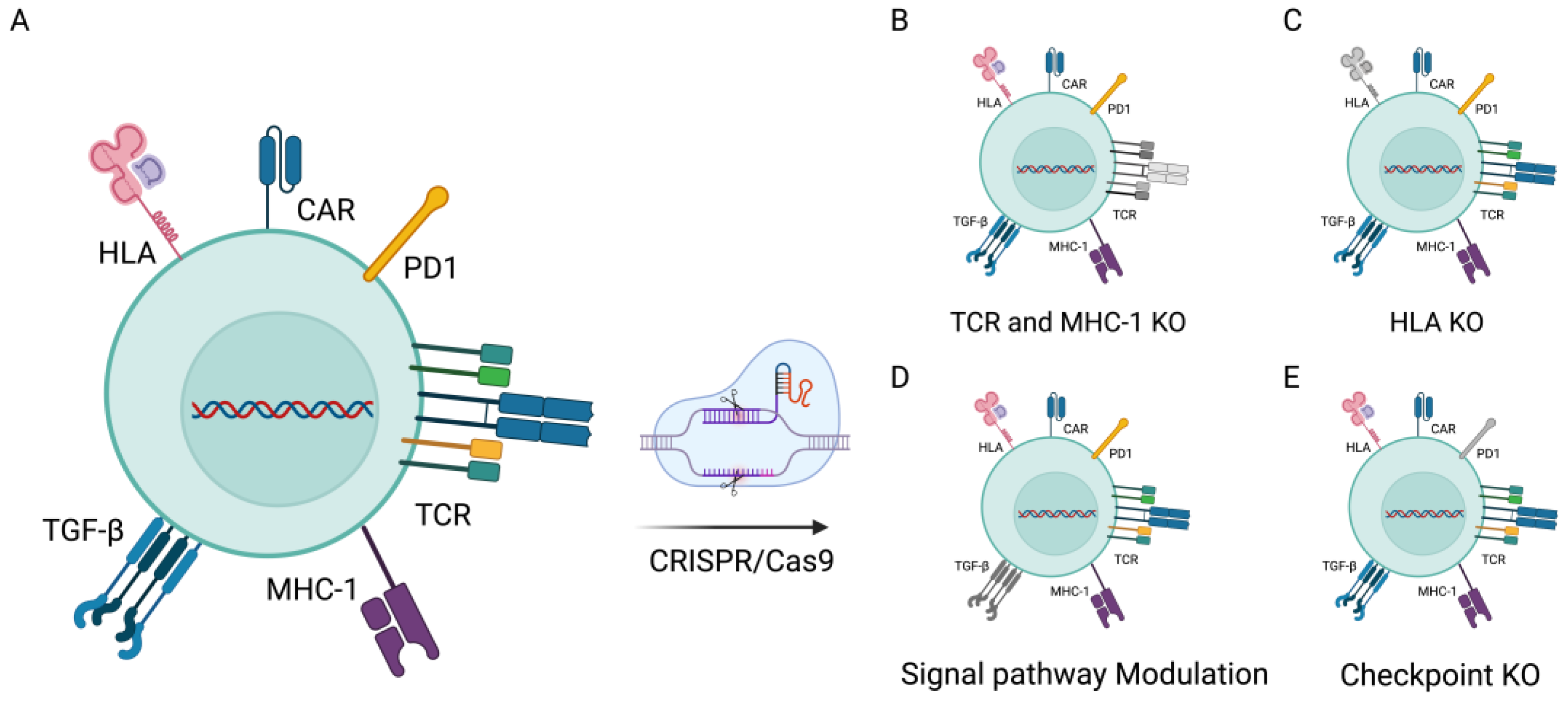
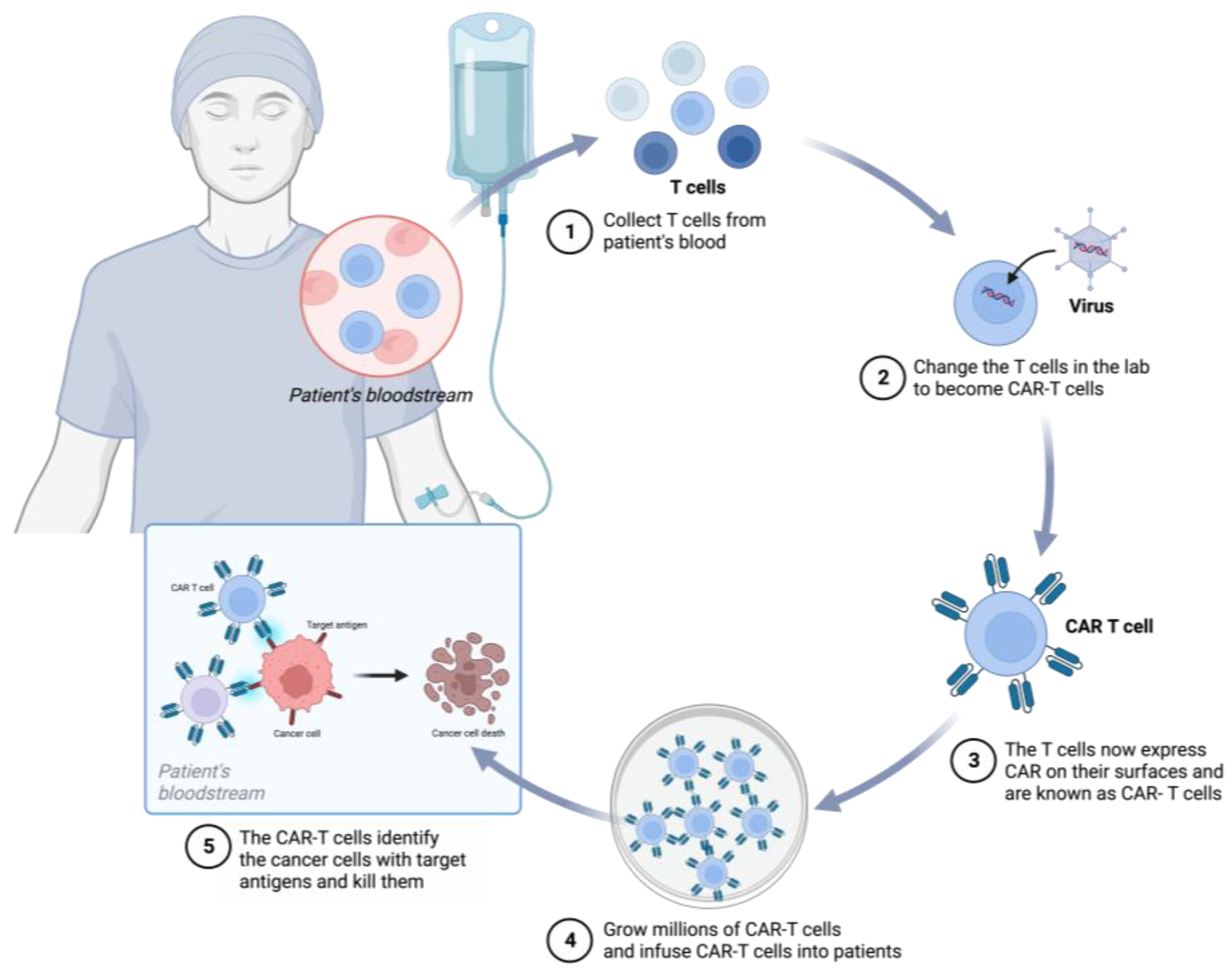
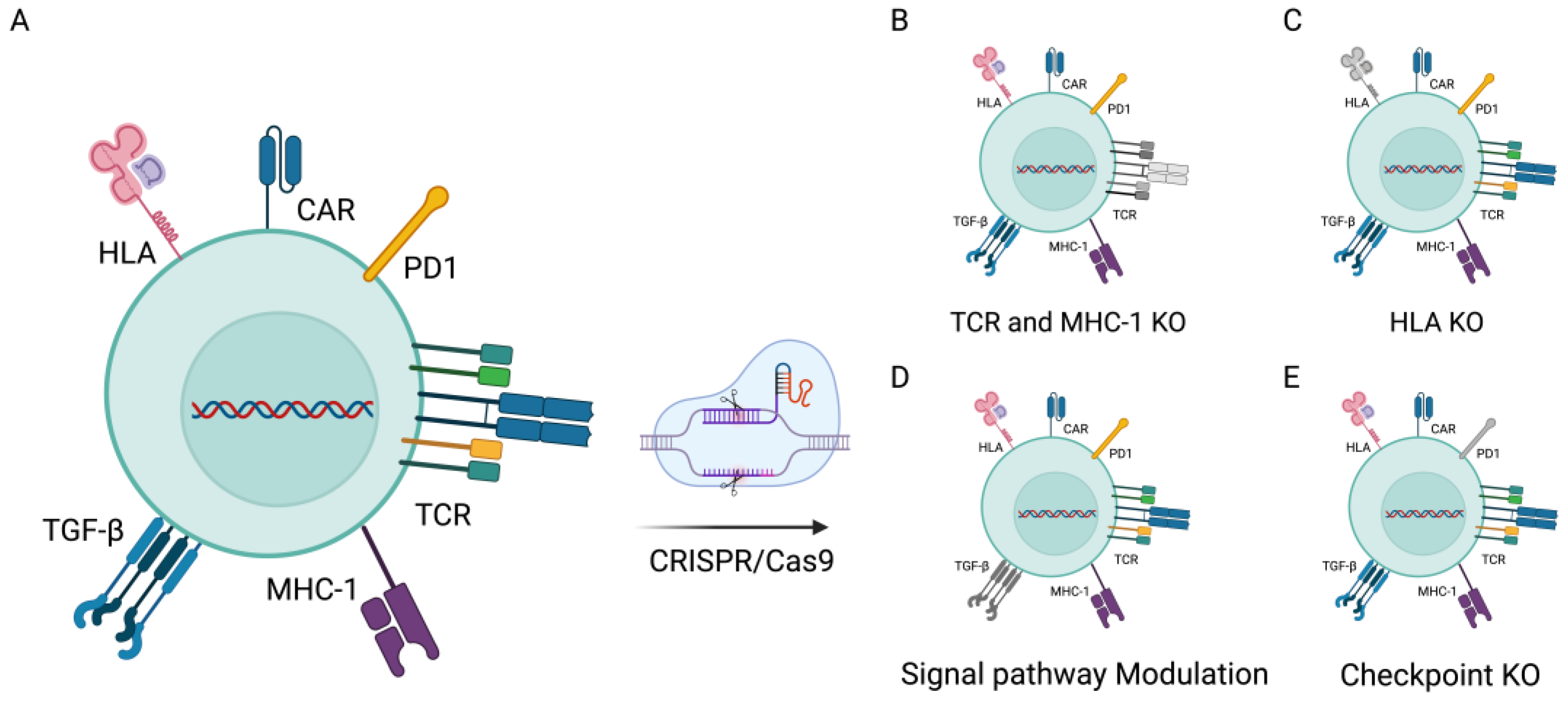
3. CRISPR/Cas9 in CAR-T-Cell Therapies
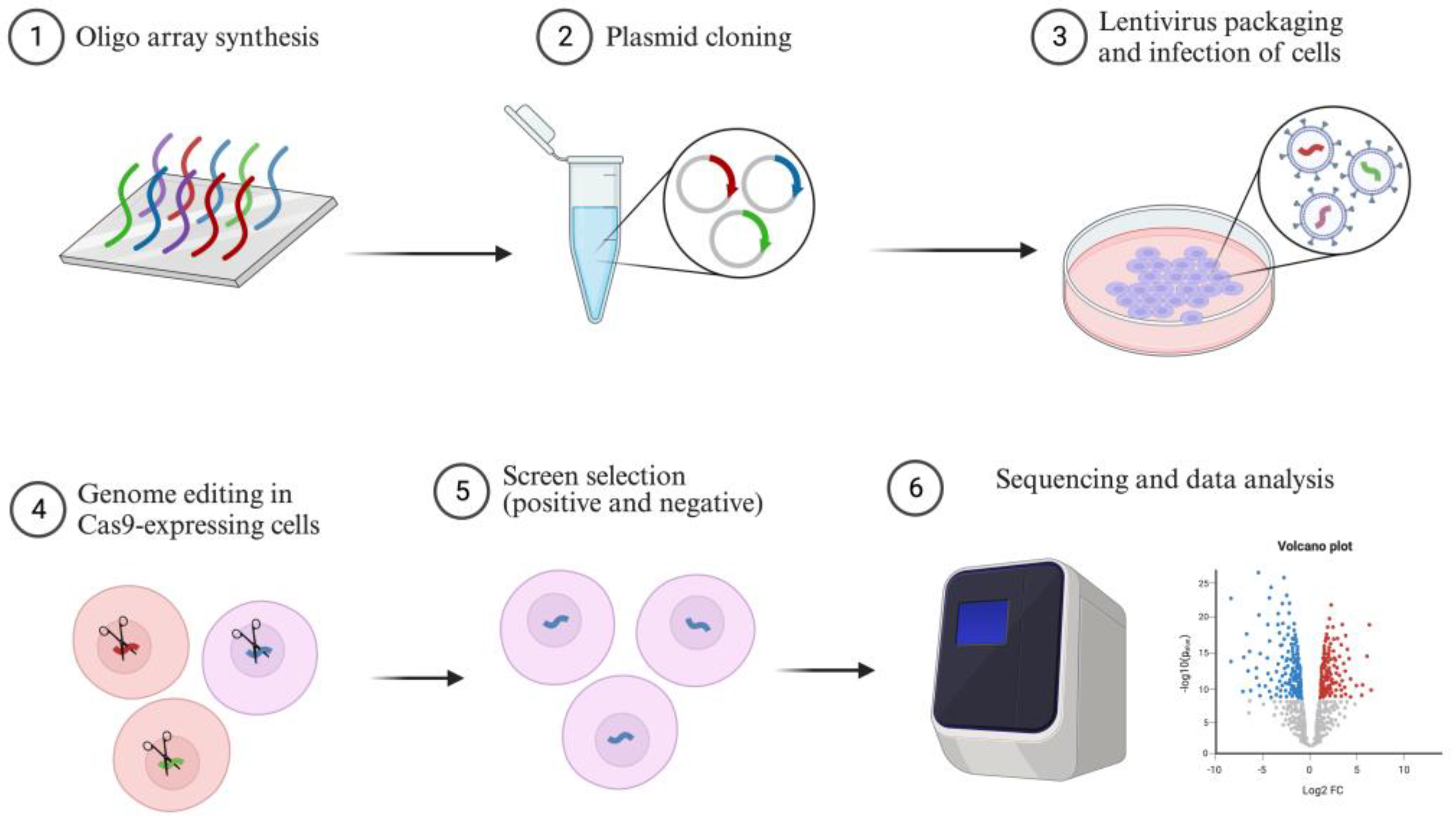
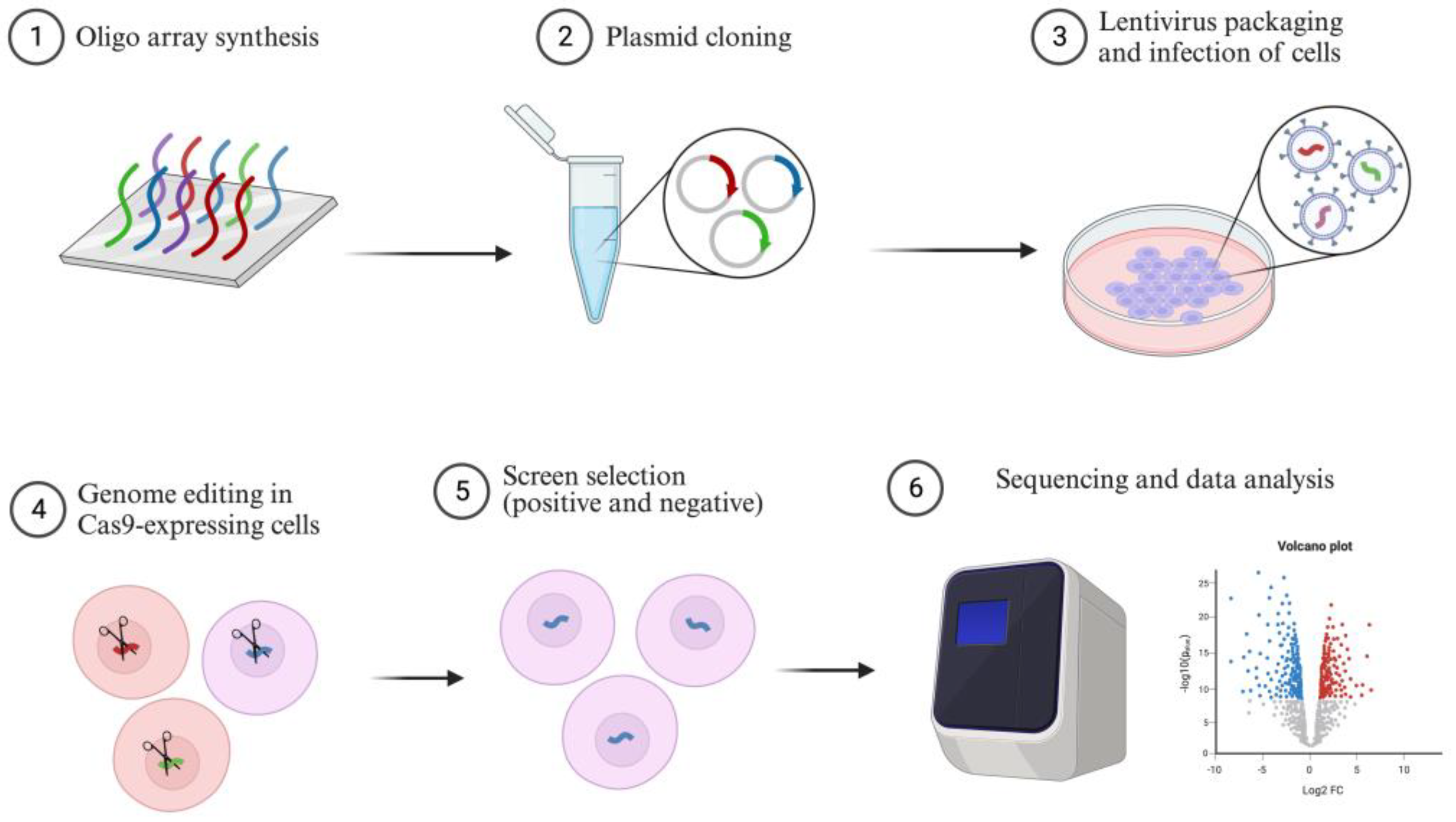
4. CRISPR Screening in Cancer
4.1. CRISPRKO Screening
4.2. CRISPRi Screening
4.3. CRISPRa Screening
5. CRISPR/Cas9 in a Cancer Model
5.1. In Vitro
5.2. In Vivo
6. Limitations of CRISPR/Cas9
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Rivera, F.J.; Jacks, T. Applications of the CRISPR-Cas9 system in cancer biology. Nat. Rev. Cancer 2015, 15, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H.; Carroll, D. Gene targeting using zinc finger nucleases. Nat. Biotechnol. 2005, 23, 967–973. [Google Scholar] [CrossRef]
- Li, T.; Huang, S.; Jiang, W.Z.; Wright, D.; Spalding, M.H.; Weeks, D.P.; Yang, B. TAL nucleases (TALNs): Hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 2011, 39, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef]
- Hendriks, D.; Clevers, H.; Artegiani, B. CRISPR-Cas Tools and Their Application in Genetic Engineering of Human Stem Cells and Organoids. Cell Stem Cell 2020, 27, 705–731. [Google Scholar] [CrossRef]
- Barrangou, R.; Marraffini, L.A. CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol. Cell 2014, 54, 234–244. [Google Scholar] [CrossRef]
- Wang, J.Y.; Doudna, J.A. CRISPR technology: A decade of genome editing is only the beginning. Science 2023, 379, eadd8643. [Google Scholar] [CrossRef]
- Fu, Y.W.; Dai, X.Y.; Wang, W.T.; Yang, Z.X.; Zhao, J.J.; Zhang, J.P.; Wen, W.; Zhang, F.; Oberg, K.C.; Zhang, L.; et al. Dynamics and competition of CRISPR-Cas9 ribonucleoproteins and AAV donor-mediated NHEJ, MMEJ and HDR editing. Nucleic Acids Res. 2021, 49, 969–985. [Google Scholar] [CrossRef]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef]
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in cancer biology and therapy. Nat. Rev. Cancer 2022, 22, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lu, H.; Ma, W.; Tian, W.; Lu, Z.; Yang, H.; Cai, Y.; Cai, P.; Sun, Y.; Zhou, Z.; et al. Genome-wide CRISPR screen identifies synthetic lethality between DOCK1 inhibition and metformin in liver cancer. Protein Cell 2022, 13, 825–841. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zheng, S.; Xie, X.; Ye, F.; Hu, X.; Tian, Z.; Yan, S.M.; Yang, L.; Kong, Y.; Tang, Y.; et al. N6-methyladenosine regulated FGFR4 attenuates ferroptotic cell death in recalcitrant HER2-positive breast cancer. Nat. Commun. 2022, 13, 2672. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Mahara, S.; Sun, C.; Doan, A.; Chua, H.K.; Xu, D.; Bian, J.; Li, Y.; Zhu, D.; Sooraj, D.; et al. Genome-scale CRISPR-Cas9 screen of Wnt/β-catenin signaling identifies therapeutic targets for colorectal cancer. Sci. Adv. 2021, 7, eabf2567. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Zhang, M.; Dong, L.; Ji, S.; Zhang, J.; Zhang, S.; Lin, Y.; Wang, X.; Ding, Z.; Qiu, S.; et al. Genome-Scale CRISPR screen identifies LAPTM5 driving lenvatinib resistance in hepatocellular carcinoma. Autophagy 2023, 19, 1184–1198. [Google Scholar] [CrossRef] [PubMed]
- Mohanraju, P.; Makarova, K.S.; Zetsche, B.; Zhang, F.; Koonin, E.V.; van der Oost, J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 2016, 353, aad5147. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013, 10, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Xu, Z.; Zhuang, Y.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Hematological Malignancies. J. Cancer 2021, 12, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Labanieh, L.; Mackall, C.L. CAR immune cells: Design principles, resistance and the next generation. Nature 2023, 614, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y.; et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat. Med. 2022, 28, 1189–1198. [Google Scholar] [CrossRef]
- Cao, B.; Liu, M.; Wang, L.; Zhu, K.; Cai, M.; Chen, X.; Feng, Y.; Yang, S.; Fu, S.; Zhi, C.; et al. Remodelling of tumour microenvironment by microwave ablation potentiates immunotherapy of AXL-specific CAR T cells against non-small cell lung cancer. Nat. Commun. 2022, 13, 6203. [Google Scholar] [CrossRef]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.F.; Bulsara, S.; et al. Glypican-3-Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef]
- Dai, H.; Wang, Y.; Lu, X.; Han, W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. J. Natl. Cancer Inst. 2016, 108, djv439. [Google Scholar] [CrossRef]
- Amini, L.; Silbert, S.K.; Maude, S.L.; Nastoupil, L.J.; Ramos, C.A.; Brentjens, R.J.; Sauter, C.S.; Shah, N.N.; Abou-El-Enein, M. Preparing for CAR T cell therapy: Patient selection, bridging therapies and lymphodepletion. Nat. Rev. Clin. Oncol. 2022, 19, 342–355. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Noh, K.E.; Lee, J.H.; Choi, S.Y.; Jung, N.C.; Nam, J.H.; Oh, J.S.; Song, J.Y.; Seo, H.G.; Wang, Y.; Lee, H.S.; et al. TGF-β/IL-7 Chimeric Switch Receptor-Expressing CAR-T Cells Inhibit Recurrence of CD19-Positive B Cell Lymphoma. Int. J. Mol. Sci. 2021, 22, 8706. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Razeghian, E.; Nasution, M.K.M.; Rahman, H.S.; Gardanova, Z.R.; Abdelbasset, W.K.; Aravindhan, S.; Bokov, D.O.; Suksatan, W.; Nakhaei, P.; Shariatzadeh, S.; et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res. Ther. 2021, 12, 428. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Li, P.J.; Blaeschke, F.; Nies, J.F.; Apathy, R.; Mowery, C.; Yu, R.; Nguyen, M.L.T.; Lee, Y.; Truong, A.; et al. Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell 2020, 181, 728–744.e721. [Google Scholar] [CrossRef] [PubMed]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Chen, D.; Ma, X.; Wang, Y.; Guo, Y.; Wei, J.; Tong, C.; Zhu, Q.; Lu, Y.; Yu, Y.; et al. CD58 loss in tumor cells confers functional impairment of CAR T cells. Blood Adv. 2022, 6, 5844–5856. [Google Scholar] [CrossRef]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef]
- Ferdosi, S.R.; Ewaisha, R.; Moghadam, F.; Krishna, S.; Park, J.G.; Ebrahimkhani, M.R.; Kiani, S.; Anderson, K.S. Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 2019, 10, 1842. [Google Scholar] [CrossRef]
- Crudele, J.M.; Chamberlain, J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 2018, 9, 3497. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef]
- Pan, K.; Farrukh, H.; Chittepu, V.; Xu, H.; Pan, C.X.; Zhu, Z. CAR race to cancer immunotherapy: From CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 2022, 41, 119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, G.; Wan, X. Challenges and new technologies in adoptive cell therapy. J. Hematol. Oncol. 2023, 16, 97. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the Human Genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Li, L.; Lin, Y.H.; Gopal, P.; Shen, S.; Zhou, K.; Yu, X.; Sharma, T.; Zhang, Y.; Siegwart, D.J.; et al. In vivo CRISPR screening identifies BAZ2 chromatin remodelers as druggable regulators of mammalian liver regeneration. Cell Stem Cell 2022, 29, 372–385.e378. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zheng, Y.; Sun, S.; Li, W.; Song, M.; Ji, Q.; Wu, Z.; Liu, Z.; Fan, Y.; Liu, F.; et al. A genome-wide CRISPR-based screen identifies KAT7 as a driver of cellular senescence. Sci. Transl. Med. 2021, 13, eabd2655. [Google Scholar] [CrossRef]
- Dubrot, J.; Du, P.P.; Lane-Reticker, S.K.; Kessler, E.A.; Muscato, A.J.; Mehta, A.; Freeman, S.S.; Allen, P.M.; Olander, K.E.; Ockerman, K.M.; et al. In vivo CRISPR screens reveal the landscape of immune evasion pathways across cancer. Nat. Immunol. 2022, 23, 1495–1506. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Liu, H.; Zhang, J.; Wang, J.; Xia, J.; Zhang, Y.; Yu, X.; Ma, J.; Huang, M.; et al. Genome-wide CRISPR/Cas9 library screen identifies PCMT1 as a critical driver of ovarian cancer metastasis. J. Exp. Clin. Cancer Res. 2022, 41, 24. [Google Scholar] [CrossRef]
- Feng, X.; Tang, M.; Dede, M.; Su, D.; Pei, G.; Jiang, D.; Wang, C.; Chen, Z.; Li, M.; Nie, L.; et al. Genome-wide CRISPR screens using isogenic cells reveal vulnerabilities conferred by loss of tumor suppressors. Sci. Adv. 2022, 8, eabm6638. [Google Scholar] [CrossRef]
- Wang, C.; Wang, G.; Feng, X.; Shepherd, P.; Zhang, J.; Tang, M.; Chen, Z.; Srivastava, M.; McLaughlin, M.E.; Navone, N.M.; et al. Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene 2019, 38, 2451–2463. [Google Scholar] [CrossRef]
- Tang, M.; Pei, G.; Su, D.; Wang, C.; Feng, X.; Srivastava, M.; Chen, Z.; Zhao, Z.; Chen, J. Genome-wide CRISPR screens reveal cyclin C as synthetic survival target of BRCA2. Nucleic Acids Res. 2021, 49, 7476–7491. [Google Scholar] [CrossRef] [PubMed]
- Awwad, S.W.; Serrano-Benitez, A.; Thomas, J.C.; Gupta, V.; Jackson, S.P. Revolutionizing DNA repair research and cancer therapy with CRISPR–Cas screens. Nat. Rev. Mol. Cell Biol. 2023, 24, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.B.; Tang, K.; Zhou, X.; Zhou, J.J.; Chen, S. Tumor immunology CRISPR screening: Present, past, and future. Trends Cancer 2022, 8, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef]
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015, 16, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Braun, C.J.; Adames, A.C.; Saur, D.; Rad, R. Tutorial: Design and execution of CRISPR in vivo screens. Nat. Protoc. 2022, 17, 1903–1925. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Wang, X.; Tokheim, C.; Gu, S.S.; Wang, B.; Tang, Q.; Li, Y.; Traugh, N.; Zeng, Z.; Zhang, Y.; Li, Z.; et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell 2021, 184, 5357–5374.e5322. [Google Scholar] [CrossRef]
- Shi, Z.D.; Hao, L.; Han, X.X.; Wu, Z.X.; Pang, K.; Dong, Y.; Qin, J.X.; Wang, G.Y.; Zhang, X.M.; Xia, T.; et al. Targeting HNRNPU to overcome cisplatin resistance in bladder cancer. Mol. Cancer 2022, 21, 37. [Google Scholar] [CrossRef]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, J.Z.; Luo, X.Q.; Wan, G.H.; Tang, Y.L.; Wang, Q.P. The application of genome-wide CRISPR-Cas9 screens to dissect the molecular mechanisms of toxins. Comput. Struct. Biotechnol. J. 2022, 20, 5076–5084. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.; Liu, L.; Taotao, S.; Tuano, N.; Chaturvedi, R.; Huang, K.K.; Itman, C.; Mandoli, A.; Qamra, A.; Hu, C.; et al. CRISPRi enables isoform-specific loss-of-function screens and identification of gastric cancer-specific isoform dependencies. Genome Biol. 2021, 22, 47. [Google Scholar]
- Liu, S.J.; Malatesta, M.; Lien, B.V.; Saha, P.; Thombare, S.S.; Hong, S.J.; Pedraza, L.; Koontz, M.; Seo, K.; Horlbeck, M.A.; et al. CRISPRi-based radiation modifier screen identifies long non-coding RNA therapeutic targets in glioma. Genome Biol. 2020, 21, 83. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; PRIyer, E.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef]
- Bester, A.C.; Lee, J.D.; Chavez, A.; Lee, Y.R.; Nachmani, D.; Vora, S.; Victor, J.; Sauvageau, M.; Monteleone, E.; Rinn, J.L.; et al. An Integrated Genome-wide CRISPRa Approach to Functionalize lncRNAs in Drug Resistance. Cell 2018, 173, 649–664.e620. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, G.; Meng, Q.; Huang, S.; Guo, P.; Leng, Q.; Sun, L.; Liu, G.; Huang, X.; Liu, J. Precise tumor immune rewiring via synthetic CRISPRa circuits gated by concurrent gain/loss of transcription factors. Nat. Commun. 2022, 13, 1454. [Google Scholar] [CrossRef]
- Joung, J.; Kirchgatterer, P.C.; Singh, A.; Cho, J.H.; Nety, S.P.; Larson, R.C.; Macrae, R.K.; Deasy, R.; Tseng, Y.-Y.; Maus, M.V.; et al. CRISPR activation screen identifies BCL-2 proteins and B3GNT2 as drivers of cancer resistance to T cell-mediated cytotoxicity. Nat. Commun. 2022, 13, 1606. [Google Scholar] [CrossRef] [PubMed]
- Tejero, R.; Huang, Y.; Katsyv, I.; Kluge, M.; Lin, J.Y.; Tome-Garcia, J.; Daviaud, N.; Wang, Y.; Zhang, B.; Tsankova, N.M.; et al. Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. EBioMedicine 2019, 42, 252–269. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Whittle, J.R.; Vaillant, F.; Chen, H.R.; Dawson, C.; Liu, K.; Geurts, M.H.; Herold, M.J.; Clevers, H.; Lindeman, G.J.; et al. Modeling Breast Cancer Using CRISPR-Cas9-Mediated Engineering of Human Breast Organoids. J. Natl. Cancer Inst. 2020, 112, 540–544. [Google Scholar] [CrossRef]
- Zhao, R.; Kaakati, R.; Liu, X.; Xu, L.; Lee, A.K.; Bachelder, R.; Li, C.Y.; Hollenbeck, S.T. CRISPR/Cas9-Mediated BRCA1 Knockdown Adipose Stem Cells Promote Breast Cancer Progression. Plast. Reconstr. Surg. 2019, 143, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Sathe, A.; Grimes, S.M.; Lau, B.T.; Chen, J.; Suarez, C.; Huang, R.J.; Poultsides, G.; Ji, H.P. Single-Cell Genomic Characterization Reveals the Cellular Reprogramming of the Gastric Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 2640–2653. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Kim, J. Autochthonous murine models for the study of smoker and never-smoker associated lung cancers. Transl. Lung Cancer Res. 2018, 7, 464–486. [Google Scholar] [CrossRef]
- Zhong, W.; Myers, J.S.; Wang, F.; Wang, K.; Lucas, J.; Rosfjord, E.; Lucas, J.; Hooper, A.T.; Yang, S.; Lemon, L.A.; et al. Comparison of the molecular and cellular phenotypes of common mouse syngeneic models with human tumors. BMC Genom. 2020, 21, 2. [Google Scholar] [CrossRef]
- Wang, J.G.; Geddings, J.E.; Aleman, M.M.; Cardenas, J.C.; Chantrathammachart, P.; Williams, J.C.; Kirchhofer, D.; Bogdanov, V.Y.; Bach, R.R.; Rak, J.; et al. Tumor-derived tissue factor activates coagulation and enhances thrombosis in a mouse xenograft model of human pancreatic cancer. Blood 2012, 119, 5543–5552. [Google Scholar] [CrossRef]
- Lai, H.; Liu, C.; Hou, L.; Lin, W.; Chen, T.; Hong, A. TRPM8-regulated calcium mobilization plays a critical role in synergistic chemosensitization of Borneol on Doxorubicin. Theranostics 2020, 10, 10154–10170. [Google Scholar] [CrossRef]
- Erstad, D.J.; Sojoodi, M.; Taylor, M.S.; Ghoshal, S.; Razavi, A.A.; Graham-O’Regan, K.A.; Bardeesy, N.; Ferrone, C.R.; Lanuti, M.; Caravan, P.; et al. Orthotopic and heterotopic murine models of pancreatic cancer and their different responses to FOLFIRINOX chemotherapy. Dis. Model. Mech. 2018, 11, dmm034793. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.P.; Chan, C.M.; Tung, L.N.; Wang, H.K.; Law, S. Tumor xenograft animal models for esophageal squamous cell carcinoma. J. Biomed. Sci. 2018, 25, 66. [Google Scholar] [CrossRef] [PubMed]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Asadzadeh Aghdaei, H.; Baghaei, K. Patient-derived xenograft (PDX) models, applications and challenges in cancer research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, E.; Schmid, J.P.; Jurinovic, V.; Becker, M.; Wirth, A.K.; Ludwig, R.; Kreissig, S.; Duque Angel, T.V.; Amend, D.; Hunt, K.; et al. Combined proteomics and CRISPR–Cas9 screens in PDX identify ADAM10 as essential for leukemia in vivo. Mol. Cancer 2023, 22, 107. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.; Hu, J.; Wang, H.; Hyle, J.; Zhang, Y.; Du, G.; Konopleva, M.Y.; Kornblau, S.M.; Djekidel, M.N.; Rosikiewicz, W.; et al. Interrogating bromodomain inhibitor resistance in KMT2A-rearranged leukemia through combinatorial CRISPR screens. Proc. Natl. Acad. Sci. USA 2023, 120, e2220134120. [Google Scholar] [CrossRef]
- Xu, W.W.; Liao, L.; Dai, W.; Zheng, C.C.; Tan, X.P.; He, Y.; Zhang, Q.H.; Huang, Z.H.; Chen, W.Y.; Qin, Y.R.; et al. Genome-wide CRISPR/Cas9 screening identifies a targetable MEST-PURA interaction in cancer metastasis. EBioMedicine 2023, 92, 104587. [Google Scholar] [CrossRef]
- Li, D.; Zhou, H.; Zeng, X. Battling CRISPR-Cas9 off-target genome editing. Cell Biol. Toxicol. 2019, 35, 403–406. [Google Scholar] [CrossRef]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef]
- Gong, S.; Yu, H.H.; Johnson, K.A.; Taylor, D.W. DNA Unwinding Is the Primary Determinant of CRISPR-Cas9 Activity. Cell Rep. 2018, 22, 359–371. [Google Scholar] [CrossRef]
- Ahmadi, S.E.; Soleymani, M.; Shahriyary, F.; Amirzargar, M.R.; Ofoghi, M.; Fattahi, M.D.; Safa, M. Viral vectors and extracellular vesicles: Innate delivery systems utilized in CRISPR/Cas-mediated cancer therapy. Cancer Gene Ther. 2023, 30, 936–954. [Google Scholar] [CrossRef]
- Chen, F.; Alphonse, M.; Liu, Q. Strategies for nonviral nanoparticle-based delivery of CRISPR/Cas9 therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1609. [Google Scholar] [CrossRef]
- Kazemian, P.; Yu, S.Y.; Thomson, S.B.; Birkenshaw, A.; Leavitt, B.R.; Ross, C.J.D. Lipid-Nanoparticle-Based Delivery of CRISPR/Cas9 Genome-Editing Components. Mol. Pharm. 2022, 19, 1669–1686. [Google Scholar] [CrossRef]
- Rosenblum, D.; Gutkin, A.; Kedmi, R.; Ramishetti, S.; Veiga, N.; Jacobi, A.M.; Schubert, M.S.; Friedmann-Morvinski, D.; Cohen, Z.R.; Behlke, M.A.; et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020, 6, eabc9450. [Google Scholar] [CrossRef]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Jiang, L.; Ingelshed, K.; Shen, Y.; Boddul, S.V.; Iyer, V.S.; Kasza, Z.; Sedimbi, S.; Lane, D.P.; Wermeling, F. CRISPR/Cas9-Induced DNA Damage Enriches for Mutations in a p53-Linked Interactome: Implications for CRISPR-Based Therapies. Cancer Res. 2022, 82, 36–45. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Autologous CAR-T-Cell Therapy | Allogeneic CAR-T-Cell Therapy |
|---|---|---|
| Definition | Patient’s own T cells | Donor T cells |
| Immunocompatibility | High | Moderate/Low |
| Preparation Time | Longer | Shorter |
| Cost | Higher | lower |
| GvHD Risks | Low | Moderate/High |
| Treatment Delay | Possible | Less |
| Indications | Most treatments | Specific cases |
| Efficacy | Variable | Higher |
| Cost Effectiveness | Moderate | Higher |
| CAR-T Cell | Tumor Type | Phase of Development | Country | Clinical Trial Identifier |
|---|---|---|---|---|
| αPD1-MSLN-CAR-T cells | Solid Tumor | Early Phase 1 | China | NCT05373147 |
| CD20/CD22 dual-targeted CAR-T cells | Lymphoid Hematological Malignancies | Early Phase 1 | China | NCT04283006 |
| bi-4SCAR CD19/79b CAR-T cells | B-Cell Malignancies | Phase 1 | China | NCT05436509 |
| CD5.CAR/28zeta CAR-T cells | T-Cell Malignancies | Phase 1 | America | NCT03081910 |
| CD19 CAR-T cells | Lymphoblastic B-Cell Lymphoma | Phase 1 | Belarus | NCT05333302 |
| GPC3 CAR-T cells | Hepatocellular Carcinoma | Phase 1 | America | NCT05003895 |
| CD44v6 CAR-T cells | Cancers Which Are CD44v6-Positive | Phase 2 | China | NCT04427449 |
| UCD19 CAR-T cells | B-cell Acute Lymphoblastic Leukemia; B-Cell Non-Hodgkin Lymphoma | Phase 2 | America | NCT04544592 |
| Target Carcinoma | Screening Purpose | CRISPR Screening Type | Experimental Setting | Candidate Gene | Literature Reference |
|---|---|---|---|---|---|
| Breast cancer | immune therapy | CRISPRKO | in vivo | Cop1 | [58] |
| Bladder cancer | drug resistance | CRISPRKO | in vitro | HNRNPU | [59] |
| Gastric cancer | isoforms function | CRISPRi | in vitro | ZFHX3 | [63] |
| Glioblastoma | lncRNA function | CRISPRi | in vitro | lncGRS-1 | [65] |
| Acute myeloid leukemia | lncRNA | CRISPRa | in vitro/vivo | GAS6-AS2 | [69] |
| Lung cancer | immune therapy | CRISPRa | in vitro | IFNG | [70] |
| Melanoma | immune therapy | CRISPRa | in vitro | BCL-2/B3GNT2 | [71] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, S.; Liu, J.; Han, X.; Tang, M. CRISPR/Cas9-Mediated Genome Editing in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 16325. https://doi.org/10.3390/ijms242216325
Ding S, Liu J, Han X, Tang M. CRISPR/Cas9-Mediated Genome Editing in Cancer Therapy. International Journal of Molecular Sciences. 2023; 24(22):16325. https://doi.org/10.3390/ijms242216325
Chicago/Turabian StyleDing, Shuai, Jinfeng Liu, Xin Han, and Mengfan Tang. 2023. "CRISPR/Cas9-Mediated Genome Editing in Cancer Therapy" International Journal of Molecular Sciences 24, no. 22: 16325. https://doi.org/10.3390/ijms242216325
APA StyleDing, S., Liu, J., Han, X., & Tang, M. (2023). CRISPR/Cas9-Mediated Genome Editing in Cancer Therapy. International Journal of Molecular Sciences, 24(22), 16325. https://doi.org/10.3390/ijms242216325