
Blood–Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies

 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
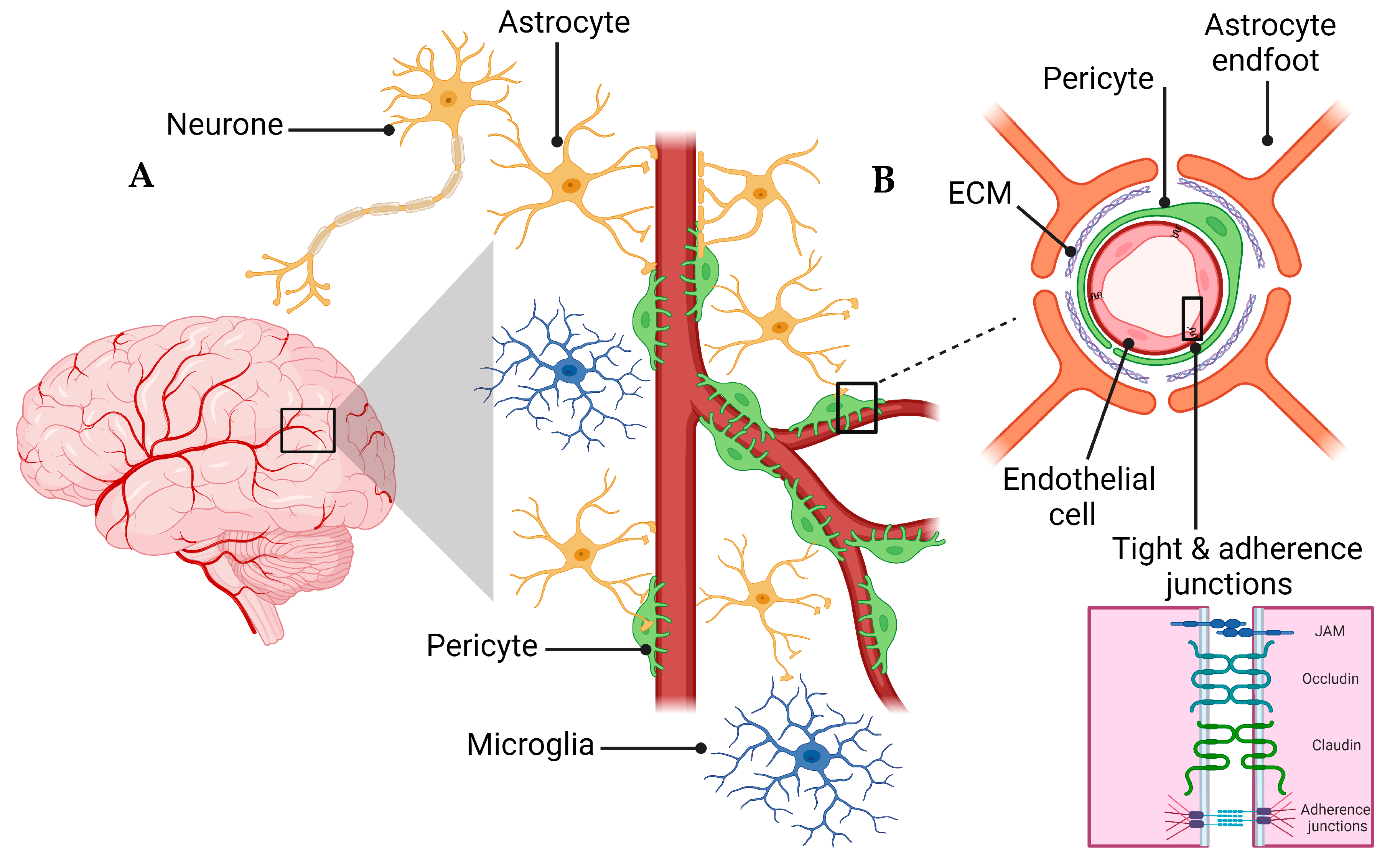
2. Overview of the NVU and the BBB
3. BBB Dysregulation with Aging
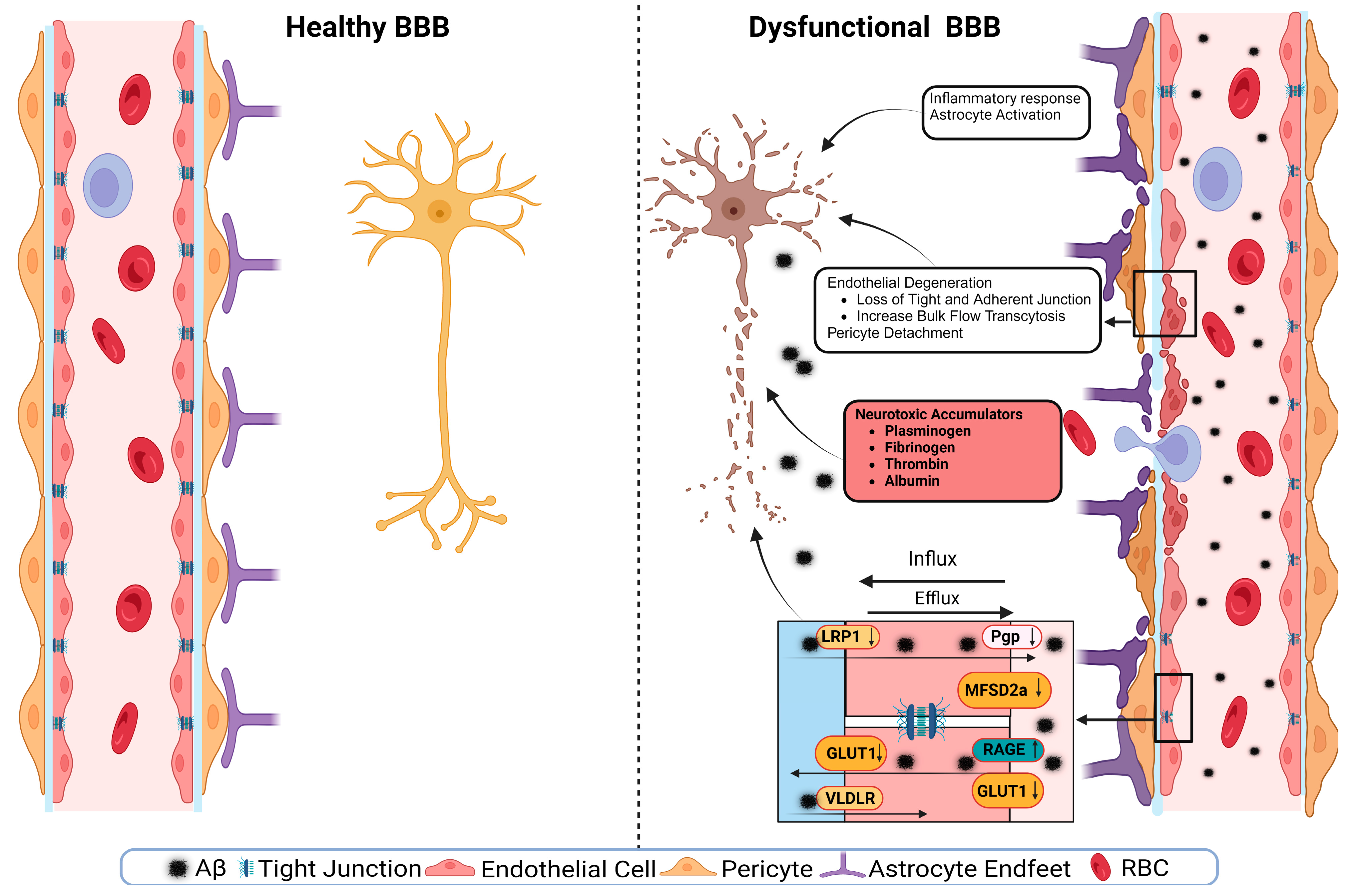
4. BBB Dysfunction in AD
4.1. Effect of Comorbidities on BBB
4.2. ApoE and BBB Dysfunction in AD
4.3. Tight Junctions’ Role in BBB Health and Disease
4.4. Transporters’ Role in BBB Health and Disease
4.4.1. SLC Transporters
4.4.2. ABC Transporters
4.4.3. MFSD2A Symporter
4.4.4. LRP1
4.4.5. RAGEs
4.4.6. Multidrug Resistance-Associated Proteins (MRPs)
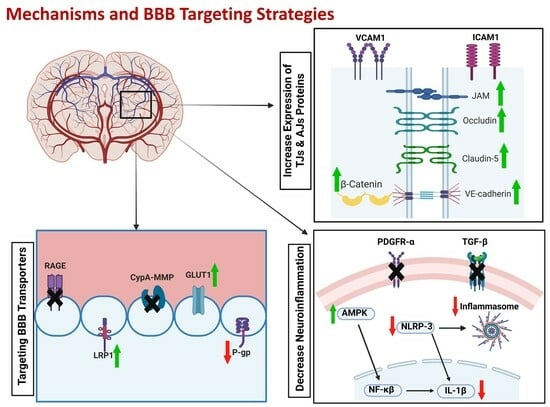
5. BBB Breakdown Mechanisms
6. The BBB as a Therapeutic Target
6.1. Enhancing Aβ Clearance across the BBB
6.1.1. RAGEs
6.1.2. LRP1
6.1.3. P-gp
6.2. Improving BBB Integrity and Function
6.2.1. Tight and Adherence Junction Proteins
6.2.2. GLUT1
6.2.3. MFSD2a
6.2.4. AQP-4
6.2.5. PCSK9
6.3. Targeting Neuroinflammation
6.3.1. TGF-β
6.3.2. VCAM and ICAM-1
6.3.3. NLRP3 Inflammasome
6.3.4. AMPK and cAMP
6.3.5. CypA-MMP-9
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar]
- Alzheimer’s association. 2023 Alzheimer’s disease facts and figures. Alzheimers Dement 2023, 19, 1598–1695. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Grants Accelerated Approval for Alzheimer’s Disease Treatment. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment (accessed on 15 May 2023).
- FDA. FDA Grants Accelerated Approval for Alzheimer’s Drug. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug (accessed on 15 May 2023).
- Marucci, G.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of Alzheimer disease: Back to the future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S. Genetics of Alzheimer disease. Contin. Lifelong Learn. Neurol. 2022, 28, 852–871. [Google Scholar] [CrossRef]
- Rabinovici, G.D. Late-onset Alzheimer Disease. Contin. Minneap. Minn. 2019, 25, 14–33. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Knopman, D.S.; Roberts, R. Vascular risk factors: Imaging and neuropathologic correlates. J. Alzheimer’s Dis. 2010, 20, 699–709. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.-d.-C.; Diaz-Cintra, S. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, C.; Wang, Z.; Tan, J. Involvement of cerebrovascular abnormalities in the pathogenesis and progression of Alzheimer’s disease: An adrenergic approach. Aging Albany NY 2021, 13, 21791–21806. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Sachs, M.C.; Zhou, Y.; Monsell, S.E.; Saver, J.L.; Vinters, H.V. Clinical predictors of severe cerebral amyloid angiopathy and influence of APOE genotype in persons with pathologically verified Alzheimer disease. JAMA Neurol. 2014, 71, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, A.; Tsantzali, I.; Kapaki, E.; Constantinides, V.C.; Voumvourakis, K.; Tsivgoulis, G.; Paraskevas, G.P. Cerebrospinal fluid biomarkers and apolipoprotein E genotype in cerebral amyloid angiopathy. A narrative review. Cereb. Circ.-Cogn. Behav. 2021, 2, 100010. [Google Scholar] [CrossRef]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Salloway, S.; Correia, S.; Peck, J.; Harrington, C. Dementia with Lewy bodies: A diagnostic and treatment challenge. Med. Health Rhode Isl. 2002, 85, 207–209. [Google Scholar]
- Montagne, A.; Nikolakopoulou, A.M.; Huuskonen, M.T.; Sagare, A.P.; Lawson, E.J.; Lazic, D.; Rege, S.V.; Grond, A.; Zuniga, E.; Barnes, S.R. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-β. Nat. Aging 2021, 1, 506–520. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and dysfunction of the blood-brain barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Guttenplan, K.A.; Liddelow, S.A. Astrocytes and microglia: Models and tools. J. Exp. Med. 2019, 216, 71–83. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M.; Hertz, L. Why are astrocytes important? Neurochem. Res. 2015, 40, 389–401. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia biology: One century of evolving concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Franze, K. The mechanical control of nervous system development. Development 2013, 140, 3069–3077. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol. Rev. 2018, 99, 21–78. [Google Scholar] [CrossRef]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Pericyte-specific expression of PDGF beta receptor in mouse models with normal and deficient PDGF beta receptor signaling. Mol. Neurodegener. 2010, 5, 32. [Google Scholar] [CrossRef]
- Alahmari, A. Blood-Brain Barrier Overview: Structural and Functional Correlation. Neural Plast 2021, 2021, 6564585. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Kaya, M.; Ahishali, B. Basic physiology of the blood-brain barrier in health and disease: A brief overview. Tissue Barriers 2021, 9, 1840913. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. The blood-brain barrier as an endocrine tissue. Nat. Rev. Endocrinol. 2019, 15, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. Brain meets body: The blood-brain barrier as an endocrine interface. Endocrinology 2012, 153, 4111–4119. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Chow, B.W.; Gu, C. The molecular constituents of the blood–brain barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Johnson, A.M.; Keep, R.F.; Andjelkovic, A.V. Junctional proteins of the blood-brain barrier: New insights into function and dysfunction. Tissue Barriers 2016, 4, e1154641. [Google Scholar] [CrossRef]
- Tietz, S.; Engelhardt, B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015, 209, 493–506. [Google Scholar] [CrossRef]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef]
- Li, W.; Chen, Z.; Chin, I.; Chen, Z.; Dai, H. The Role of VE-cadherin in Blood-brain Barrier Integrity Under Central Nervous System Pathological Conditions. Curr. Neuropharmacol. 2018, 16, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature 2020, 583, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.A.; Ryu, J.K.; Akassoglou, K. Fibrinogen in neurological diseases: Mechanisms, imaging and therapeutics. Nat. Rev. Neurosci. 2018, 19, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef]
- Vardy, E.R.; Kellett, K.A.; Cocklin, S.L.; Hooper, N.M. Alkaline phosphatase is increased in both brain and plasma in Alzheimer’s disease. Neurodegener. Dis. 2011, 9, 31–37. [Google Scholar] [CrossRef]
- Erdő, F.; Denes, L.; de Lange, E. Age-associated physiological and pathological changes at the blood-brain barrier: A review. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 4–24. [Google Scholar] [CrossRef]
- Wen, J.; Ding, Y.; Wang, L.; Xiao, Y. Gut microbiome improves postoperative cognitive function by decreasing permeability of the blood-brain barrier in aged mice. Brain Res. Bull. 2020, 164, 249–256. [Google Scholar] [CrossRef]
- Bowman, G.L.; Dayon, L.; Kirkland, R.; Wojcik, J.; Peyratout, G.; Severin, I.C.; Henry, H.; Oikonomidi, A.; Migliavacca, E.; Bacher, M.; et al. Blood-brain barrier breakdown, neuroinflammation, and cognitive decline in older adults. Alzheimer’s Dement. 2018, 14, 1640–1650. [Google Scholar] [CrossRef]
- Toth, A.N.; Tarantini, S.; DelFavero, J.; Yan, F.; Balasubramanian, P.; Tang, Q.; Csiszar, A.; Ungvari, Z. Demonstration Of Age-Related Increase In Blood-Brain Barrier Permeability By Longitudinal Intravital Microscopy. Innov. Aging 2021, 5, 663. [Google Scholar] [CrossRef]
- Knox, E.G.; Aburto, M.R.; Clarke, G.; Cryan, J.F.; O’Driscoll, C.M. The blood-brain barrier in aging and neurodegeneration. Mol. Psychiatry 2022, 27, 2659–2673. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Reed, M.J.; Logsdon, A.F.; Rhea, E.M.; Erickson, M.A. Healthy aging and the blood-brain barrier. Nat. Aging 2021, 1, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- Vutukuri, R.; Brunkhorst, R.; Kestner, R.-I.; Hansen, L.; Bouzas, N.F.; Pfeilschifter, J.; Devraj, K.; Pfeilschifter, W. Alteration of sphingolipid metabolism as a putative mechanism underlying LPS-induced BBB disruption. J. Neurochem. 2018, 144, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Stahr, N.; Galkina, E.V. Immune Response at the Crossroads of Atherosclerosis and Alzheimer’s Disease. Front. Cardiovasc. Med. 2022, 9, 870144. [Google Scholar] [CrossRef]
- Iadecola, C. Atherosclerosis and neurodegeneration: Unexpected conspirators in Alzheimer’s dementia. Arterioscler Thromb Vasc Biol 2003, 23, 1951–1953. [Google Scholar] [CrossRef]
- Dolan, H.; Crain, B.; Troncoso, J.; Resnick, S.M.; Zonderman, A.B.; Obrien, R.J. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of Aging cohort. Ann. Neurol. 2010, 68, 231–240. [Google Scholar] [CrossRef]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef]
- de Bruijn, R.F.; Ikram, M.A. Cardiovascular risk factors and future risk of Alzheimer’s disease. BMC Med. 2014, 12, 130. [Google Scholar] [CrossRef]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef] [PubMed]
- van Sloten, T.T.; Sedaghat, S.; Carnethon, M.R.; Launer, L.J.; Stehouwer, C.D. Cerebral microvascular complications of type 2 diabetes: Stroke, cognitive dysfunction, and depression. Lancet Diabetes Endocrinol. 2020, 8, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, R.J.; Soiza, R.L. Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’sa vascular disorder? Am. J. Cardiovasc. Dis. 2013, 3, 197. [Google Scholar] [PubMed]
- Rhea, E.M.; Salameh, T.S.; Logsdon, A.F.; Hanson, A.J.; Erickson, M.A.; Banks, W.A. Blood-brain barriers in obesity. AAPS J. 2017, 19, 921–930. [Google Scholar] [CrossRef]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef]
- Ghribi, O. Potential mechanisms linking cholesterol to Alzheimer’s disease-like pathology in rabbit brain, hippocampal organotypic slices, and skeletal muscle. J. Alzheimer’s Dis. 2008, 15, 673–684. [Google Scholar] [CrossRef]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef]
- Santiago, J.A.; Potashkin, J.A. The Impact of Disease Comorbidities in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 631770. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009, 8, 205–216. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Thompson, J.; Taheri, S.; Grossetete, M.; Adair, J.C.; Edmonds, E.; Prestopnik, J.; Wills, J.; Rosenberg, G.A. Matrix metalloproteinases are associated with increased blood–brain barrier opening in vascular cognitive impairment. Stroke 2011, 42, 1345–1350. [Google Scholar] [CrossRef]
- Holland, P.R.; Searcy, J.L.; Salvadores, N.; Scullion, G.; Chen, G.; Lawson, G.; Scott, F.; Bastin, M.E.; Ihara, M.; Kalaria, R. Gliovascular disruption and cognitive deficits in a mouse model with features of small vessel disease. J. Cereb. Blood Flow Metab. 2015, 35, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Liu, C.-C.; Qiao, W.; Bu, G. Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol. Psychiatry 2018, 83, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Alagarsamy, J.; Jaeschke, A.; Hui, D.Y. Apolipoprotein E in Cardiometabolic and Neurological Health and Diseases. Int. J. Mol. Sci. 2022, 23, 9892. [Google Scholar] [CrossRef]
- Vecchio, F.L.; Bisceglia, P.; Imbimbo, B.P.; Lozupone, M.; Latino, R.R.; Resta, E.; Leone, M.; Solfrizzi, V.; Greco, A.; Daniele, A. Are apolipoprotein E fragments a promising new therapeutic target for Alzheimer’s disease? Ther. Adv. Chronic Dis. 2022, 13, 20406223221081605. [Google Scholar] [CrossRef]
- Bu, G. APOE targeting strategy in Alzheimer’s disease: Lessons learned from protective variants. Mol. Neurodegener. 2022, 17, 51. [Google Scholar] [CrossRef]
- Fernández-Calle, R.; Konings, S.C.; Frontiñán-Rubio, J.; García-Revilla, J.; Camprubí-Ferrer, L.; Svensson, M.; Martinson, I.; Boza-Serrano, A.; Venero, J.L.; Nielsen, H.M. APOE in the bullseye of neurodegenerative diseases: Impact of the APOE genotype in Alzheimer’s disease pathology and brain diseases. Mol. Neurodegener. 2022, 17, 62. [Google Scholar] [CrossRef]
- Liu, C.-C.; Zhao, J.; Fu, Y.; Inoue, Y.; Ren, Y.; Chen, Y.; Doss, S.V.; Shue, F.; Jeevaratnam, S.; Bastea, L. Peripheral apoE4 enhances Alzheimer’s pathology and impairs cognition by compromising cerebrovascular function. Nat. Neurosci. 2022, 25, 1020–1033. [Google Scholar] [CrossRef]
- Jackson, R.J.; Meltzer, J.C.; Nguyen, H.; Commins, C.; Bennett, R.E.; Hudry, E.; Hyman, B.T. APOE4 derived from astrocytes leads to blood-brain barrier impairment. Brain 2022, 145, 3582–3593. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Hays, C.C.; Zlatar, Z.Z.; Meloy, M.J.; Bondi, M.W.; Gilbert, P.E.; Liu, T.T.; Helm, J.L.; Wierenga, C.E. APOE modifies the interaction of entorhinal cerebral blood flow and cortical thickness on memory function in cognitively normal older adults. Neuroimage 2019, 202, 116162. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Campbell, M. Tight junction modulation of the blood brain barrier: CNS delivery of small molecules. Tissue Barriers 2016, 4, e1138017. [Google Scholar] [CrossRef] [PubMed]
- González-Mariscal, L.; Betanzos, A.; Nava, P.; Jaramillo, B.E. Tight junction proteins. Prog. Biophys. Mol. Biol. 2003, 81, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef]
- Rempe, R.G.; Hartz, A.M.; Soldner, E.L.; Sokola, B.S.; Alluri, S.R.; Abner, E.L.; Kryscio, R.J.; Pekcec, A.; Schlichtiger, J.; Bauer, B. Matrix metalloproteinase-mediated blood-brain barrier dysfunction in epilepsy. J. Neurosci. 2018, 38, 4301–4315. [Google Scholar] [CrossRef]
- Raeeszadeh-Sarmazdeh, M.; Do, L.D.; Hritz, B.G. Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells 2020, 9, 1313. [Google Scholar] [CrossRef]
- Sharma, C.; Woo, H.; Kim, S.R. Addressing Blood-Brain Barrier Impairment in Alzheimer’s Disease. Biomedicines 2022, 10, 742. [Google Scholar] [CrossRef]
- Ayka, A.; Şehirli, A. The Role of the SLC Transporters Protein in the Neurodegenerative Disorders. Clin. Psychopharmacol. Neurosci. Off. Sci. J. Korean Coll. Neuropsychopharmacol. 2020, 18, 174–187. [Google Scholar] [CrossRef]
- Gil-Martins, E.; Barbosa, D.J.; Silva, V.; Remião, F.; Silva, R. Dysfunction of ABC transporters at the blood-brain barrier: Role in neurological disorders. Pharmacol. Ther. 2020, 213, 107554. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood–brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9, S3. [Google Scholar] [CrossRef]
- Al-Majdoub, Z.M.; Al Feteisi, H.; Achour, B.; Warwood, S.; Neuhoff, S.; Rostami-Hodjegan, A.; Barber, J. Proteomic Quantification of Human Blood–Brain Barrier SLC and ABC Transporters in Healthy Individuals and Dementia Patients. Mol. Pharm. 2019, 16, 1220–1233. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Wang, L.; Fan, Q.; Xiao, G.; Wang, X.; Zhong, Y.; Zhou, B. Genetic Inhibition of Solute-Linked Carrier 39 Family Transporter 1 Ameliorates Aβ Pathology in a Drosophila Model of Alzheimer’s Disease. PLoS Genet. 2012, 8, e1002683. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int. J. Mol. Sci. 2019, 20, 5671. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflügers Archiv. Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- Głuchowska, K.; Pliszka, M.; Szablewski, L. Expression of glucose transporters in human neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2021, 540, 8–15. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Chen, Y.; Joo, J.; Chu, J.M.; Chang, R.C.; Wong, G.T. Downregulation of the glucose transporter GLUT 1 in the cerebral microvasculature contributes to postoperative neurocognitive disorders in aged mice. J. Neuroinflammation 2023, 20, 237. [Google Scholar] [CrossRef]
- Ardanaz, C.G.; Gil, M.J.R.; Smerdou, C.; Solas, M. Astrocytic GLUT1 ablation improves systemic glucose metabolism and promotes cognition. Alzheimer’s Dement. 2021, 17, e058650. [Google Scholar] [CrossRef]
- Puris, E.; Saveleva, L.; de Sousa Maciel, I.; Kanninen, K.M.; Auriola, S.; Fricker, G. Protein Expression of Amino Acid Transporters Is Altered in Isolated Cerebral Microvessels of 5xFAD Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2023, 60, 732–748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cheng, X.; Dang, R.; Zhang, W.; Zhang, J.; Yao, Z. Lactate Deficit in an Alzheimer Disease Mouse Model: The Relationship With Neuronal Damage. J. Neuropathol. Exp. Neurol. 2018, 77, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.D.; Martins, F.; Wiltfang, J.; da Cruz, E.S.O.A.B.; Rebelo, S. ABC Transporters Are Key Players in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 463–485. [Google Scholar] [CrossRef]
- Wolf, A.; Bauer, B.; Hartz, A.M. ABC Transporters and the Alzheimer’s Disease Enigma. Front. Psychiatry 2012, 3, 54. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.B.; Leung, G.K.F.; Callaghan, R.; Gelissen, I.C. P-glycoprotein: A role in the export of amyloid-β in Alzheimer’s disease? FEBS J. 2020, 287, 612–625. [Google Scholar] [CrossRef]
- Storelli, F.; Billington, S.; Kumar, A.R.; Unadkat, J.D. Abundance of P-Glycoprotein and Other Drug Transporters at the Human Blood-Brain Barrier in Alzheimer’s Disease: A Quantitative Targeted Proteomic Study. Clin. Pharmacol. Ther. 2021, 109, 667–675. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef]
- Puris, E.; Auriola, S.; Petralla, S.; Hartman, R.; Gynther, M.; de Lange, E.C.M.; Fricker, G. Altered protein expression of membrane transporters in isolated cerebral microvessels and brain cortex of a rat Alzheimer’s disease model. Neurobiol. Dis. 2022, 169, 105741. [Google Scholar] [CrossRef]
- Xiong, H.; Callaghan, D.; Jones, A.; Bai, J.; Rasquinha, I.; Smith, C.; Pei, K.; Walker, D.; Lue, L.F.; Stanimirovic, D.; et al. ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Abeta(1-40) peptides. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 5463–5475. [Google Scholar] [CrossRef]
- Krohn, M.; Lange, C.; Hofrichter, J.; Scheffler, K.; Stenzel, J.; Steffen, J.; Schumacher, T.; Brüning, T.; Plath, A.-S.; Alfen, F.; et al. Cerebral amyloid-β proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Investig. 2011, 121, 3924–3931. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Li, X. The Role of Mfsd2a in Nervous System Diseases. Front. Neurosci. 2021, 15, 730534. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Campillo, M.; Ruiz-Pastor, M.J.; Gázquez, A.; Marín-Muñoz, J.; Noguera-Perea, F.; Ruiz-Alcaraz, A.J.; Manzanares-Sánchez, S.; Antúnez, C.; Larqué, E. Decreased Blood Level of MFSD2a as a Potential Biomarker of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 21, 70. [Google Scholar] [CrossRef]
- Nkiliza, A.; Evans, J.; Ringland, C.; Daniel, N.; Huguenard, C.; David, R.; Ojo, J.; Crawford, F.; Mullan, M.; Bachmeier, C.; et al. APOE e4 dependent deficits in brain DHA phospholipids and mfsd2a in Alzheimer’s disease patients with severe cerebral amyloid angiopathy. Alzheimer’s Dement. 2022, 18, e067434. [Google Scholar] [CrossRef]
- Wood, C.A.P.; Zhang, J.; Aydin, D.; Xu, Y.; Andreone, B.J.; Langen, U.H.; Dror, R.O.; Gu, C.; Feng, L. Structure and mechanism of blood-brain-barrier lipid transporter MFSD2A. Nature 2021, 596, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.W.; Gu, C. Gradual Suppression of Transcytosis Governs Functional Blood-Retinal Barrier Formation. Neuron 2017, 93, 1325–1333.e1323. [Google Scholar] [CrossRef]
- Qu, C.; Song, H.; Shen, J.; Xu, L.; Li, Y.; Qu, C.; Li, T.; Zhang, J. Mfsd2a Reverses Spatial Learning and Memory Impairment Caused by Chronic Cerebral Hypoperfusion via Protection of the Blood–Brain Barrier. Front. Neurosci. 2020, 14, 461. [Google Scholar] [CrossRef]
- Ma, Q.; Zhao, Z.; Sagare, A.P.; Wu, Y.; Wang, M.; Owens, N.C.; Verghese, P.B.; Herz, J.; Holtzman, D.M.; Zlokovic, B.V. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 2018, 13, 57. [Google Scholar] [CrossRef]
- Storck, S.E.; Hartz, A.M.S.; Bernard, J.; Wolf, A.; Kachlmeier, A.; Mahringer, A.; Weggen, S.; Pahnke, J.; Pietrzik, C.U. The concerted amyloid-beta clearance of LRP1 and ABCB1/P-gp across the blood-brain barrier is linked by PICALM. Brain Behav. Immun. 2018, 73, 21–33. [Google Scholar] [CrossRef]
- Storck, S.E.; Kurtyka, M.; Pietrzik, C.U. Brain endothelial LRP1 maintains blood–brain barrier integrity. Fluids Barriers CNS 2021, 18, 27. [Google Scholar] [CrossRef]
- Nikolakopoulou, A.M.; Wang, Y.; Ma, Q.; Sagare, A.P.; Montagne, A.; Huuskonen, M.T.; Rege, S.V.; Kisler, K.; Dai, Z.; Körbelin, J.; et al. Endothelial LRP1 protects against neurodegeneration by blocking cyclophilin A. J. Exp. Med. 2021, 218, e20202207. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: Evidence from clinical and preclinical studies. J. Lipid Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Storck, S.E.; Meister, S.; Nahrath, J.; Meißner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.E.; Bouter, Y.; Prikulis, I. Endothelial LRP1 transports amyloid-β 1–42 across the blood-brain barrier. J. Clin. Investig. 2016, 126, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, B.; Storck, S.E.; Reekmans, S.M.; Lechat, B.; Gordts, P.L.; Pradier, L.; Pietrzik, C.U.; Roebroek, A.J. LRP1 has a predominant role in production over clearance of Aβ in a mouse model of Alzheimer’s disease. Mol. Neurobiol. 2019, 56, 7234–7245. [Google Scholar] [CrossRef] [PubMed]
- Gorina, Y.V.; Osipova, E.D.; Morgun, A.V.; Boytsova, E.B.; Lopatina, O.L.; Salmina, A.B. Assessment of the Level of Rage in Cells Blood-Brain Barrier in Experimental Alzheimer’s Disease. Cell Tissue Biol. 2021, 15, 280–286. [Google Scholar] [CrossRef]
- Wan, W.; Cao, L.; Liu, L.; Zhang, C.; Kalionis, B.; Tai, X.; Li, Y.; Xia, S. Aβ1–42 oligomer-induced leakage in an in vitro blood–brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J. Neurochem. 2015, 134, 382–393. [Google Scholar] [CrossRef]
- Wang, H.; Chen, F.; Du, Y.-F.; Long, Y.; Reed, M.N.; Hu, M.; Suppiramaniam, V.; Hong, H.; Tang, S.-S. Targeted inhibition of RAGE reduces amyloid-β influx across the blood-brain barrier and improves cognitive deficits in db/db mice. Neuropharmacology 2018, 131, 143–153. [Google Scholar] [CrossRef]
- Dallas, S.; Miller, D.S.; Bendayan, R. Multidrug resistance-associated proteins: Expression and function in the central nervous system. Pharmacol. Rev. 2006, 58, 140–161. [Google Scholar] [CrossRef]
- Nies, A.; Jedlitschky, G.; König, J.; Herold-Mende, C.; Steiner, H.; Schmitt, H.-P.; Keppler, D. Expression and immunolocalization of the multidrug resistance proteins, MRP1–MRP6 (ABCC1–ABCC6), in human brain. Neuroscience 2004, 129, 349–360. [Google Scholar] [CrossRef]
- Vellonen, K.S.; Ihalainen, J.; Boucau, M.C.; Gosselet, F.; Picardat, T.; Gynther, M.; Kanninen, K.M.; White, A.R.; Malm, T.; Koistinaho, J.; et al. Disease-Induced Alterations in Brain Drug Transporters in Animal Models of Alzheimer’s Disease: Theme: Drug Discovery, Development and Delivery in Alzheimer’s Disease Guest Editor: Davide Brambilla. Pharm. Res. 2017, 34, 2652–2662. [Google Scholar] [CrossRef]
- Kook, S.-Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. Aβ1–42-RAGE Interaction Disrupts Tight Junctions of the Blood–Brain Barrier Via Ca2+-Calcineurin Signaling. J. Neurosci. 2012, 32, 8845–8854. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Shinohara, M.; Shinohara, M.; Yamazaki, A.; Murray, M.E.; Liesinger, A.M.; Heckman, M.G.; Lesser, E.R.; Parisi, J.E.; Petersen, R.C.; et al. Selective loss of cortical endothelial tight junction proteins during Alzheimer’s disease progression. Brain J. Neurol. 2019, 142, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Chen, L.L.; Yang, H.; Li, L.; Liu, J.; Chen, L.; Hong, W.J.; Wang, C.G.; Ma, J.J.; Huang, J.; et al. Effect of High Cholesterol Regulation of LRP1 and RAGE on Aβ Transport Across the Blood-Brain Barrier in Alzheimer’s Disease. Curr. Alzheimer Res. 2021, 18, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J. Molecular regulation of the blood-brain barrier GLUT1 glucose transporter by brain-derived factors. In Ageing and Dementia; Springer: Vienna, Austria, 1998; pp. 323–331. [Google Scholar]
- Deane, R.; Sagare, A.; Zlokovic, B.V. The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 1601–1605. [Google Scholar] [CrossRef]
- Boucher, P.; Herz, J. Signaling through LRP1: Protection from atherosclerosis and beyond. Biochem. Pharmacol. 2011, 81, 1–5. [Google Scholar] [CrossRef]
- Sita, G.; Graziosi, A.; Hrelia, P.; Morroni, F. NLRP3 and Infections: β-Amyloid in Inflammasome beyond Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 6984. [Google Scholar] [CrossRef]
- Merlini, M.; Rafalski, V.A.; Rios Coronado, P.E.; Gill, T.M.; Ellisman, M.; Muthukumar, G.; Subramanian, K.S.; Ryu, J.K.; Syme, C.A.; Davalos, D.; et al. Fibrinogen Induces Microglia-Mediated Spine Elimination and Cognitive Impairment in an Alzheimer’s Disease Model. Neuron 2019, 101, 1099–1108.e1096. [Google Scholar] [CrossRef]
- Al-Ghraiybah, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 10572. [Google Scholar] [CrossRef]
- Heithoff, B.P.; George, K.K.; Phares, A.N.; Zuidhoek, I.A.; Munoz-Ballester, C.; Robel, S. Astrocytes are necessary for blood–brain barrier maintenance in the adult mouse brain. Glia 2021, 69, 436–472. [Google Scholar] [CrossRef]
- Silva, I.; Silva, J.; Ferreira, R.; Trigo, D. Glymphatic system, AQP4, and their implications in Alzheimer’s disease. Neurol. Res. Pract. 2021, 3, 5. [Google Scholar] [CrossRef]
- Jeon, H.; Kim, M.; Park, W.; Lim, J.S.; Lee, E.; Cha, H.; Ahn, J.S.; Kim, J.H.; Hong, S.H.; Park, J.E.; et al. Upregulation of AQP4 Improves Blood–Brain Barrier Integrity and Perihematomal Edema Following Intracerebral Hemorrhage. Neurotherapeutics 2021, 18, 2692–2706. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Zhang, Y.; Wu, S.; Chen, Q.; Wang, L. The Role of NLRP3 Inflammasome in Alzheimer’s Disease and Potential Therapeutic Targets. Front. Pharmacol. 2022, 13, 845185. [Google Scholar] [CrossRef] [PubMed]
- Al Rihani, S.B.; Darakjian, L.I.; Kaddoumi, A. Oleocanthal-Rich Extra-Virgin Olive Oil Restores the Blood-Brain Barrier Function through NLRP3 Inflammasome Inhibition Simultaneously with Autophagy Induction in TgSwDI Mice. ACS Chem. Neurosci. 2019, 10, 3543–3554. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative Stress-Mediated Blood-Brain Barrier (BBB) Disruption in Neurological Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4356386. [Google Scholar] [CrossRef]
- Lehner, C.; Gehwolf, R.; Tempfer, H.; Krizbai, I.; Hennig, B.; Bauer, H.C.; Bauer, H. Oxidative stress and blood-brain barrier dysfunction under particular consideration of matrix metalloproteinases. Antioxid. Redox Signal. 2011, 15, 1305–1323. [Google Scholar] [CrossRef]
- Chung, T.D.; Linville, R.M.; Guo, Z.; Ye, R.; Jha, R.; Grifno, G.N.; Searson, P.C. Effects of acute and chronic oxidative stress on the blood–brain barrier in 2D and 3D in vitro models. Fluids Barriers CNS 2022, 19, 33. [Google Scholar] [CrossRef]
- Alcendor, D.J. Interactions between Amyloid-Β Proteins and Human Brain Pericytes: Implications for the Pathobiology of Alzheimer’s Disease. J. Clin. Med. 2020, 9, 1490. [Google Scholar] [CrossRef]
- Ding, R.; Hase, Y.; Ameen-Ali, K.E.; Ndung’u, M.; Stevenson, W.; Barsby, J.; Gourlay, R.; Akinyemi, T.; Akinyemi, R.; Uemura, M.T.; et al. Loss of capillary pericytes and the blood–brain barrier in white matter in poststroke and vascular dementias and Alzheimer’s disease. Brain Pathol. 2020, 30, 1087–1101. [Google Scholar] [CrossRef]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef]
- Blanchard, J.W.; Bula, M.; Davila-Velderrain, J.; Akay, L.A.; Zhu, L.; Frank, A.; Victor, M.B.; Bonner, J.M.; Mathys, H.; Lin, Y.-T.; et al. Reconstruction of the human blood–brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat. Med. 2020, 26, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.C.D.; Highet, B.; Jansson, D.; Wu, J.; Rustenhoven, J.; Aalderink, M.; Tan, A.; Li, S.; Johnson, R.; Coppieters, N.; et al. Characterisation of PDGF-BB:PDGFRβ signalling pathways in human brain pericytes: Evidence of disruption in Alzheimer’s disease. Commun. Biol. 2022, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wan, W.; Xia, S.; Kalionis, B.; Li, Y. Dysfunctional Wnt/β-catenin signaling contributes to blood–brain barrier breakdown in Alzheimer’s disease. Neurochem. Int. 2014, 75, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huang, X.; Su, Y.; Yin, G.; Wang, S.; Yu, B.; Li, H.; Qi, J.; Chen, H.; Zeng, W.; et al. Activation of Wnt/β-catenin pathway mitigates blood–brain barrier dysfunction in Alzheimer’s disease. Brain J. Neurol. 2022, 145, 4474–4488. [Google Scholar] [CrossRef]
- Gastfriend, B.D.; Nishihara, H.; Canfield, S.G.; Foreman, K.L.; Engelhardt, B.; Palecek, S.P.; Shusta, E.V. Wnt signaling mediates acquisition of blood–brain barrier properties in naïve endothelium derived from human pluripotent stem cells. eLife 2021, 10, e70992. [Google Scholar] [CrossRef]
- Hussain, B.; Fang, C.; Huang, X.; Feng, Z.; Yao, Y.; Wang, Y.; Chang, J. Endothelial β-Catenin Deficiency Causes Blood-Brain Barrier Breakdown via Enhancing the Paracellular and Transcellular Permeability. Front. Mol. Neurosci. 2022, 15, 895429. [Google Scholar] [CrossRef]
- Yang, A.C.; Vest, R.T.; Kern, F.; Lee, D.P.; Agam, M.; Maat, C.A.; Losada, P.M.; Chen, M.B.; Schaum, N.; Khoury, N. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 2022, 603, 885–892. [Google Scholar] [CrossRef]
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; Winkler, E.A.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987. [Google Scholar] [CrossRef]
- Juul Rasmussen, I.; Tybjærg-Hansen, A.; Rasmussen, K.L.; Nordestgaard, B.G.; Frikke-Schmidt, R. Blood-brain barrier transcytosis genes, risk of dementia and stroke: A prospective cohort study of 74,754 individuals. Eur. J. Epidemiol. 2019, 34, 579–590. [Google Scholar] [CrossRef]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S. Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef]
- Tian, Y.; Chang, J.C.; Fan, E.Y.; Flajolet, M.; Greengard, P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 17071–17076. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Kajiho, H.; Saito, K.; Tsujita, K.; Kontani, K.; Araki, Y.; Kurosu, H.; Katada, T. RIN3: A novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J. Cell Sci. 2003, 116, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.C.; Daly, S.B.; Urquhart, J.E.; Black, G.C.; Pilz, D.T.; Brockmann, K.; McEntagart, M.; Abdel-Salam, G.; Zaki, M.; Wolf, N.I. Recessive mutations in the gene encoding the tight junction protein occludin cause band-like calcification with simplified gyration and polymicrogyria. Am. J. Hum. Genet. 2010, 87, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Wyss, L.; Schäfer, J.; Liebner, S.; Mittelbronn, M.; Deutsch, U.; Enzmann, G.; Adams, R.H.; Aurrand-Lions, M.; Plate, K.H.; Imhof, B.A. Junctional adhesion molecule (JAM)-C deficient C57BL/6 mice develop a severe hydrocephalus. PLoS ONE 2012, 7, e45619. [Google Scholar] [CrossRef] [PubMed]
- Akawi, N.A.; Canpolat, F.E.; White, S.M.; Quilis-Esquerra, J.; Morales Sanchez, M.; Gamundi, M.J.; Mochida, G.H.; Walsh, C.A.; Ali, B.R.; Al-Gazali, L. Delineation of the clinical, molecular and cellular aspects of novel JAM 3 mutations underlying the autosomal recessive hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. Hum. Mutat. 2013, 34, 498–505. [Google Scholar] [CrossRef]
- Chabriat, H.; Joutel, A.; Dichgans, M.; Tournier-Lasserve, E.; Bousser, M.-G. Cadasil. Lancet Neurol. 2009, 8, 643–653. [Google Scholar] [CrossRef]
- Ghosh, M.; Balbi, M.; Hellal, F.; Dichgans, M.; Lindauer, U.; Plesnila, N. Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Ann. Neurol. 2015, 78, 887–900. [Google Scholar] [CrossRef]
- Guemez-Gamboa, A.; Nguyen, L.N.; Yang, H.; Zaki, M.S.; Kara, M.; Ben-Omran, T.; Akizu, N.; Rosti, R.O.; Rosti, B.; Scott, E. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat. Genet. 2015, 47, 809–813. [Google Scholar] [CrossRef]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood–brain barrier. Nature 2014, 509, 507–511. [Google Scholar] [CrossRef]
- Zhao, Z.; Zlokovic, B.V. Blood-brain barrier: A dual life of MFSD2A? Neuron 2014, 82, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Winfree, R.L.; Dumitrescu, L.; Blennow, K.; Zetterberg, H.; Gifford, K.A.; Pechman, K.R.; Jefferson, A.L.; Hohman, T.J. Biological correlates of elevated soluble TREM2 in cerebrospinal fluid. Neurobiol. Aging 2022, 118, 88–98. [Google Scholar] [CrossRef]
- Wu, R.; Li, X.; Xu, P.; Huang, L.; Cheng, J.; Huang, X.; Jiang, J.; Wu, L.-J.; Tang, Y. TREM2 protects against cerebral ischemia/reperfusion injury. Mol. Brain 2017, 10, 20. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, W.; Zhang, J.; Zhao, Y.; Xu, Y. TREM2 overexpression attenuates cognitive deficits in experimental models of vascular dementia. Neural Plast. 2020, 2020. [Google Scholar] [CrossRef]
- Taylor, X.; Cisternas, P.; You, Y.; You, Y.; Xiang, S.; Marambio, Y.; Zhang, J.; Vidal, R.; Lasagna-Reeves, C.A. A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J. Neuroinflam. 2020, 17, 223. [Google Scholar] [CrossRef]
- Rauchmann, B.-S.; Sadlon, A.; Perneczky, R.; Initiative, A.s.D.N. Soluble TREM2 and inflammatory proteins in Alzheimer’s disease cerebrospinal fluid. J. Alzheimer’s Dis. 2020, 73, 1615–1626. [Google Scholar] [CrossRef]
- Zuliani, G.; Cavalieri, M.; Galvani, M.; Passaro, A.; Munari, M.; Bosi, C.; Zurlo, A.; Fellin, R. Markers of endothelial dysfunction in older subjects with late onset Alzheimer’s disease or vascular dementia. J. Neurol. Sci. 2008, 272, 164–170. [Google Scholar] [CrossRef]
- Lee, A.J.; Raghavan, N.S.; Bhattarai, P.; Siddiqui, T.; Sariya, S.; Reyes-Dumeyer, D.; Flowers, X.E.; Cardoso, S.A.L.; De Jager, P.L.; Bennett, D.A.; et al. FMNL2 regulates gliovascular interactions and is associated with vascular risk factors and cerebrovascular pathology in Alzheimer’s disease. Acta Neuropathol. 2022, 144, 59–79. [Google Scholar] [CrossRef]
- Sousa, J.A.; Bernardes, C.; Bernardo-Castro, S.; Lino, M.; Albino, I.; Ferreira, L.; Brás, J.; Guerreiro, R.; Tábuas-Pereira, M.; Baldeiras, I. Reconsidering the role of blood-brain barrier in Alzheimer’s disease: From delivery to target. Front. Aging Neurosci. 2023, 15, 1102809. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, N.S.; Sariya, S.; Lee, A.J.; Gao, Y.; Reyes-Dumeyer, D.; De Jager, P.L.; Bennett, D.A.; Menon, V.; Lantigua, R.A.; Kukull, W.A. FMNL2 interacts with cerebrovascular risk factors to alter Alzheimer’s disease risk. medRxiv 2020, 2030.20184879.
- Boyé, K.; Geraldo, L.H.; Furtado, J.; Pibouin-Fragner, L.; Poulet, M.; Kim, D.; Nelson, B.; Xu, Y.; Jacob, L.; Maissa, N. Endothelial Unc5B controls blood-brain barrier integrity. Nat. Commun. 2022, 13, 1169. [Google Scholar] [CrossRef] [PubMed]
- Mora, P.; Hollier, P.-L.; Guimbal, S.; Abelanet, A.; Diop, A.; Cornuault, L.; Couffinhal, T.; Horng, S.; Gadeau, A.-P.; Renault, M.-A. Blood–brain barrier genetic disruption leads to protective barrier formation at the Glia Limitans. PLoS Biol. 2020, 18, e3000946. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhao, J.; Liu, P.; Ji, D.; Zhang, L.; Zhang, M.; Li, Y.; Xiao, Y. Preparation and in vitro evaluation of multi-target-directed selenium-chondroitin sulfate nanoparticles in protecting against the Alzheimer’s disease. Int. J. Biol. Macromol. 2020, 142, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Blood–brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood–brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef]
- Deane, R.J. Is RAGE still a therapeutic target for Alzheimer’s disease? Future Med. Chem. 2012, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N.; Agro, A.; Bell, J.; Aisen, P.S.; Schweizer, E.; Galasko, D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer’s disease. Alzheimer Dis. Assoc. Disord. 2011, 25, 206. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J. Clearance of Alzheimer’s amyloid-β 1-40 peptide from brain by LDL receptor–related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Sagare, A.P.; Friedman, A.E.; Bedi, G.S.; Holtzman, D.M.; Deane, R.; Zlokovic, B.V. Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab. 2007, 27, 909–918. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef]
- Sagare, A.P.; Deane, R.; Zlokovic, B.V. Low-density lipoprotein receptor-related protein 1: A physiological Aβ homeostatic mechanism with multiple therapeutic opportunities. Pharmacol. Ther. 2012, 136, 94–105. [Google Scholar] [CrossRef]
- Storck, S.E.; Pietrzik, C.U. Endothelial LRP1–a potential target for the treatment of Alzheimer’s disease: Theme: Drug discovery, development and delivery in Alzheimer’s disease Guest Editor: Davide Brambilla. Pharm. Res. 2017, 34, 2637–2651. [Google Scholar] [CrossRef]
- Cicero, A.F.; Ruscica, M.; Banach, M. Resveratrol and cognitive decline: A clinician perspective. Arch. Med. Sci. 2019, 15, 936–943. [Google Scholar] [CrossRef]
- Batarseh, Y.S.; Bharate, S.S.; Kumar, V.; Kumar, A.; Vishwakarma, R.A.; Bharate, S.B.; Kaddoumi, A. Crocus sativus Extract Tightens the Blood-Brain Barrier, Reduces Amyloid β Load and Related Toxicity in 5XFAD Mice. ACS Chem. Neurosci. 2017, 8, 1756–1766. [Google Scholar] [CrossRef]
- Tzekaki, E.E.; Tsolaki, M.; Pantazaki A, A.; Geromichalos, G.; Lazarou, E.; Kozori, M.; Sinakos, Z. The pleiotropic beneficial intervention of olive oil intake on the Alzheimer’s disease onset via fibrinolytic system. Exp. Gerontol. 2021, 150, 111344. [Google Scholar] [CrossRef]
- Q Yulug, B.; Hanoglu, L.; Kilic, E.; Schabitz, W.R. RIFAMPICIN: An antibiotic with brain protective function. Brain Res. Bull. 2014, 107, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Qosa, H.; Abuznait, A.H.; Hill, R.A.; Kaddoumi, A. Enhanced brain amyloid-β clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 31, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Bodles-Brakhop, A.M.; Barger, S.W. A Role for P-Glycoprotein in Clearance of Alzheimer Amyloid β -Peptide from the Brain. Curr. Alzheimer Res. 2016, 13, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.; Bates, S.E. Tariquidar (XR9576): A P-glycoprotein drug efflux pump inhibitor. Expert Rev. Anticancer. Ther. 2007, 7, 447–459. [Google Scholar] [CrossRef]
- Abdallah, I.M.; Al-Shami, K.M.; Yang, E.; Kaddoumi, A. Blood-brain barrier disruption increases amyloid-related pathology in TgSwDI mice. Int. J. Mol. Sci. 2021, 22, 1231. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier and delivery of protein and gene therapeutics to brain. Front. Aging Neurosci. 2020, 11, 373. [Google Scholar] [CrossRef]
- Abuznait, A.H.; Qosa, H.; Busnena, B.A.; El Sayed, K.A.; Kaddoumi, A. Olive-oil-derived oleocanthal enhances β-amyloid clearance as a potential neuroprotective mechanism against Alzheimer’s disease: In vitro and in vivo studies. ACS Chem. Neurosci. 2013, 4, 973–982. [Google Scholar] [CrossRef]
- Limtrakul, P.; Khantamat, O.; Pintha, K. Inhibition of P-glycoprotein function and expression by kaempferol and quercetin. J. Chemother. 2005, 17, 86–95. [Google Scholar] [CrossRef]
- Ren, X.; Lv, F.; Fang, B.; Liu, S.; Lv, H.; He, G.; Ma, H.; Cao, Y.; Wang, Y. Anesthetic agent propofol inhibits myeloid differentiation factor 88-dependent and independent signaling and mitigates lipopolysaccharide-mediated reactive oxygen species production in human neutrophils in vitro. Eur. J. Pharmacol. 2014, 744, 164–172. [Google Scholar] [CrossRef]
- Hartz, A.M.; Miller, D.S.; Bauer, B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-β in a mouse model of Alzheimer’s disease. Mol. Pharmacol. 2010, 77, 715–723. [Google Scholar] [CrossRef]
- Luissint, A.-C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.-O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Haines, D.E.; Mihailoff, G.A. Fundamental Neuroscience for Basic and Clinical Applications E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Gavard, J.; Gutkind, J.S. VE-cadherin and claudin-5: It takes two to tango. Nat. Cell Biol. 2008, 10, 883–885. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Y.; Choi, J.; Ting, K.K.; Coleman, P.; Chen, J.; Cogger, V.C.; Wan, L.; Shi, Z.; Moller, T.; et al. Targeting miR-27a/VE-cadherin interactions rescues cerebral cavernous malformations in mice. PLoS Biol. 2020, 18, e3000734. [Google Scholar] [CrossRef]
- Abdallah, I.M.; Al-Shami, K.M.; Yang, E.; Wang, J.; Guillaume, C.; Kaddoumi, A. Oleuropein-rich olive leaf extract attenuates neuroinflammation in the Alzheimer’s disease mouse model. ACS Chem. Neurosci. 2022, 13, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, I.M.; Al-Shami, K.M.; Alkhalifa, A.E.; Al-Ghraiybah, N.F.; Guillaume, C.; Kaddoumi, A. Comparison of Oleocanthal-Low EVOO and Oleocanthal against Amyloid-β and Related Pathology in a Mouse Model of Alzheimer’s Disease. Molecules 2023, 28, 1249. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5–deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef]
- Ito, S.; Ueno, T.; Ohtsuki, S.; Terasaki, T. Lack of brain-to-blood efflux transport activity of low-density lipoprotein receptor-related protein-1 (LRP-1) for amyloid-β peptide (1–40) in mouse: Involvement of an LRP-1-independent pathway. J. Neurochem. 2010, 113, 1356–1363. [Google Scholar] [CrossRef]
- Al Rihani, S.B.; Lan, R.S.; Kaddoumi, A. Granisetron Alleviates Alzheimer’s Disease Pathology in TgSwDI Mice Through Calmodulin-Dependent Protein Kinase II/cAMP-Response Element Binding Protein Pathway. J. Alzheimers Dis. 2019, 72, 1097–1117. [Google Scholar] [CrossRef]
- Elfakhri, K.H.; Abdallah, I.M.; Brannen, A.D.; Kaddoumi, A. Multi-faceted therapeutic strategy for treatment of Alzheimer’s disease by concurrent administration of etodolac and α-tocopherol. Neurobiol. Dis. 2019, 125, 123–134. [Google Scholar] [CrossRef]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Møller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Brændgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef]
- Nørgaard, C.H.; Friedrich, S.; Hansen, C.T.; Gerds, T.; Ballard, C.; Møller, D.V.; Knudsen, L.B.; Kvist, K.; Zinman, B.; Holm, E. Treatment with glucagon-like peptide-1 receptor agonists and incidence of dementia: Data from pooled double-blind randomized controlled trials and nationwide disease and prescription registers. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2022, 8, e12268. [Google Scholar] [CrossRef] [PubMed]
- Edison, P.; Femminella, G.D.; Ritchie, C.W.; Holmes, C.; Walker, Z.; Ridha, B.H.; Raza, S.; Livingston, N.R.; Nowell, J.; Busza, G. Evaluation of liraglutide in the treatment of Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, e057848. [Google Scholar] [CrossRef]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.; Silver, D.L. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.B.; Gu, C. Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron 2017, 94, 581–594.e585. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Sovio, U.; Goulding, N.; McBride, N.; Cook, E.; Gaccioli, F.; Charnock-Jones, D.S.; Lawlor, D.A.; Smith, G.C. A maternal serum metabolite ratio predicts fetal growth restriction at term. Nat. Med. 2020, 26, 348–353. [Google Scholar] [CrossRef]
- Gupta, R.K.; Kanungo, M. Glial molecular alterations with mouse brain development and aging: Up-regulation of the Kir4. 1 and aquaporin-4. Age 2013, 35, 59–67. [Google Scholar] [CrossRef][Green Version]
- Zeppenfeld, D.M.; Simon, M.; Haswell, J.D.; D’Abreo, D.; Murchison, C.; Quinn, J.F.; Grafe, M.R.; Woltjer, R.L.; Kaye, J.; Iliff, J.J. Association of perivascular localization of aquaporin-4 with cognition and Alzheimer disease in aging brains. JAMA Neurol. 2017, 74, 91–99. [Google Scholar] [CrossRef]
- Gao, M.; Zhu, S.-Y.; Tan, C.-B.; Xu, B.; Zhang, W.-C.; Du, G.-H. Pinocembrin protects the neurovascular unit by reducing inflammation and extracellular proteolysis in MCAO rats. J. Asian Nat. Prod. Res. 2010, 12, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.S.; Fan, Y.Y.; Ye, G.; Li, J.; Feng, X.P.; Lin, K.; Dong, M.; Wang, Z. Curcumin alleviates brain edema by lowering AQP4 expression levels in a rat model of hypoxia-hypercapnia-induced brain damage. Exp. Ther. Med. 2016, 11, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Zhu, Y.; Diao, Y.; Tao, L.; Yuan, W.; Xiong, X.-c. Anti-edema effect of epigallocatechin gallate on spinal cord injury in rats. Brain Res. 2013, 1527, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Pirici, I.; Balsanu, T.A.; Bogdan, C.; Margaritescu, C.; Divan, T.; Vitalie, V.; Mogoanta, L.; Pirici, D.; Carare, R.O.; Muresanu, D.F. Inhibition of aquaporin-4 improves the outcome of ischaemic stroke and modulates brain paravascular drainage pathways. Int. J. Mol. Sci. 2017, 19, 46. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.-J.; Dai, T.-M.; Shen, Y.-Y.; He, J.-L.; Li, J.; Tu, J.-L. Atorvastatin pretreatment attenuates ischemic brain edema by suppressing aquaporin 4. J. Stroke Cerebrovasc. Dis. 2018, 27, 3247–3255. [Google Scholar] [CrossRef]
- Wallisch, J.S.; Janesko-Feldman, K.; Alexander, H.; Jha, R.M.; Farr, G.W.; McGuirk, P.R.; Kline, A.E.; Jackson, T.C.; Pelletier, M.F.; Clark, R.S.B.; et al. The aquaporin-4 inhibitor AER-271 blocks acute cerebral edema and improves early outcome in a pediatric model of asphyxial cardiac arrest. Pediatr. Res. 2019, 85, 511–517. [Google Scholar] [CrossRef]
- Farr, G.W.; Hall, C.H.; Farr, S.M.; Wade, R.; Detzel, J.M.; Adams, A.G.; Buch, J.M.; Beahm, D.L.; Flask, C.A.; Xu, K. Functionalized phenylbenzamides inhibit aquaporin-4 reducing cerebral edema and improving outcome in two models of CNS injury. Neuroscience 2019, 404, 484–498. [Google Scholar] [CrossRef]
- Bell, A.S.; Wagner, J.; Rosoff, D.B.; Lohoff, F.W. Proprotein convertase subtilisin/kexin type 9 (PCSK9) in the central nervous system. Neurosci. Biobehav. Rev. 2023, 2023, 105155. [Google Scholar] [CrossRef]
- Mazura, A.D.; Ohler, A.; Storck, S.E.; Kurtyka, M.; Scharfenberg, F.; Weggen, S.; Becker-Pauly, C.; Pietrzik, C.U. PCSK9 acts as a key regulator of Aβ clearance across the blood–brain barrier. Cell. Mol. Life Sci. 2022, 79, 212. [Google Scholar] [CrossRef]
- Ardura-Fabregat, A.; Boddeke, E.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzériat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T. Targeting neuroinflammation to treat Alzheimer’s disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef]
- Hamano, T.; Hayashi, K.; Shirafuji, N.; Nakamoto, Y. The implications of autophagy in Alzheimer’s disease. Curr. Alzheimer Res. 2018, 15, 1283–1296. [Google Scholar] [CrossRef] [PubMed]
- Valero, J.; Bernardino, L.; Cardoso, F.L.; Silva, A.P.; Fontes-Ribeiro, C.; Ambrósio, A.F.; Malva, J.O. Impact of neuroinflammation on hippocampal neurogenesis: Relevance to aging and Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 60, S161–S168. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.; Kaye, J.; Moore, M.; Waichunas, D.; Carlson, N.; Quinn, J. Blood–brain barrier impairment in Alzheimer disease: Stability and functional significance. Neurology 2007, 68, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Menard, C.; Pfau, M.L.; Hodes, G.E.; Kana, V.; Wang, V.X.; Bouchard, S.; Takahashi, A.; Flanigan, M.E.; Aleyasin, H.; LeClair, K.B. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 2017, 20, 1752–1760. [Google Scholar] [CrossRef]
- Li, J.; Zheng, M.; Shimoni, O.; Banks, W.A.; Bush, A.I.; Gamble, J.R.; Shi, B. Development of novel therapeutics targeting the blood–brain barrier: From barrier to carrier. Adv. Sci. 2021, 8, 2101090. [Google Scholar] [CrossRef]
- Lanz, T.V.; Ding, Z.; Ho, P.P.; Luo, J.; Agrawal, A.N.; Srinagesh, H.; Axtell, R.; Zhang, H.; Platten, M.; Wyss-Coray, T. Angiotensin II sustains brain inflammation in mice via TGF-β. J. Clin. Investig. 2010, 120, 2782–2794. [Google Scholar] [CrossRef]
- Shen, W.; Li, S.; Chung, S.H.; Zhu, L.; Stayt, J.; Su, T.; Couraud, P.-O.; Romero, I.A.; Weksler, B.; Gillies, M.C. Tyrosine phosphorylation of VE-cadherin and claudin-5 is associated with TGF-β1-induced permeability of centrally derived vascular endothelium. Eur. J. Cell Biol. 2011, 90, 323–332. [Google Scholar] [CrossRef]
- Senatorov, V.V., Jr.; Friedman, A.R.; Milikovsky, D.Z.; Ofer, J.; Saar-Ashkenazy, R.; Charbash, A.; Jahan, N.; Chin, G.; Mihaly, E.; Lin, J.M. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFβ signaling and chronic yet reversible neural dysfunction. Sci. Transl. Med. 2019, 11, eaaw8283. [Google Scholar] [CrossRef]
- Zhang, D.-D.; Jin, C.; Zhang, Y.-T.; Gan, X.-D.; Zou, M.-J.; Wang, Y.-Y.; Fu, W.-L.; Xu, T.; Xing, W.-W.; Xia, W.-R. A novel IL-1RA-PEP fusion protein alleviates blood-brain barrier disruption after ischemia-reperfusion in male rats. J. Neuroinflam. 2018, 15, 16. [Google Scholar] [CrossRef]
- Pillai, J.A.; Bena, J.; Bekris, L.; Kodur, N.; Kasumov, T.; Leverenz, J.B.; Kashyap, S.R. Metabolic syndrome biomarkers relate to rate of cognitive decline in MCI and dementia stages of Alzheimer’s disease. Alzheimer’s Res. Ther. 2023, 15, 54. [Google Scholar] [CrossRef]
- van Buul, J.D.; van Rijssel, J.; van Alphen, F.P.; van Stalborch, A.M.; Mul, E.P.; Hordijk, P.L. ICAM-1 clustering on endothelial cells recruits VCAM-1. J. Biomed. Biotechnol. 2010, 2010, 120328. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.P.; Simonsen, H.J.; Varatharaj, A.; Galea, I.; Frederiksen, J.L.; Larsson, H.B. Permeability of the blood–brain barrier predicts no evidence of disease activity at 2 years after natalizumab or fingolimod treatment in relapsing–remitting multiple sclerosis. Ann. Neurol. 2018, 83, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Stroke Trail, E.A. Investigators. Use of anti-ICAM-1 therapy in Ischemic stroke: Results of the Enlimonab Acute stroke Trail. Neurology 2001, 57, 1428–1434. [Google Scholar]
- Furuya, K.; Takeda, H.; Azhar, S.; McCarron, R.M.; Chen, Y.; Ruetzler, C.A.; Wolcott, K.M.; DeGraba, T.J.; Rothlein, R.; Hugli, T.E. Examination of several potential mechanisms for the negative outcome in a clinical stroke trial of enlimomab, a murine anti-human intercellular adhesion molecule-1 antibody: A bedside-to-bench study. Stroke 2001, 32, 2665–2674. [Google Scholar] [CrossRef]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Shi, W.; Li, F.; Xin, T.; Pang, Q. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb. Blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef]
- Takata, F.; Dohgu, S.; Matsumoto, J.; Machida, T.; Kaneshima, S.; Matsuo, M.; Sakaguchi, S.; Takeshige, Y.; Yamauchi, A.; Kataoka, Y. Metformin induces up-regulation of blood–brain barrier functions by activating AMP-activated protein kinase in rat brain microvascular endothelial cells. Biochem. Biophys. Res. Commun. 2013, 433, 586–590. [Google Scholar] [CrossRef]
- Wang, X.; Xue, G.X.; Liu, W.C.; Shu, H.; Wang, M.; Sun, Y.; Liu, X.; Sun, Y.E.; Liu, C.F.; Liu, J. Melatonin alleviates lipopolysaccharide-compromised integrity of blood–brain barrier through activating AMP-activated protein kinase in old mice. Aging Cell 2017, 16, 414–421. [Google Scholar] [CrossRef]
- Adzemovic, M.Z.; Zeitelhofer, M.; Eriksson, U.; Olsson, T.; Nilsson, I. Imatinib ameliorates neuroinflammation in a rat model of multiple sclerosis by enhancing blood-brain barrier integrity and by modulating the peripheral immune response. PLoS ONE 2013, 8, e56586. [Google Scholar] [CrossRef]
- Ishizaki, T.; Chiba, H.; Kojima, T.; Fujibe, M.; Soma, T.; Miyajima, H.; Nagasawa, K.; Wada, I.; Sawada, N. Cyclic AMP induces phosphorylation of claudin-5 immunoprecipitates and expression of claudin-5 gene in blood–brain-barrier endothelial cells via protein kinase A-dependent and-independent pathways. Exp. Cell Res. 2003, 290, 275–288. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-brain barrier dysfunction amplifies the development of neuroinflammation: Understanding of cellular events in brain microvascular endothelial cells for prevention and treatment of BBB dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, K.; Garvert, L.; Kühn, L.; Bonk, S.; Grabe, H.J.; Van der Auwera, S. Detrimental Effects of ApoE ε4 on Blood–Brain Barrier Integrity and Their Potential Implications on the Pathogenesis of Alzheimer’s Disease. Cells 2023, 12, 2512. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkhalifa, A.E.; Al-Ghraiybah, N.F.; Odum, J.; Shunnarah, J.G.; Austin, N.; Kaddoumi, A. Blood–Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies. Int. J. Mol. Sci. 2023, 24, 16288. https://doi.org/10.3390/ijms242216288
Alkhalifa AE, Al-Ghraiybah NF, Odum J, Shunnarah JG, Austin N, Kaddoumi A. Blood–Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies. International Journal of Molecular Sciences. 2023; 24(22):16288. https://doi.org/10.3390/ijms242216288
Chicago/Turabian StyleAlkhalifa, Amer E., Nour F. Al-Ghraiybah, Julia Odum, John G. Shunnarah, Nataleigh Austin, and Amal Kaddoumi. 2023. "Blood–Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies" International Journal of Molecular Sciences 24, no. 22: 16288. https://doi.org/10.3390/ijms242216288
APA StyleAlkhalifa, A. E., Al-Ghraiybah, N. F., Odum, J., Shunnarah, J. G., Austin, N., & Kaddoumi, A. (2023). Blood–Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies. International Journal of Molecular Sciences, 24(22), 16288. https://doi.org/10.3390/ijms242216288