
Prdx6 Regulates Nlrp3 Inflammasome Activation-Driven Inflammatory Response in Lens Epithelial Cells




Abstract
:1. Introduction
2. Results
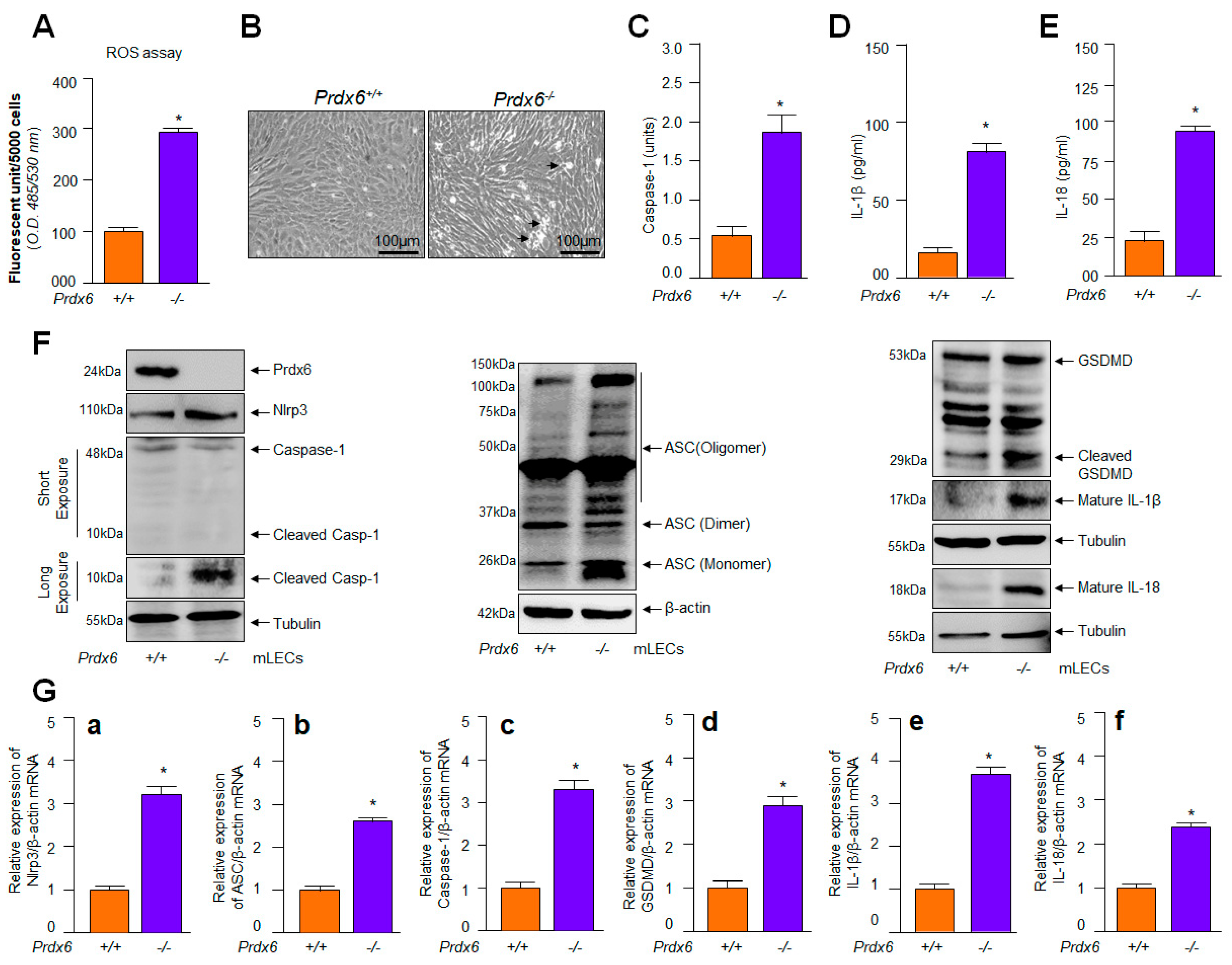
2.1. Prdx6-Deficient mLECs Having Increased ROS Accumulation Displayed Aberrant Expression and Activation of Nlrp3 Inflammasome, Bioactive Caspase-1, IL-1β, IL-18 and GSDMD
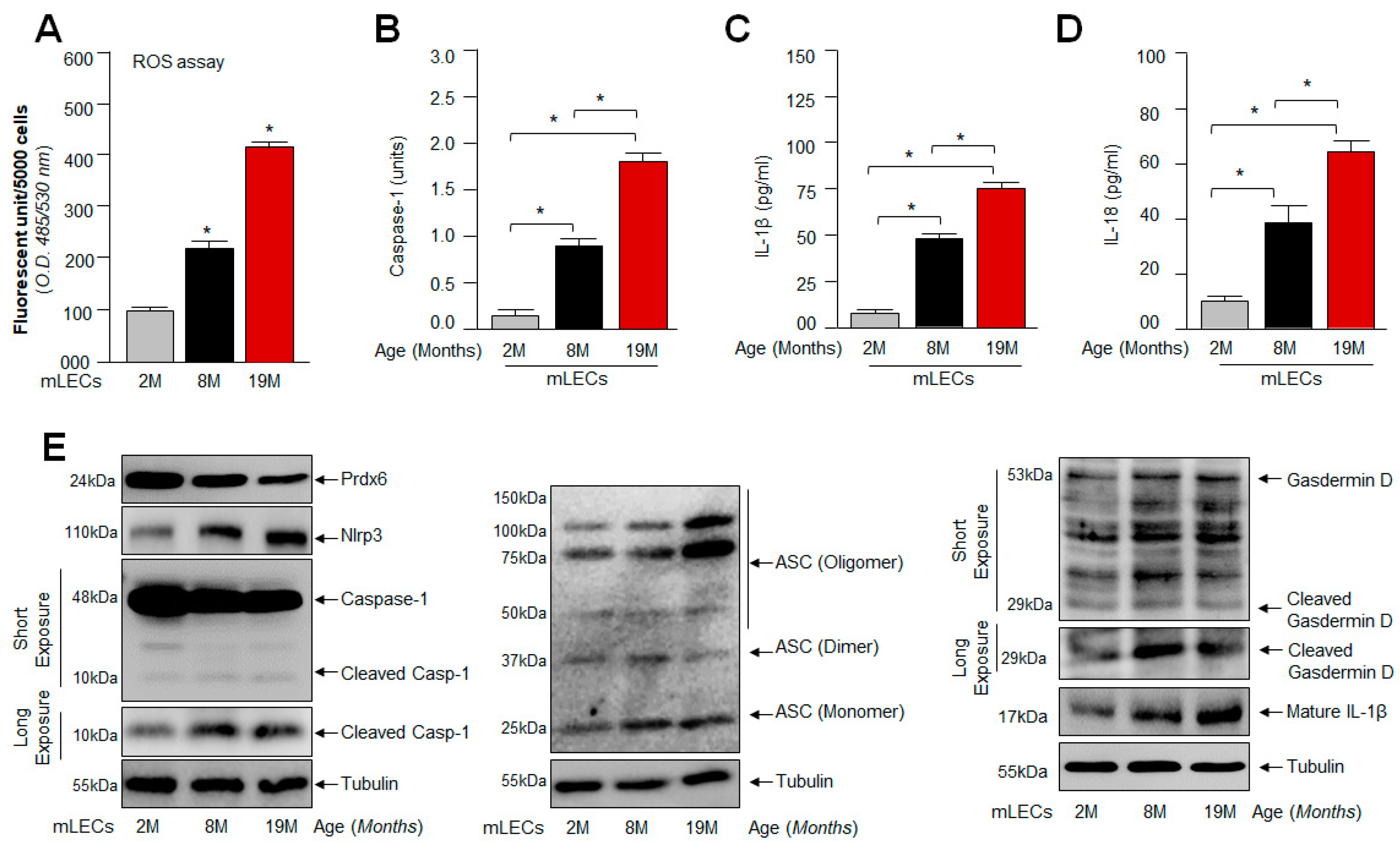
2.2. Age-Dependent Increased Levels of Bioactive Nlrp3, ASC, Cleaved Casp-1 and Inflammatory Cytokines Were Related to Progressive Loss of Prdx6 with Increased Accumulation of ROS
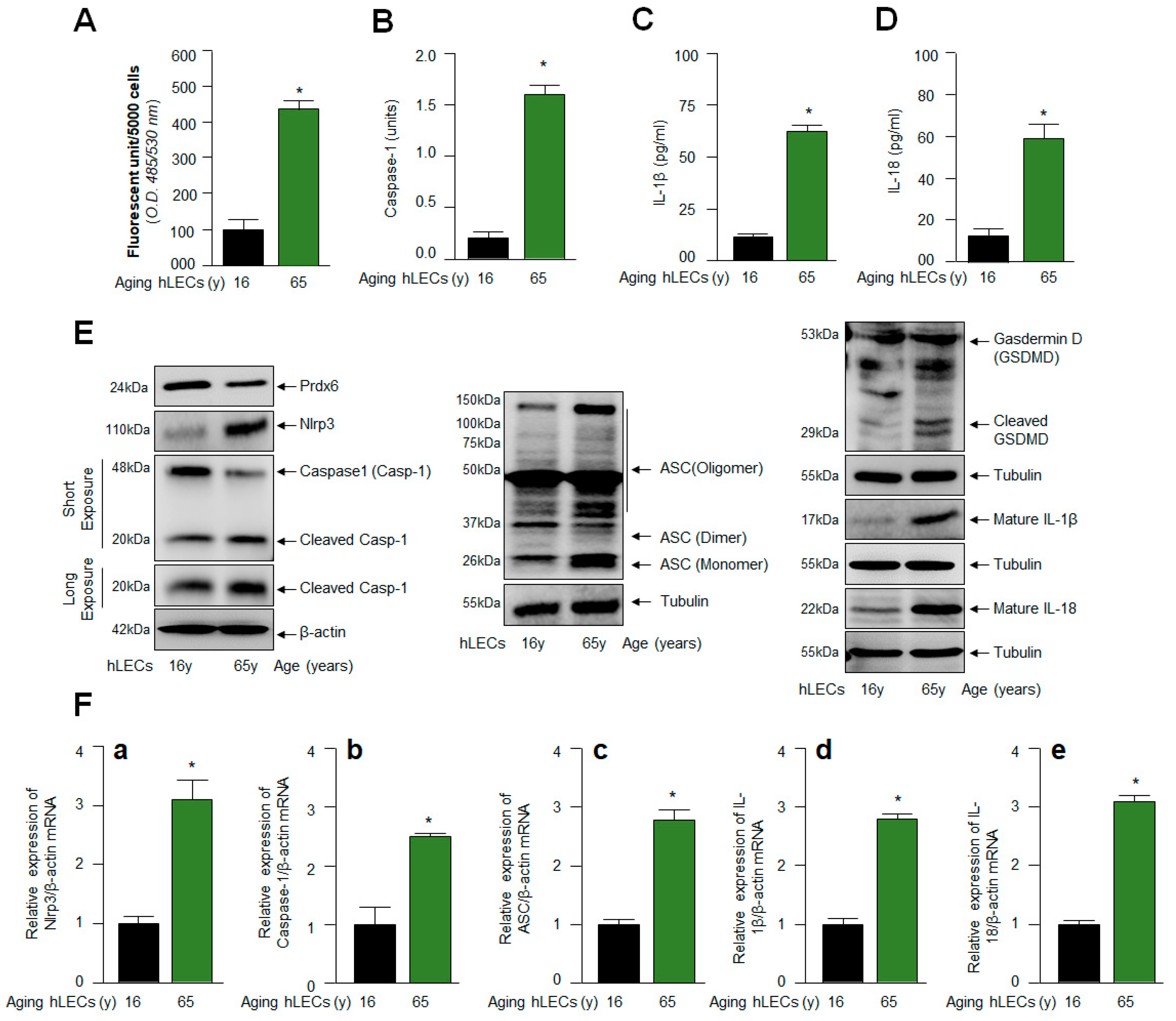
2.3. Similar to mLECs, Aged hLECs Had Increased Expression and Activation of Nlrp3 Inflammasome Components, Including Inflammatory Bioactive Molecules, Casp-1 and IL-1β and IL-18 and GSDMD
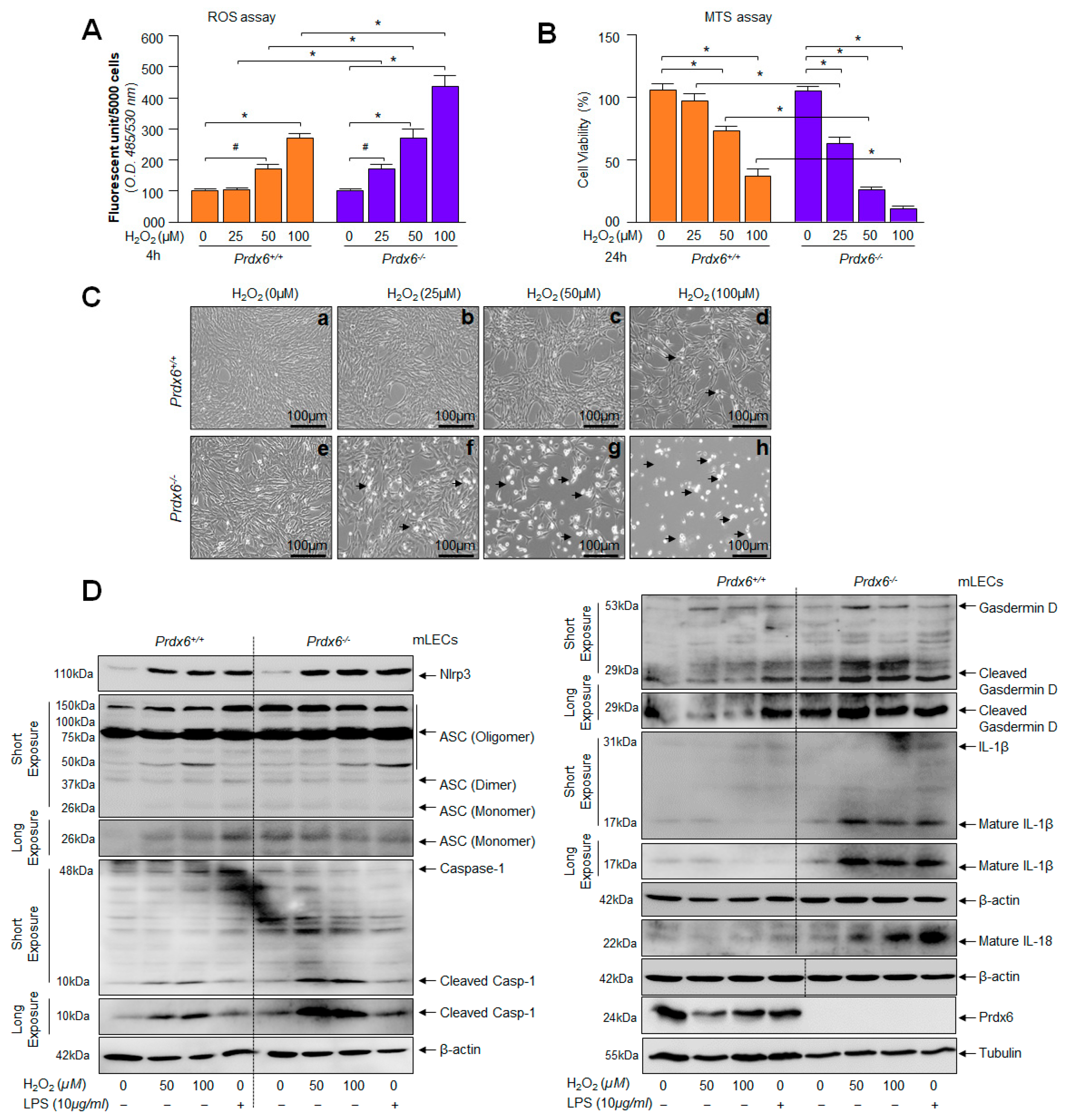
2.4. Prdx6-deficient mLECs and Prdx6 Expressing mLECs under Extrinsic Oxidative Stimulus Showed Aberrant Nlrp3 Inflammasome Activation-Mediated Signaling
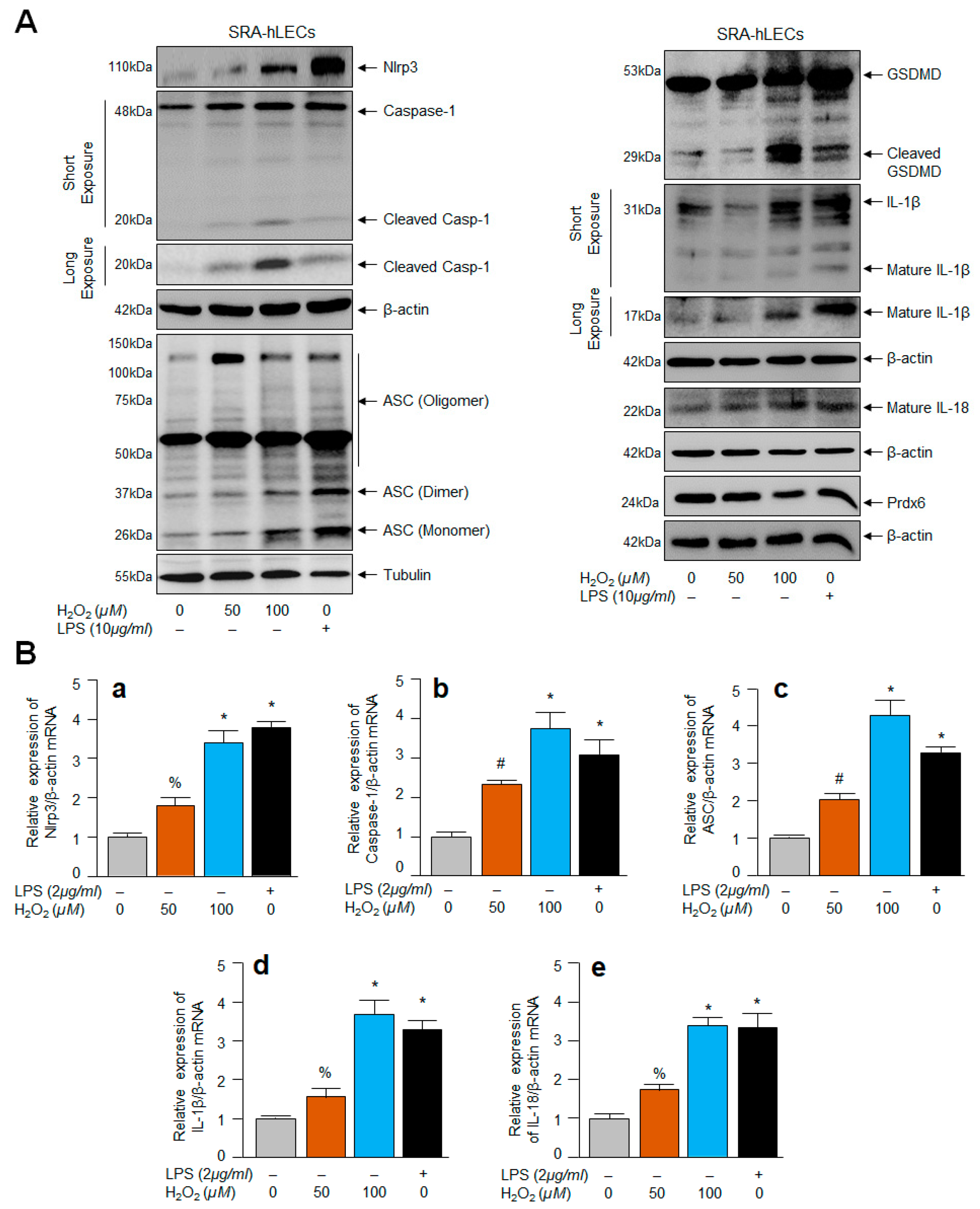
2.5. SRA-hLECs under Oxidative Stress Showed Aberrant Nlrp3 Inflammasome Activation and Its Components, Bioactive ASC, Activated Casp-1 and GSDMD, and Enhanced Release of IL-1β and IL-18
2.6. Enhanced Klf9 Levels in Aging LECs and Prdx6−/− mLECs Were Related to Antioxidant Gene Repression
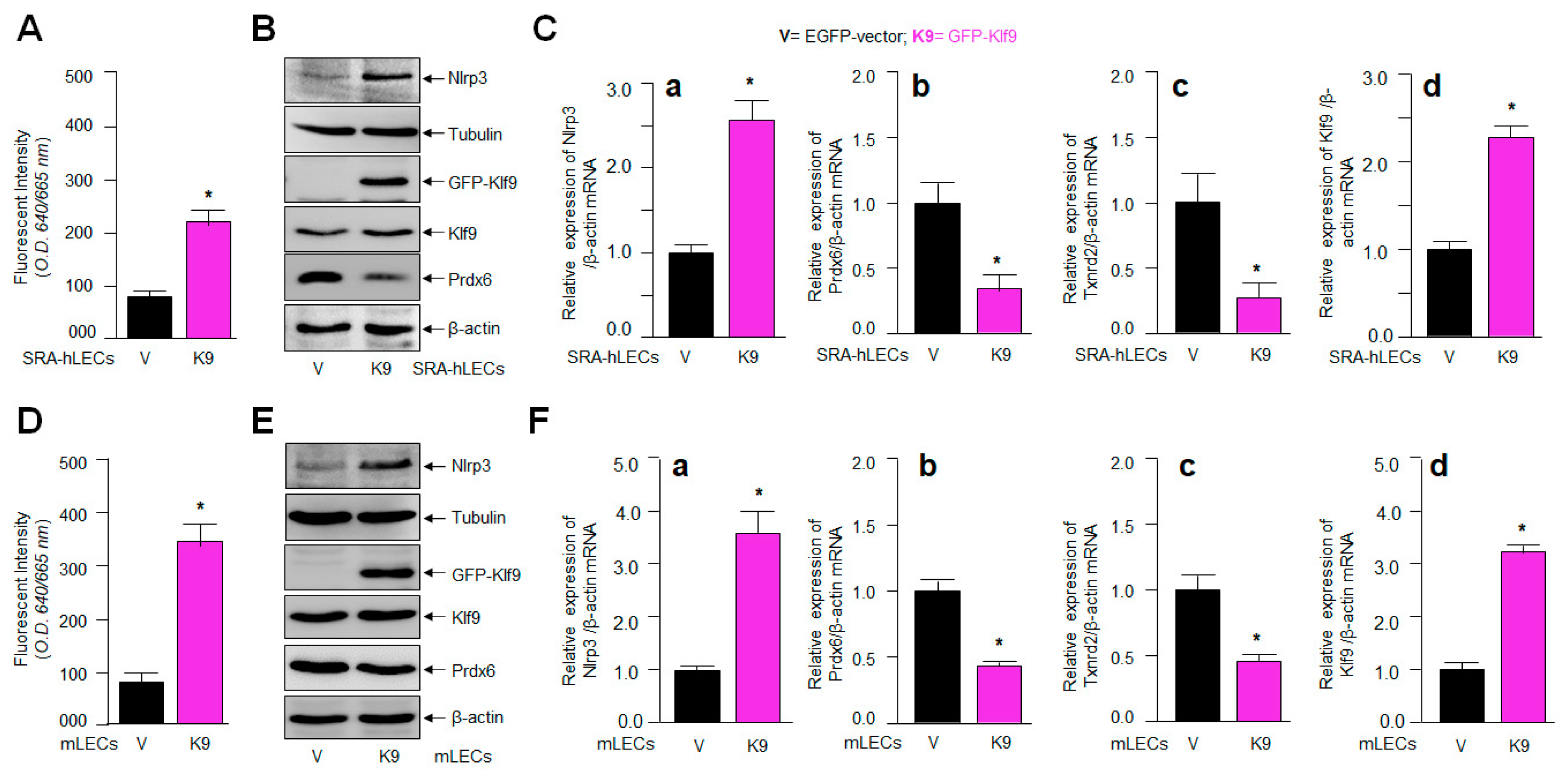
2.7. Klf9 Overexpression in LECs Caused Elevated Oxidative Load, and the Aberrant Expression of Nlrp3 with Reduced Expression of Antioxidant Genes
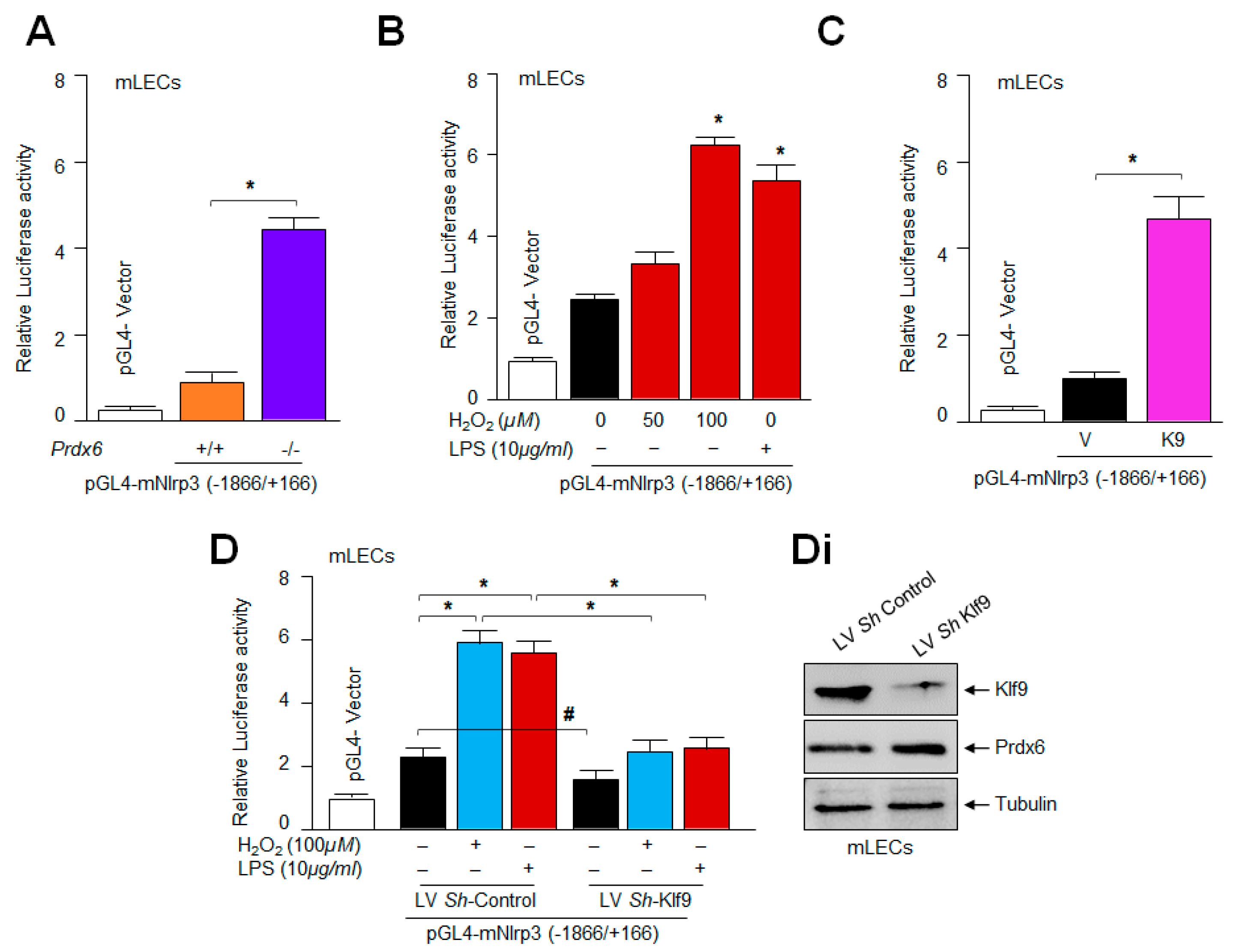
2.8. Loss and Gain Experimentation with Klf9 Disclosed That the Nlrp3 Gene Could Be a New Target for Klf9-Mediated Transcription
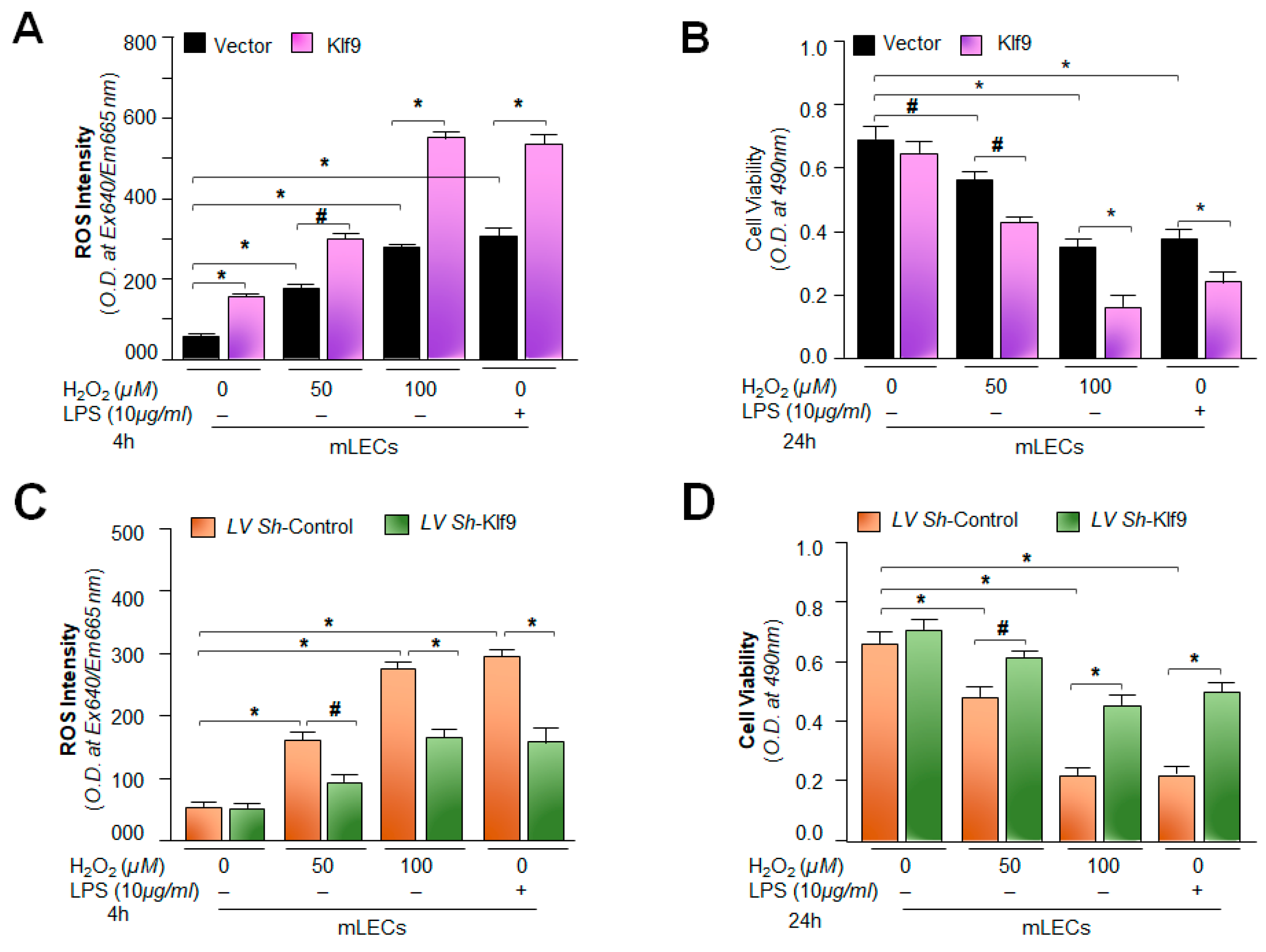
2.9. Physiological Expression of Klf9 Determined the Fate of mLECs during Oxidative Stress by Controlling Antioxidant Gene Prdx6 Expression
2.10. Prdx6 Expression Inhibited Nlrp3 Inflammasome and Its Inflammatory Components Expression/Activation via Alleviating ROS
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.1.1. Mouse Lens Epithelial Cells (mLECs) Isolated from Lenses of C57BL/6 Mice
4.1.2. SRA-Human Lens Epithelial Cells (hLECs) and Primary hLECs Culture and Maintenance
4.2. Quantitation of Intracellular Reactive Oxygen Species (ROS)
4.2.1. ROS Level by H2-DCF-DA in LECs
4.2.2. Quantitation of Intracellular ROS Level by CellROX® Deep Red Reagent
4.3. Protein Isolation and Western Blotting
4.4. RNA Isolation and mRNA Analysis of Mouse or Human LECs Using RT-qPCR
4.5. Mouse and Human Caspase-1 ELISA Assay
4.6. Mouse and Human IL-1β ELISA Assay
4.7. Mouse and Human IL-18 ELISA Assay
4.8. Plasmids or Constructs and Lentiviral (LV) Infection
4.9. Construction of Nlrp3 Promoter—Luciferase and Promoter Activity
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yerramothu, P.; Vijay, A.K.; Willcox, M.D.P. Inflammasomes, the eye and anti-inflammasome therapy. Eye 2018, 32, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, T.C.; Hasegawa, Y.; Iguchi, R.; Cheung, A.; Caffrey, D.R.; Thatcher, E.J.; Mao, W.; Germain, G.; Tamburro, N.D.; Okabe, S.; et al. Inflammasome-derived cytokine IL18 suppresses amyloid-induced seizures in Alzheimer-prone mice. Proc. Natl. Acad. Sci. USA 2018, 115, 9002–9007. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Gao, J.; Van, J.; To, E.; Wang, A.; Cao, S.; Cui, J.Z.; Guo, J.P.; Lee, M.; McGeer, P.L.; et al. Age-related increases in amyloid beta and membrane attack complex: Evidence of inflammasome activation in the rodent eye. J. Neuroinflamm. 2015, 12, 121. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Obligatory Role of AMPK Activation and Antioxidant Defense Pathway in the Regulatory Effects of Metformin on Cellular Protection and Prevention of Lens Opacity. Cells 2022, 11, 3021. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Kubo, E.; Krueger, R.R.; Singh, D.P. Hydralazine Revives Cellular and Ocular Lens Health-Span by Ameliorating the Aging and Oxidative-Dependent Loss of the Nrf2-Activated Cellular Stress Response. Antioxidants 2023, 12, 140. [Google Scholar] [CrossRef]
- Chung, H.Y.; Cesari, M.; Anton, S.; Marzetti, E.; Giovannini, S.; Seo, A.Y.; Carter, C.; Yu, B.P.; Leeuwenburgh, C. Molecular inflammation: Underpinnings of aging and age-related diseases. Ageing Res. Rev. 2009, 8, 18–30. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef]
- Jin, X.; Jin, H.; Shi, Y.; Guo, Y.; Zhang, H. Pyroptosis, a novel mechanism implicated in cataracts. Mol. Med. Rep. 2018, 18, 2277–2285. [Google Scholar] [CrossRef]
- Kauppinen, A.; Niskanen, H.; Suuronen, T.; Kinnunen, K.; Salminen, A.; Kaarniranta, K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells--implications for age-related macular degeneration (AMD). Immunol. Lett. 2012, 147, 29–33. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Wang, Y.X.; Jiang, C.L. Inflammation: The Common Pathway of Stress-Related Diseases. Front. Hum. Neurosci. 2017, 11, 316. [Google Scholar] [CrossRef]
- Pronin, A.; Pham, D.; An, W.; Dvoriantchikova, G.; Reshetnikova, G.; Qiao, J.; Kozhekbaeva, Z.; Reiser, A.E.; Slepak, V.Z.; Shestopalov, V.I. Inflammasome Activation Induces Pyroptosis in the Retina Exposed to Ocular Hypertension Injury. Front. Mol. Neurosci. 2019, 12, 36. [Google Scholar] [CrossRef]
- Dostert, C.; Petrilli, V. Asbestos triggers inflammation by activating the Nalp3 inflammasome. Med. Sci. 2008, 24, 916–918. [Google Scholar] [CrossRef]
- Dostert, C.; Petrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Hogquist, K.A.; Nett, M.A.; Unanue, E.R.; Chaplin, D.D. Interleukin 1 is processed and released during apoptosis. Proc. Natl. Acad. Sci. USA 1991, 88, 8485–8489. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef]
- Dominic, A.; Le, N.T.; Takahashi, M. Loop between NLRP3 Inflammasome and Reactive Oxygen Species. Antioxid. Redox Signal 2022, 36, 784–796. [Google Scholar] [CrossRef]
- Kanneganti, T.D.; Body-Malapel, M.; Amer, A.; Park, J.H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Perregaux, D.; Gabel, C.A. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 1994, 269, 15195–15203. [Google Scholar] [CrossRef]
- Yu, M.; Levine, S.J. Toll-like receptor, RIG-I-like receptors and the NLRP3 inflammasome: Key modulators of innate immune responses to double-stranded RNA viruses. Cytokine Growth Factor. Rev. 2011, 22, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; He, W.T.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef]
- Kurokawa, M.; Kornbluth, S. Caspases and kinases in a death grip. Cell 2009, 138, 838–854. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef]
- Adams, S.; Green, P.; Claxton, R.; Simcox, S.; Williams, M.V.; Walsh, K.; Leeuwenburgh, C. Reactive carbonyl formation by oxidative and non-oxidative pathways. Front. Biosci. 2001, 6, A17–A24. [Google Scholar] [CrossRef]
- Youm, Y.H.; Adijiang, A.; Vandanmagsar, B.; Burk, D.; Ravussin, A.; Dixit, V.D. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 2011, 152, 4039–4045. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Fatma, N.; Kubo, E.; Takamura, Y.; Ishihara, K.; Garcia, C.; Beebe, D.C.; Singh, D.P. Loss of NF-kappaB control and repression of Prdx6 gene transcription by reactive oxygen species-driven SMAD3-mediated transforming growth factor beta signaling. J. Biol. Chem. 2009, 284, 22758–22772. [Google Scholar] [CrossRef] [PubMed]
- Fatma, N.; Singh, P.; Chhunchha, B.; Kubo, E.; Shinohara, T.; Bhargavan, B.; Singh, D.P. Deficiency of Prdx6 in lens epithelial cells induces ER stress response-mediated impaired homeostasis and apoptosis. Am. J. Physiol. Cell Physiol. 2011, 301, C954–C967. [Google Scholar] [CrossRef]
- Chhunchha, B.; Fatma, N.; Kubo, E.; Rai, P.; Singh, S.P.; Singh, D.P. Curcumin abates hypoxia-induced oxidative stress based-ER stress-mediated cell death in mouse hippocampal cells (HT22) by controlling Prdx6 and NF-kappaB regulation. Am. J. Physiol. Cell Physiol. 2013, 304, C636–C655. [Google Scholar] [CrossRef]
- Honda, T.S.B.; Ku, J.; Anders, H.J. Cell type-specific roles of NLRP3, inflammasome-dependent and -independent, in host defense, sterile necroinflammation, tissue repair, and fibrosis. Front. Immunol. 2023, 14, 1214289. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef]
- McKee, C.M.; Coll, R.C. NLRP3 inflammasome priming: A riddle wrapped in a mystery inside an enigma. J. Leukoc. Biol. 2020, 108, 937–952. [Google Scholar] [CrossRef]
- Takahashi, M. NLRP3 inflammasome as a novel player in myocardial infarction. Int. Heart J. 2014, 55, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Sulforaphane-Induced Klf9/Prdx6 Axis Acts as a Molecular Switch to Control Redox Signaling and Determines Fate of Cells. Cells 2019, 8, 1159. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Switching of Redox Signaling by Prdx6 Expression Decides Cellular Fate by Hormetic Phenomena Involving Nrf2 and Reactive Oxygen Species. Cells 2022, 11, 1266. [Google Scholar] [CrossRef]
- Fink, E.E.; Moparthy, S.; Bagati, A.; Bianchi-Smiraglia, A.; Lipchick, B.C.; Wolff, D.W.; Roll, M.V.; Wang, J.; Liu, S.; Bakin, A.V.; et al. XBP1-KLF9 Axis Acts as a Molecular Rheostat to Control the Transition from Adaptive to Cytotoxic Unfolded Protein Response. Cell Rep. 2018, 25, 212–223.e4. [Google Scholar] [CrossRef]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef]
- Gu, P.; Wang, Z.; Yu, X.; Wu, N.; Wu, L.; Li, Y.; Hu, X. Mechanism of KLF9 in airway inflammation in chronic obstructive pulmonary. Immun. Inflamm. Dis. 2023, 11, e1043. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Y.; Zhong, X.; Xia, H.; Zhao, M.; Zhao, M.; Xu, L.; Guo, X.; You, C.G. Lipoxin A4 attenuates MSU-crystal-induced NLRP3 inflammasome activation through suppressing Nrf2 thereby increasing TXNRD2. Front. Immunol. 2022, 13, 1060441. [Google Scholar] [CrossRef] [PubMed]
- Bagati, A.; Moparthy, S.; Fink, E.E.; Bianchi-Smiraglia, A.; Yun, D.H.; Kolesnikova, M.; Udartseva, O.O.; Wolff, D.W.; Roll, M.V.; Lipchick, B.C.; et al. KLF9-dependent ROS regulate melanoma progression in stage-specific manner. Oncogene 2019, 38, 3585–3597. [Google Scholar] [CrossRef] [PubMed]
- Imataka, H.; Sogawa, K.; Yasumoto, K.; Kikuchi, Y.; Sasano, K.; Kobayashi, A.; Hayami, M.; Fujii-Kuriyama, Y. Two regulatory proteins that bind to the basic transcription element (BTE), a GC box sequence in the promoter region of the rat P-4501A1 gene. EMBO J. 1992, 11, 3663–3671. [Google Scholar] [CrossRef]
- Mannava, S.; Zhuang, D.; Nair, J.R.; Bansal, R.; Wawrzyniak, J.A.; Zucker, S.N.; Fink, E.E.; Moparthy, K.C.; Hu, Q.; Liu, S.; et al. KLF9 is a novel transcriptional regulator of bortezomib- and LBH589-induced apoptosis in multiple myeloma cells. Blood 2012, 119, 1450–1458. [Google Scholar] [CrossRef]
- Qu, R.; Liu, J.; Feng, L.; Li, L.; Liu, J.; Sun, F.; Sun, L. Down-regulation of KLF9 ameliorates LPS-caused acute lung injury and inflammation in mice via reducing GSDMD expression. Autoimmunity 2022, 55, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Eismann, T.; Huber, N.; Shin, T.; Kuboki, S.; Galloway, E.; Wyder, M.; Edwards, M.J.; Greis, K.D.; Shertzer, H.G.; Fisher, A.B.; et al. Peroxiredoxin-6 protects against mitochondrial dysfunction and liver injury during ischemia-reperfusion in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G266–G274. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A(2) activities. Antioxid. Redox Signal 2011, 15, 831–844. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6 in the repair of peroxidized cell membranes and cell signaling. Arch. Biochem. Biophys. 2017, 617, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Dodia, C.; Feinstein, S.I. Identification of Small Peptides that Inhibit NADPH Oxidase (Nox2) Activation. Antioxidants 2018, 7, 181. [Google Scholar] [CrossRef]
- Fisher, A.B.; Dodia, C.; Sorokina, E.M.; Li, H.; Zhou, S.; Raabe, T.; Feinstein, S.I. A novel lysophosphatidylcholine acyl transferase activity is expressed by peroxiredoxin 6. J. Lipid Res. 2016, 57, 587–596. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef]
- Li, Y.; Huang, H.; Liu, B.; Zhang, Y.; Pan, X.; Yu, X.Y.; Shen, Z.; Song, Y.H. Inflammasomes as therapeutic targets in human diseases. Signal Transduct. Target. Ther. 2021, 6, 247. [Google Scholar] [CrossRef]
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxid. Med. Cell Longev. 2016, 2016, 2183026. [Google Scholar] [CrossRef]
- Sebastian-Valverde, M.; Pasinetti, G.M. The NLRP3 Inflammasome as a Critical Actor in the Inflammaging Process. Cells 2020, 9, 1552. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed]
- Abderrazak, A.; Couchie, D.; Mahmood, D.F.; Elhage, R.; Vindis, C.; Laffargue, M.; Mateo, V.; Buchele, B.; Ayala, M.R.; El Gaafary, M.; et al. Anti-inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a high-fat diet. Circulation 2015, 131, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef]
- Chhunchha, B.; Kubo, E.; Fatma, N.; Singh, D.P. Sumoylation-deficient Prdx6 gains protective function by amplifying enzymatic activity and stability and escapes oxidative stress-induced aberrant Sumoylation. Cell Death Dis. 2017, 8, e2525. [Google Scholar] [CrossRef]
- Sun, Y.; Rong, X.; Li, D.; Jiang, Y.; Lu, Y.; Ji, Y. Down-regulation of CRTAC1 attenuates UVB-induced pyroptosis in HLECs through inhibiting ROS production. Biochem. Biophys. Res. Commun. 2020, 532, 159–165. [Google Scholar] [CrossRef]
- Szeliga, M. Peroxiredoxins in Neurodegenerative Diseases. Antioxidants 2020, 9, 1203. [Google Scholar] [CrossRef]
- Wahlig, S.; Lovatt, M.; Mehta, J.S. Functional role of peroxiredoxin 6 in the eye. Free Radic. Biol. Med. 2018, 126, 210–220. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiao, Y.; Li, X.; Gao, S.; Zhou, N.; Duan, J.; Zhang, M. Pyroptosis: A New Insight Into Eye Disease Therapy. Front. Pharmacol. 2021, 12, 797110. [Google Scholar] [CrossRef]
- Ambruso, D.R.; Ellison, M.A.; Thurman, G.W.; Leto, T.L. Peroxiredoxin 6 translocates to the plasma membrane during neutrophil activation and is required for optimal NADPH oxidase activity. Biochim. Biophys. Acta 2012, 1823, 306–315. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, X.; Zheng, L.; Li, Z.; Zhao, X.; Lai, W.; Shen, H.; Lv, J.; Yang, G.; Wang, Q.; et al. Peroxiredoxin 6 Is a Crucial Factor in the Initial Step of Mitochondrial Clearance and Is Upstream of the PINK1-Parkin Pathway. Antioxid. Redox Signal 2016, 24, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Manevich, Y.; Hutchens, S.; Halushka, P.V.; Tew, K.D.; Townsend, D.M.; Jauch, E.C.; Borg, K. Peroxiredoxin VI oxidation in cerebrospinal fluid correlates with traumatic brain injury outcome. Free Radic. Biol. Med. 2014, 72, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, E.M.; Feinstein, S.I.; Zhou, S.; Fisher, A.B. Intracellular targeting of peroxiredoxin 6 to lysosomal organelles requires MAPK activity and binding to 14-3-3epsilon. Am. J. Physiol. Cell Physiol. 2011, 300, C1430–C1441. [Google Scholar] [CrossRef]
- Gritsenko, A.; Green, J.P.; Brough, D.; Lopez-Castejon, G. Mechanisms of NLRP3 priming in inflammaging and age related diseases. Cytokine Growth Factor. Rev. 2020, 22, 15–25. [Google Scholar] [CrossRef]
- Lopez-Grueso, M.J.; Lagal, D.J.; Garcia-Jimenez, A.F.; Tarradas, R.M.; Carmona-Hidalgo, B.; Peinado, J.; Requejo-Aguilar, R.; Barcena, J.A.; Padilla, C.A. Knockout of PRDX6 induces mitochondrial dysfunction and cell cycle arrest at G2/M in HepG2 hepatocarcinoma cells. Redox Biol. 2020, 37, 101737. [Google Scholar] [CrossRef] [PubMed]
- Barlan, A.U.; Danthi, P.; Wiethoff, C.M. Lysosomal localization and mechanism of membrane penetration influence nonenveloped virus activation of the NLRP3 inflammasome. Virology 2011, 412, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; den Hartigh, A.B.; Loomis, W.P.; Cookson, B.T. Coordinated host responses during pyroptosis: Caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. J. Immunol. 2011, 187, 2748–2754. [Google Scholar] [CrossRef] [PubMed]
- Lima, H., Jr.; Jacobson, L.S.; Goldberg, M.F.; Chandran, K.; Diaz-Griffero, F.; Lisanti, M.P.; Brojatsch, J. Role of lysosome rupture in controlling Nlrp3 signaling and necrotic cell death. Cell Cycle 2013, 12, 1868–1878. [Google Scholar] [CrossRef]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Weber, K.; Schilling, J.D. Distinct lysosome phenotypes influence inflammatory function in peritoneal and bone marrow-derived macrophages. Int. J. Inflam. 2014, 2014, 154936. [Google Scholar] [CrossRef]
- Zhuang, Y.; Yasinta, M.; Hu, C.; Zhao, M.; Ding, G.; Bai, M.; Yang, L.; Ni, J.; Wang, R.; Jia, Z.; et al. Mitochondrial dysfunction confers albumin-induced NLRP3 inflammasome activation and renal tubular injury. Am. J. Physiol. Renal Physiol. 2015, 308, F857–F866. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, S.; Pelegrin, P. Pyroptosis and Redox Balance in Kidney Diseases. Antioxid. Redox Signal 2021, 35, 40–60. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, C.; Pelegrin, P.; Baroja-Mazo, A.; Cuevas, S. Emerging Role of the Inflammasome and Pyroptosis in Hypertension. Int. J. Mol. Sci. 2021, 22, 1064. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Wang, X.; Phelan, S.A.; Forsman-Semb, K.; Taylor, E.F.; Petros, C.; Brown, A.; Lerner, C.P.; Paigen, B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem. 2003, 278, 25179–25190. [Google Scholar] [CrossRef]
- Piippo, N.; Korhonen, E.; Hytti, M.; Kinnunen, K.; Kaarniranta, K.; Kauppinen, A. Oxidative Stress is the Principal Contributor to Inflammasome Activation in Retinal Pigment Epithelium Cells with Defunct Proteasomes and Autophagy. Cell Physiol. Biochem. 2018, 49, 359–367. [Google Scholar] [CrossRef]
- Sharma, A.; Tate, M.; Mathew, G.; Vince, J.E.; Ritchie, R.H.; de Haan, J.B. Oxidative Stress and NLRP3-Inflammasome Activity as Significant Drivers of Diabetic Cardiovascular Complications: Therapeutic Implications. Front. Physiol. 2018, 9, 114. [Google Scholar] [CrossRef]
- Gao, J.; Liu, R.T.; Cao, S.; Cui, J.Z.; Wang, A.; To, E.; Matsubara, J.A. NLRP3 inflammasome: Activation and regulation in age-related macular degeneration. Mediators Inflamm. 2015, 2015, 690243. [Google Scholar] [CrossRef]
- Han, S.; Cai, W.; Yang, X.; Jia, Y.; Zheng, Z.; Wang, H.; Li, J.; Li, Y.; Gao, J.; Fan, L.; et al. ROS-Mediated NLRP3 Inflammasome Activity Is Essential for Burn-Induced Acute Lung Injury. Mediators Inflamm. 2015, 2015, 720457. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, Y.T.; Wang, L.Q.; Li, L.Y.; Bao, Q.; Tian, S.; Chen, M.X.; Chen, H.X.; Cui, J.; Li, C.W. NOD-like receptor family, pyrin domain containing 3 (NLRP3) contributes to inflammation, pyroptosis, and mucin production in human airway epithelium on rhinovirus infection. J. Allergy Clin. Immunol. 2019, 144, 777–787.e9. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Boswell, B.A.; Korol, A.; West-Mays, J.A.; Musil, L.S. Dual function of TGFbeta in lens epithelial cell fate: Implications for secondary cataract. Mol. Biol. Cell 2017, 28, 907–921. [Google Scholar] [CrossRef]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; Lopez-Giraldez, F.; Dash, B.C.; Munoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018, 362, eaar2971. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- McCormick, M.A.; Promislow, D.E.L. Recent Advances in the Systems Biology of Aging. Antioxid. Redox Signal 2018, 29, 973–984. [Google Scholar] [CrossRef]
- Zou, Y.; Cui, B.; Liang, P.; Tian, X.; Ma, Y.; Zhao, S. Inhibition of NLRP3 Protects Human Lens Epithelial Cells against Oxidative Stress-Induced Apoptosis by NF-kappaB Signaling. Ophthalmic Res. 2020, 63, 174–181. [Google Scholar] [CrossRef]
- Gans, I.; Hartig, E.I.; Zhu, S.; Tilden, A.R.; Hutchins, L.N.; Maki, N.J.; Graber, J.H.; Coffman, J.A. Klf9 is a key feedforward regulator of the transcriptomic response to glucocorticoid receptor activity. Sci. Rep. 2020, 10, 11415. [Google Scholar] [CrossRef]
- Gans, I.M.; Grendler, J.; Babich, R.; Jayasundara, N.; Coffman, J.A. Glucocorticoid-Responsive Transcription Factor Kruppel-Like Factor 9 Regulates fkbp5 and Metabolism. Front. Cell Dev. Biol. 2021, 9, 727037. [Google Scholar] [CrossRef]
- Boaru, S.G.; Borkham-Kamphorst, E.; Van de Leur, E.; Lehnen, E.; Liedtke, C.; Weiskirchen, R. NLRP3 inflammasome expression is driven by NF-kappaB in cultured hepatocytes. Biochem. Biophys. Res. Commun. 2015, 458, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Grenier, J.M.; Wang, L.; Manji, G.A.; Huang, W.J.; Al-Garawi, A.; Kelly, R.; Carlson, A.; Merriam, S.; Lora, J.M.; Briskin, M.; et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS Lett. 2002, 530, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Dai, Z.; Li, Y.; Zhu, H.; Zhao, L. TLR9 regulates NLRP3 inflammasome activation via the NF-kB signaling pathway in diabetic nephropathy. Diabetol. Metab. Syndr. 2022, 14, 26. [Google Scholar] [CrossRef]
- Minton, K. Inflammation: Inflammasome-related ageing. Nat. Rev. Immunol. 2017, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Flavell, R.A. Molecular mechanism of NLRP3 inflammasome activation. J. Clin. Immunol. 2010, 30, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Flavell, R.A. Inflammasome activation. The missing link: How the inflammasome senses oxidative stress. Immunol. Cell Biol. 2010, 88, 510–512. [Google Scholar] [CrossRef]
- Jin, X.; Wang, C.; Wu, W.; Liu, T.; Ji, B.; Zhou, F. Cyanidin-3-glucoside Alleviates 4-Hydroxyhexenal-Induced NLRP3 Inflammasome Activation via JNK-c-Jun/AP-1 Pathway in Human Retinal Pigment Epithelial Cells. J. Immunol. Res. 2018, 2018, 5604610. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef]
- Xiong, W.; MacColl Garfinkel, A.E.; Li, Y.; Benowitz, L.I.; Cepko, C.L. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J. Clin. Investig. 2015, 125, 1433–1445. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Harijith, A.; Ebenezer, D.L.; Natarajan, V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 2014, 5, 352. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611. [Google Scholar] [CrossRef]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef]
- Kubo, E.; Miyazawa, T.; Fatma, N.; Akagi, Y.; Singh, D.P. Development- and age-associated expression pattern of peroxiredoxin 6, and its regulation in murine ocular lens. Mech. Ageing Dev. 2006, 127, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.X.; Werstuck, G.; Chen, Y. Oxidative stress and aging diseases. Oxid. Med. Cell Longev. 2014, 2014, 569146. [Google Scholar] [CrossRef] [PubMed]
- Akhter, J.; Khan, J.; Baghel, M.; Beg, M.M.A.; Goswami, P.; Afjal, M.A.; Ahmad, S.; Habib, H.; Najmi, A.K.; Raisuddin, S. NLRP3 inflammasome in rosmarinic acid-afforded attenuation of acute kidney injury in mice. Sci. Rep. 2022, 12, 1313. [Google Scholar] [CrossRef]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Lim, J.; Luderer, U. Oxidative damage increases and antioxidant gene expression decreases with aging in the mouse ovary. Biol. Reprod. 2011, 84, 775–782. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Peng, J.; Feng, H.; Yang, Y.; Gao, J.; Liu, C.; Xu, J.; Zhao, Y.; Pan, S.; Wang, Y.; et al. KLF9 Aggravates Streptozotocin-Induced Diabetic Cardiomyopathy by Inhibiting PPARgamma/NRF2 Signalling. Cells 2022, 11, 3393. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Weng, Y.; Lai, L.; Lei, H.; Xu, S.; Zhang, Y.; Li, L. KLF9 regulates PRDX6 expression in hyperglycemia-aggravated bupivacaine neurotoxicity. Mol. Cell Biochem. 2021, 476, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Li, H. KLF9 deficiency protects the heart from inflammatory injury triggered by myocardial infarction. Korean J. Physiol. Pharmacol. 2023, 27, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.C.; Shastri, M.D.; Peterson, G.M.; Patel, R.P.; Pathinayake, P.S.; Dua, K.; Hansbro, N.G.; Hsu, A.C.; Wark, P.A.; Shukla, S.D.; et al. The complex interplay between endoplasmic reticulum stress and the NLRP3 inflammasome: A potential therapeutic target for inflammatory disorders. Clin. Transl. Immunology 2021, 10, e1247. [Google Scholar] [CrossRef]
- Li, W.; Cao, T.; Luo, C.; Cai, J.; Zhou, X.; Xiao, X.; Liu, S. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl. Microbiol. Biotechnol. 2020, 104, 6129–6140. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Tezcan, G.; Garanina, E.E.; Alsaadi, M.; Gilazieva, Z.E.; Martinova, E.V.; Markelova, M.I.; Arkhipova, S.S.; Hamza, S.; McIntyre, A.; Rizvanov, A.A.; et al. Therapeutic Potential of Pharmacological Targeting NLRP3 Inflammasome Complex in Cancer. Front. Immunol. 2020, 11, 607881. [Google Scholar] [CrossRef]
- Yu, J.W.; Lee, M.S. Mitochondria and the NLRP3 inflammasome: Physiological and pathological relevance. Arch. Pharm. Res. 2016, 39, 1503–1518. [Google Scholar] [CrossRef]
- Rani, N.; Arya, D.S. Chrysin rescues rat myocardium from ischemia-reperfusion injury via PPAR-gamma/Nrf2 activation. Eur. J. Pharmacol. 2020, 883, 173389. [Google Scholar] [CrossRef]
- Yan, Q.; He, B.; Hao, G.; Liu, Z.; Tang, J.; Fu, Q.; Jiang, C.X. KLF9 aggravates ischemic injury in cardiomyocytes through augmenting oxidative stress. Life Sci. 2019, 233, 116641. [Google Scholar] [CrossRef]
- Ye, C.; Li, S.; Yao, W.; Xu, L.; Qiu, Y.; Liu, Y.; Wu, Z.; Hou, Y. The anti-inflammatory effects of baicalin through suppression of NLRP3 inflammasome pathway in LPS-challenged piglet mononuclear phagocytes. Innate Immun. 2016, 22, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, H.; Lu, X.; Zhao, S. N-acetylcysteine alleviates ocular surface damage in STZ-induced diabetic mice by inhibiting the ROS/NLRP3/Caspase-1/IL-1beta signaling pathway. Exp. Eye Res. 2021, 209, 108654. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yao, W.; Xu, J.; Qiu, Y.; Cao, F.; Li, S.; Yang, S.; Yang, H.; Wu, Z.; Hou, Y. The anti-inflammatory effects of acetaminophen and N-acetylcysteine through suppression of the NLRP3 inflammasome pathway in LPS-challenged piglet mononuclear phagocytes. Innate Immun. 2015, 21, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, F.J.; Yang, W.L.; Qiao, H.Z.; Zhang, S.J. Quercetin improves cognitive disorder in aging mice by inhibiting NLRP3 inflammasome activation. Food Funct. 2021, 12, 717–725. [Google Scholar] [CrossRef]
- Ibaraki, N.; Chen, S.C.; Lin, L.R.; Okamoto, H.; Pipas, J.M.; Reddy, V.N. Human lens epithelial cell line. Exp. Eye Res. 1998, 67, 577–585. [Google Scholar] [CrossRef]
- McAvoy, J.W.; Chamberlain, C.G.; de Iongh, R.U.; Hales, A.M.; Lovicu, F.J. Lens development. Eye 1999, 13 Pt 3b, 425–437. [Google Scholar] [CrossRef]
- Piatigorsky, J.; Rothschild, S.S. Loss during development of the ability of chick embryonic lens cells to elongate in culture: Inverse relationship between cell division and elongation. Dev. Biol. 1972, 28, 382–389. [Google Scholar] [CrossRef]

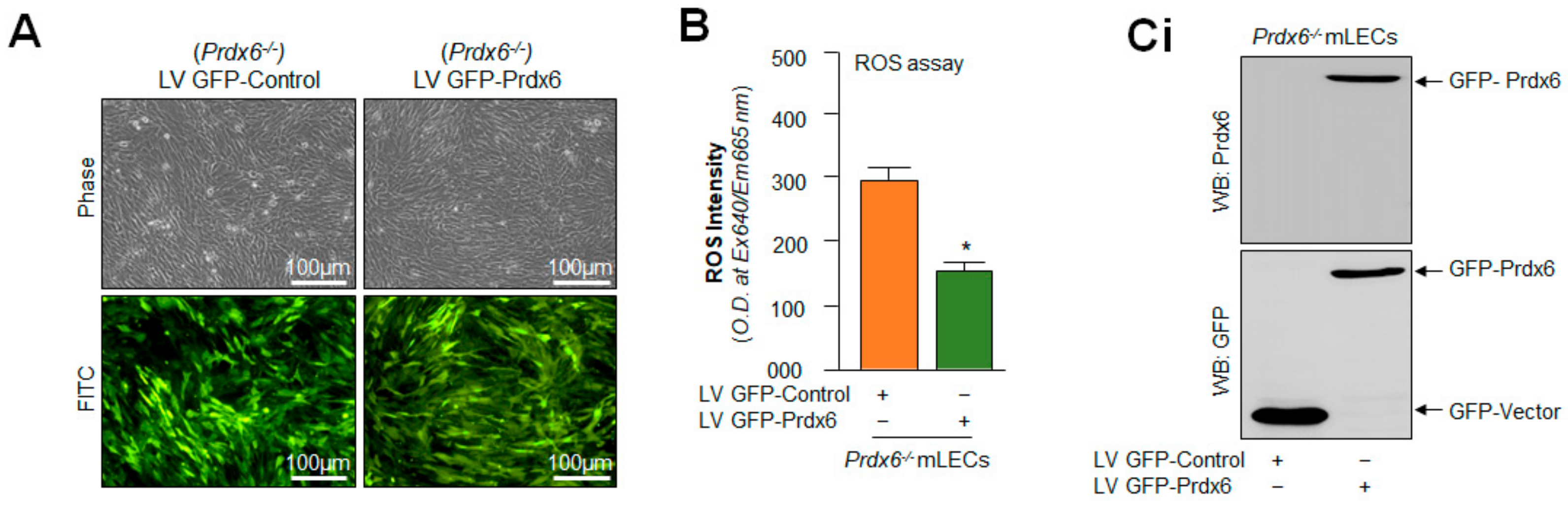
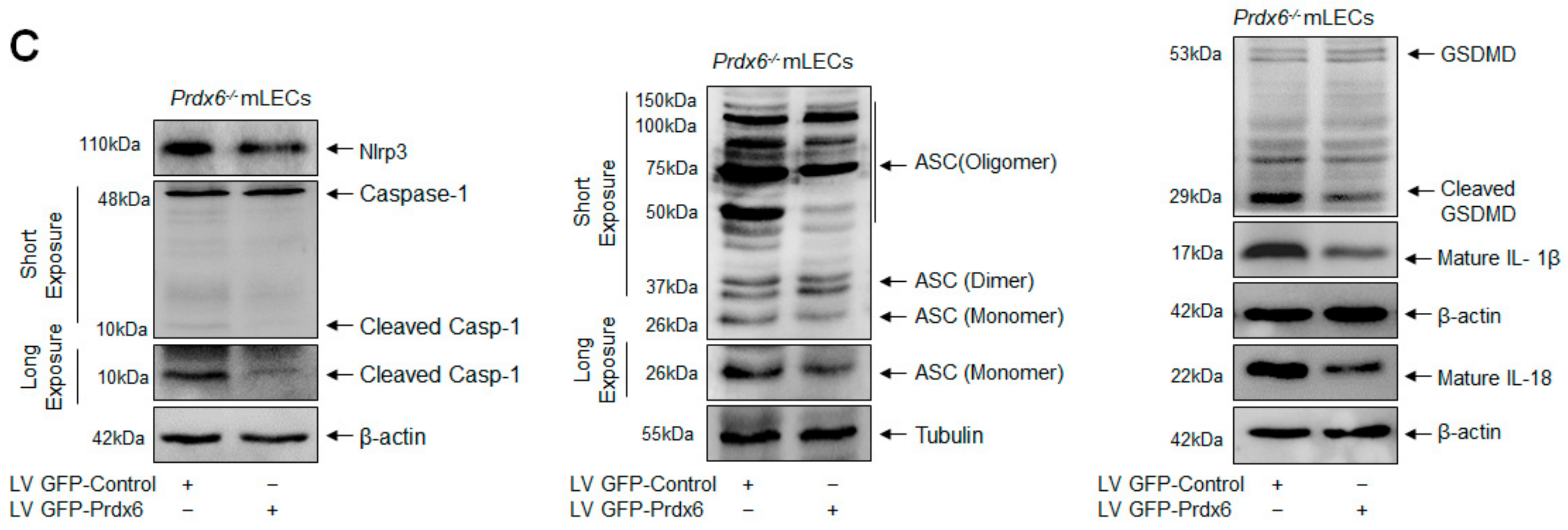


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| mNlrp3 | TCACAACTCGCCCAAGGAGGAA | AAGAGACCACGGCAGAAGCTAG |
| mASC | CTGCTCAGAGTACAGCCAGAAC | CTGTCCTTCAGTCAGCACACTG |
| mCaspase-1 | GGCACATTTCCAGGACTGACTG | GCAAGACGTGTACGAGTGGTTG |
| mIL-1β | TGGACCTTCCAGGATGAGGACA | GTTCATCTCGGAGCCTGTAGTG |
| mIL-18 | GACAGCCTGTGTTCGAGGATATG | TGTTCTTACAGGAGAGGGTAGAC |
| mGSDMD | GGTGCTTGACTCTGGAGAACTG | GCTGCTTTGACAGCACCGTTGT |
| mPrdx6 | TTCAATAGACAGTGTTGAGGATCA | CGTGGGTGTTTCACCATTG |
| mKlf9 | CTACAGTGGCTGTGGGAAAGTC | CTCATCCGAGCGCGAGAACTTT |
| mTxnrd2 | GCCATTGGAGATGTTGCTGAGG | CACAGCCATACTCCAGTGGTGT |
| Mβ-actin | CTAAGGCCAACCGTGAAAAG | ACCAGAGGCATACAGGGACA |
| hNlrp3 | GGACTGAAGCACCTGTTGTGCA | TCCTGAGTCTCCCAAGGCATTC |
| hASC | AGCTCACCGCTAACGTGCTGC | GCTTGGCTGCCGACTGAGGAG |
| hCaspase-1 | GCTGAGGTTGACATCACAGGCA | TGCTGTCAGAGGTCTTGTGCTC |
| hIL-1β | CCACAGACCTTCCAGGAGAATG | CCTTGATGTTATCAGGAGGATTCA |
| hIL-18 | GATAGCCAGCCTAGAGGTATGG | GATAGCCAGCCTAGAGGTATGG |
| hPrdx6 | GCATCCGTTTCCACGACT | TGCACACTGGGGTAAAGTCC |
| hKlf9 | CTGGTTGCTGGGACTGTAGC | GTTTTCCAGCTCCCAAACAG |
| hTxnrd2 | GCACCTTTGACACCGTCCTGTG | CACCAGGATCTTCTGAGTGTCG |
| Hβ-actin | CCAACCGCGAGAAGATGA | CCAGAGGCGTACAGGGATAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chhunchha, B.; Kumar, R.; Kubo, E.; Thakur, P.; Singh, D.P. Prdx6 Regulates Nlrp3 Inflammasome Activation-Driven Inflammatory Response in Lens Epithelial Cells. Int. J. Mol. Sci. 2023, 24, 16276. https://doi.org/10.3390/ijms242216276
Chhunchha B, Kumar R, Kubo E, Thakur P, Singh DP. Prdx6 Regulates Nlrp3 Inflammasome Activation-Driven Inflammatory Response in Lens Epithelial Cells. International Journal of Molecular Sciences. 2023; 24(22):16276. https://doi.org/10.3390/ijms242216276
Chicago/Turabian StyleChhunchha, Bhavana, Rakesh Kumar, Eri Kubo, Priyanka Thakur, and Dhirendra P. Singh. 2023. "Prdx6 Regulates Nlrp3 Inflammasome Activation-Driven Inflammatory Response in Lens Epithelial Cells" International Journal of Molecular Sciences 24, no. 22: 16276. https://doi.org/10.3390/ijms242216276
APA StyleChhunchha, B., Kumar, R., Kubo, E., Thakur, P., & Singh, D. P. (2023). Prdx6 Regulates Nlrp3 Inflammasome Activation-Driven Inflammatory Response in Lens Epithelial Cells. International Journal of Molecular Sciences, 24(22), 16276. https://doi.org/10.3390/ijms242216276