


Endocrinopathies in Hemoglobinopathies: What Is the Role of Iron?

 ,
,  and
and  on behalf of the International Hemoglobinopathy Research Network (INHERENT)
on behalf of the International Hemoglobinopathy Research Network (INHERENT)
Abstract
1. Introduction
2. Methods
3. Impaired Glucose Metabolism and Diabetes Mellitus

{kind=link}
| Endocrine Disorder | Prevalence | References | Risk Factors–Pathogenesis |
|---|---|---|---|
| Diabetes mellitus | 0–35% | [10,11,12,13,14,54] | Older age, higher BMI, splenectomy, iron overload, HCV infection, chronic liver disease, zinc deficiency |
| Hypothyroidism | 6.2–52% | [15,55,56,57,58,59,60,61,62,63] | Older age, splenectomy, chronic anemia, iron overload 1 |
| Hypoparathyroidism | 1–38% | [15,61,64,65,66] | Iron accumulation in parathyroid glands, increased serum ferritin levels, chronic anemia, magnesium deficiency |
| Osteoporosis | 12.5–62% | [67] | Vitamin D deficiency, iron overload, bone marrow hyperplasia, endocrine disorders |
| Hypogonadism | 20–77.5% | [68,69,70,71] | Iron accumulation in the pituitary gland, liver, ovaries, and testes; chronic anemia; HCV infection |
| Growth hormone deficiency | 26.6% | [72] | Increased serum ferritin levels, hepatic iron overload |
| Adrenal insufficiency | 13–46% | [73,74] | Iron overload, chronic anemia |
| Glucose Metabolism Disorder | Criteria for the Diagnosis |
|---|---|
| DM | Fasting serum glucose > 126 mg/dL |
| Serum glucose at 2 h > 200 mg/dL in 2 h OGTT | |
| IGT | Serum glucose at 2 h > 140 mg/dL in 2 h OGTT |
| Endocrine Disorder | Prevalence | References | Risk Factors–Pathogenesis |
|---|---|---|---|
| Diabetes mellitus | 1.46–19% | [36,37,38,39,40,41,42] | Older age, higher BMI, iron overload |
| Hypothyroidism | 0–18.1% | [75,76,77,78,79,80,81] | Hepatic iron overload, thyroid microcirculation dysfunction, iron overload1 1 |
| Secondary hyperparathyroidism | 8.7–71.4% | [82,83,84,85] | Vitamin D deficiency |
| Osteoporosis | 7–40% | [86,87,88,89,90,91] | Calcium and vitamin D deficiency, bone vaso-occlusion-induced ischemia |
| Hypogonadism | 24% 2 | [92] | Vaso-occlusive events in vessels of hypothalamic–pituitary system, iron overload |
| Adrenal insufficiency | 19.4% 3 | [93] | Vaso-occlusive events in vessels of hypothalamic–pituitary system and adrenal glands, iron overload, chronic use of opioid analgesics |
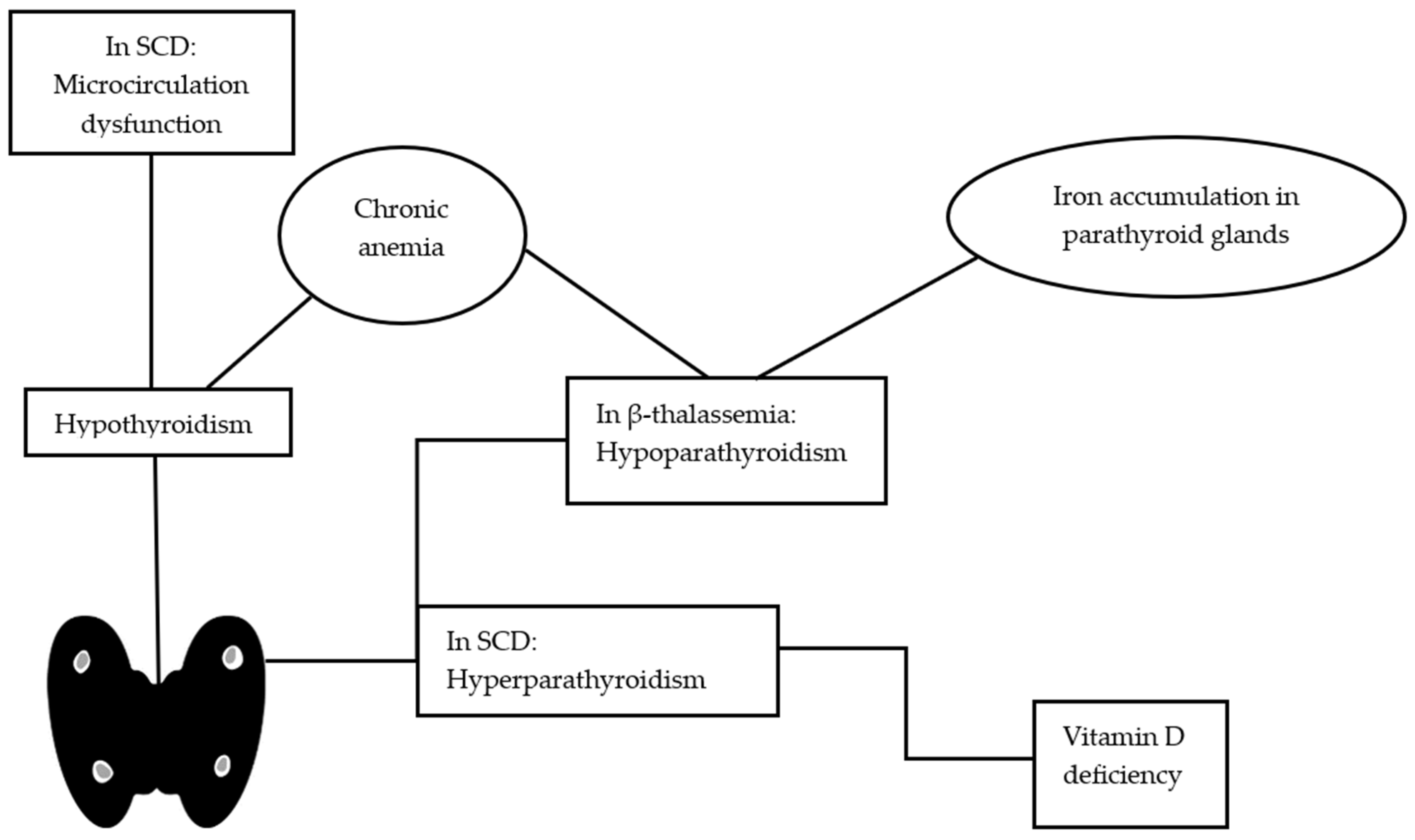
4. Hypothyroidism
5. Parathyroid Dysfunction
6. Bone Mineral Density
7. Gonadal Failure
8. Growth Failure
9. Adrenal Insufficiency
10. Genetic Modifiers of Endocrinopathies in Hemoglobinopathies
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kohne, E. Hemoglobinopathies. Dtsch. Ärzteblatt Int. 2011, 108, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Carsote, M.; Vasiliu, C.; Trandafir, A.I.; Albu, S.E.; Dumitrascu, M.-C.; Popa, A.; Mehedintu, C.; Petca, R.-C.; Petca, A.; Sandru, F. New Entity—Thalassemic Endocrine Disease: Major Beta-Thalassemia and Endocrine Involvement. Diagnostics 2022, 12, 1921. [Google Scholar] [CrossRef] [PubMed]
- Viprakasit, V.; Ekwattanakit, S. Clinical Classification, Screening and Diagnosis for Thalassemia. Hematol. Oncol. Clin. N. Am. 2018, 32, 193–211. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V. The icet-a recommendations for the diagnosis and management of disturbances of glucose homeostasis in thalassemia major patients. Mediterr. J. Hematol. Infect. Dis. 2016, 8, 2016058. [Google Scholar] [CrossRef]
- Bunn, H.F. Pathogenesis and Treatment of Sickle Cell Disease. N. Engl. J. Med. 1997, 337, 762–769. [Google Scholar] [CrossRef]
- Tampaki, A.; Gavriilaki, E.; Varelas, C.; Anagnostopoulos, A.; Vlachaki, E. Complement in Sickle Cell Disease and Targeted Therapy: I Know One Thing, That I Know Nothing. Blood Rev. 2021, 48, 100805. [Google Scholar] [CrossRef]
- Varelas, C.; Tampaki, A.; Sakellari, I.; Anagnostopoulos, A.; Gavriilaki, E.; Vlachaki, E. Complement in Sickle Cell Disease: Are We Ready for Prime Time? J. Blood Med. 2021, 12, 177–187. [Google Scholar] [CrossRef]
- Gavriilaki, E.; Mainou, M.; Christodoulou, I.; Koravou, E.-E.; Paleta, A.; Touloumenidou, T.; Papalexandri, A.; Athanasiadou, A.; Apostolou, C.; Klonizakis, P.; et al. In Vitro Evidence of Complement Activation in Patients with Sickle Cell Disease. Haematologica 2017, 102, e481–e482. [Google Scholar] [CrossRef]
- Smiley, D.; Dagogo-Jack, S.; Umpierrez, G. Therapy Insight: Metabolic and Endocrine Disorders in Sickle Cell Disease. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 102–109. [Google Scholar] [CrossRef]
- Chern, J.P.S.; Lin, K.-H.; Lu, M.-Y.; Lin, D.-T.; Lin, K.-S.; Chen, J.-D.; Fu, C.-C. Abnormal Glucose Tolerance in Transfusion-Dependent β-Thalassemic Patients. Diabetes Care 2001, 24, 850–854. [Google Scholar] [CrossRef]
- Hafez, M.; Youssry, I.; El-Hamed, F.A.; Ibrahim, A. Abnormal Glucose Tolerance in β-Thalassemia: Assessment of Risk Factors. Hemoglobin 2009, 33, 101–108. [Google Scholar] [CrossRef] [PubMed]
- El-Samahy, M.H.; Tantawy, A.A.; Adly, A.A.; Abdelmaksoud, A.A.; Ismail, E.A.; Salah, N.Y. Evaluation of Continuous Glucose Monitoring System for Detection of Alterations in Glucose Homeostasis in Pediatric Patients with β-Thalassemia Major. Pediatr. Diabetes 2019, 20, 65–72. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.; Daar, S.; Tzoulis, P.; Di Maio, S.; Kattamis, C. Glucose homeostasis and αssessment of β-cell function by 3-hour oral glucose tolerance (ogtt) in patients with β-thalassemia major with serum ferritin below 1000 ng/dl: Results from a single icet-a centre. Mediterr. J. Hematol. Infect. Dis. 2023, 15, e2023006. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.T.; Daar, S.; Tzoulis, P.; Di Maio, S.; Kattamis, C. Longitudinal Study of ICET-A on Glucose Tolerance, Insulin Sensitivity and β-Cell Secretion in Eleven β-Thalassemia Major Patients with Mild Iron Overload. Acta Bio-Medica Atenei Parm. 2023, 94, e2023011. [Google Scholar] [CrossRef]
- Multicentre Study on Prevalence of Endocrine Complications in Thalassaemia Major. Italian Working Group on Endocrine Complications in Non-Endocrine Diseases. Clin. Endocrinol. 1995, 42, 581–586. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Abd El-Fatah, A.H.; Abd El-Halim, A.F.; Mohamed, F.F. Serum Ferritin Levels and Other Associated Parameters with Diabetes Mellitus in Adult Patients Suffering from Beta Thalassemia Major. J. Blood Med. 2023, 14, 67–81. [Google Scholar] [CrossRef]
- Meloni, A.; Pistoia, L.; Gamberini, M.; Ricchi, P.; Cecinati, V.; Sorrentino, F.; Cuccia, L.; Allò, M.; Righi, R.; Fina, P.; et al. The Link of Pancreatic Iron with Glucose Metabolism and Cardiac Iron in Thalassemia Intermedia: A Large, Multicenter Observational Study. J. Clin. Med. 2021, 10, 5561. [Google Scholar] [CrossRef]
- Pinto, V.M.; Bacigalupo, L.; Gianesin, B.; Balocco, M.; De Franceschi, L.; Malagò, R.; Wood, J.; Forni, G.L. Lack of Correlation between Heart, Liver and Pancreas MRI-R2*: Results from Long-Term Follow-up in a Cohort of Adult β-Thalassemia Major Patients. Am. J. Hematol. 2018, 93, E79–E82. [Google Scholar] [CrossRef]
- Cario, H.; Holl, R.W.; Debatin, K.-M.; Kohne, E. Insulin Sensitivity and β-Cell Secretion in Thalassaemia Major with Secondary Haemochromatosis: Assessment by Oral Glucose Tolerance Test. Eur. J. Pediatr. 2003, 162, 139–146. [Google Scholar] [CrossRef]
- Chatterjee, R.; Bajoria, R. New Concept in Natural History and Management of Diabetes Mellitus in Thalassemia Major. Hemoglobin 2009, 33, S127–S130. [Google Scholar] [CrossRef]
- Suvarna, J.; Ingle, H.; Deshmukh, C.T. Insulin Resistance and Beta Cell Function in Chronically Transfused Patients of Thalassemia Major. Indian Pediatr. 2006, 43, 393–400. [Google Scholar] [PubMed]
- De Sanctis, V.; Daar, S.; Soliman, A.T.; Tzoulis, P.; Karimi, M.; Di Maio, S.; Kattamis, C. Screening for Glucose Dysregulation in β-Thalassemia Major (β-TM): An Update of Current Evidences and Personal Experience. Acta Bio-Medica Atenei Parm. 2022, 93, e2022158. [Google Scholar] [CrossRef]
- Hashemieh, M.; Radfar, M.; Azarkeivan, A.; Noghabaei, G.; Sheibani, K. T2* Magnetic Resonance Imaging Study of Pancreatic Iron Overload and Its Relation With the Diabetic State in Thalassemic Patients. J. Pediatr. Hematol. Oncol. 2017, 39, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Labropoulou-Karatza, C.; Goritsas, C.; Fragopanagou, H.; Repandi, M.; Matsouka, P.; Alexandrides, T. High Prevalence of Diabetes Mellitus among Adult Beta-Thalassaemic Patients with Chronic Hepatitis C. Eur. J. Gastroenterol. Hepatol. 1999, 11, 1033–1036. [Google Scholar] [CrossRef]
- Farmakis, D.; Porter, J.; Taher, A.; Domenica Cappellini, M.; Angastiniotis, M.; Eleftheriou, A. 2021 Thalassaemia International Federation Guidelines for the Management of Transfusion-Dependent Thalassemia. HemaSphere 2022, 6, e732. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, Z.; Jiang, Z.; Liu, Z.; Hou, L.; Cai, G.; Ou, H.; Huang, S.; Song, Q.; Fang, J.; et al. Indicators of Glucose Dysregulation and the Relationship with Iron Overload in Chinese Children with Beta Thalassemia Major. Pediatr. Diabetes 2022, 23, 562–568. [Google Scholar] [CrossRef]
- Warncke, K.; Konrad, K.; Kohne, E.; Hammer, E.; Ohlenschläger, U.; Herrlinger, S.; Jäger, A.; Holl, R. Diabetes in Patients with SS-Thalassemia or Other Hemoglobinopathies—Analysis from the DPV Database. Klin. Pädiatrie 2016, 228, 307–312. [Google Scholar] [CrossRef]
- Noetzli, L.J.; Mittelman, S.D.; Watanabe, R.M.; Coates, T.D.; Wood, J.C. Pancreatic Iron and Glucose Dysregulation in Thalassemia Major. Am. J. Hematol. 2012, 87, 155–160. [Google Scholar] [CrossRef]
- Ricchi, P.; Meloni, A.; Pistoia, L.; Gamberini, M.R.; Cuccia, L.; Allò, M.; Putti, M.C.; Spasiano, A.; Rosso, R.; Cecinati, V.; et al. Longitudinal Prospective Comparison of Pancreatic Iron by Magnetic Resonance in Thalassemia Patients Transfusion-Dependent since Early Childhood Treated with Combination Deferiprone-Desferrioxamine vs. Deferiprone or Deferasirox Monotherapy. Blood Transfus. Trasfus. Del Sangue 2023. [Google Scholar] [CrossRef]
- Pepe, A.; Pistoia, L.; Gamberini, M.R.; Cuccia, L.; Peluso, A.; Messina, G.; Spasiano, A.; Allò, M.; Bisconte, M.G.; Putti, M.C.; et al. The Close Link of Pancreatic Iron With Glucose Metabolism and With Cardiac Complications in Thalassemia Major: A Large, Multicenter Observational Study. Diabetes Care 2020, 43, 2830–2839. [Google Scholar] [CrossRef]
- Christoforidis, A.; Perifanis, V.; Athanassiou-Metaxa, M. Combined Chelation Therapy Improves Glucose Metabolism in Patients with β-Thalassaemia Major. Br. J. Haematol. 2006, 135, 271–272. [Google Scholar] [CrossRef]
- Farmaki, K.; Angelopoulos, N.; Anagnostopoulos, G.; Gotsis, E.; Rombopoulos, G.; Tolis, G. Effect of Enhanced Iron Chelation Therapy on Glucose Metabolism in Patients with β-Thalassaemia Major. Br. J. Haematol. 2006, 134, 438–444. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.T.; Elsedfy, H.; Pepe, A.; Kattamis, C.; El Kholy, M.; Yassin, M. Diabetes and Glucose Metabolism in Thalassemia Major: An Update. Expert Rev. Hematol. 2016, 9, 401–408. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.; Tzoulis, P.; Daar, S.; Kattamis, A.; Delaporta, P.; Karimi, M.; Yassin, M.A.; Zarei, T.; Saki, F.; et al. The The Use of Oral Glucose-Lowering Agents (GLAs) in β-Thalassemia Patients with Diabetes: Preliminary Data from a Retrospective Study of ICET-A Network. Acta Bio-Medica Atenei Parm. 2022, 93, e2022162. [Google Scholar] [CrossRef]
- Stefánsson, B.V.; Heerspink, H.J.L.; Wheeler, D.C.; Sjöström, C.D.; Greasley, P.J.; Sartipy, P.; Cain, V.; Correa-Rotter, R. Correction of Anemia by Dapagliflozin in Patients with Type 2 Diabetes. J. Diabetes Complicat. 2020, 34, 107729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, W.; Saraf, S.L.; Nouraie, M.; Han, J.; Gowhari, M.; Hassan, J.; Miasnikova, G.; Sergueeva, A.; Nekhai, S.; et al. Genetic Polymorphism of APOB Is Associated with Diabetes Mellitus in Sickle Cell Disease. Hum. Genet. 2015, 134, 895–904. [Google Scholar] [CrossRef][Green Version]
- Suwaidan, A.A.; Al-Qurashi, F.; Whitford, D.L. Does Sickle Cell Disease Protect Against Diabetes Mellitus?: Cross-Sectional Study. Sultan Qaboos Univ. Med. J. 2015, 15, e116–e119. [Google Scholar]
- Prusty, B.; Soren, T.; Choudhury, A.; Biswal, R.; Pradhan, D.; Thatoi, P. Sickle Cell Disease Prevents Diabetes Mellitus Occurrence: A Hospital Based Cross-Sectional Study. J. Fam. Med. Prim. Care 2019, 8, 361. [Google Scholar] [CrossRef]
- Soliman, A.T.; De Sanctis, V.; Yassin, M.; Alshurafa, A.; Ata, F.; Nashwan, A. Blood Transfusion and Iron Overload in Patients with Sickle Cell Disease (SCD): Personal Experience and a Short Update of Diabetes Mellitus Occurrence. Acta Biomed. 2022, 93, e2022291. [Google Scholar] [CrossRef]
- Jang, T.; Mo, G.; Stewart, C.; Khoury, L.; Ferguson, N.; Egini, O.; Muthu, J.; Dutta, D.; Salifu, M.; Lim, S.H. Obesity and Diabetes Mellitus in Patients with Sickle Cell Disease. Ann. Hematol. 2021, 100, 2203–2205. [Google Scholar] [CrossRef]
- Morrison, J.C.; Schneider, J.M.; Kraus, A.P.; Kitabchi, A.E. The Prevalence of Diabetes Mellitus in Sickle Cell Hemoglobinopathies*. J. Clin. Endocrinol. Metab. 1979, 48, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Han, J.; Nutescu, E.A.; Galanter, W.L.; Walton, S.M.; Gordeuk, V.R.; Saraf, S.L.; Calip, G.S. Similar Burden of Type 2 Diabetes among Adult Patients with Sickle Cell Disease Relative to African Americans in the U.S. Population: A Six-year Population-based Cohort Analysis. Br. J. Haematol. 2019, 185, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Fung, E.B.; Harmatz, P.R.; Lee, P.D.K.; Milet, M.; Bellevue, R.; Jeng, M.R.; Kalinyak, K.A.; Hudes, M.; Bhatia, S.; Vichinsky, E.P. Increased Prevalence of Iron-Overload Associated Endocrinopathy in Thalassaemia versus Sickle-Cell Disease. Br. J. Haematol. 2006, 135, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Lanzkron, S.; Carroll, C.P.; Haywood, C. Mortality Rates and Age at Death from Sickle Cell Disease: U.S., 1979–2005. Public Health Rep. 2013, 128, 110–116. [Google Scholar] [CrossRef]
- Skinner, S.C.; Diaw, M.; Pialoux, V.; Mbaye, M.N.; Mury, P.; Lopez, P.; Bousquet, D.; Gueye, F.; Diedhiou, D.; Joly, P.; et al. Increased Prevalence of Type 2 Diabetes–Related Complications in Combined Type 2 Diabetes and Sickle Cell Trait. Diabetes Care 2018, 41, 2595–2602. [Google Scholar] [CrossRef]
- Ajayi, A.A.; Kolawole, B.A. Sickle Cell Trait and Gender Influence Type 2 Diabetic Complications in African Patients. Eur. J. Intern. Med. 2004, 15, 312–315. [Google Scholar] [CrossRef]
- Yavropoulou, M.P.; Pikilidou, M.; Pantelidou, D.; Tsalikakis, D.G.; Mousiolis, A.; Chalkia, P.; Yovos, J.G.; Zebekakis, P. Insulin Secretion and Resistance in Normoglycemic Patients with Sickle Cell Disease. Hemoglobin 2017, 41, 6–11. [Google Scholar] [CrossRef]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes. Diabetes Care 2015, 38, S8–S16. [Google Scholar] [CrossRef]
- McLean, A.; Wright, F.; DeJong, N.; Skinner, S.; Loughlin, C.E.; Levenson, A.; Carden, M.A. Hemoglobin A 1c and Fructosamine Correlate in a Patient with Sickle Cell Disease and Diabetes on Chronic Transfusion Therapy. Pediatr. Blood Cancer 2020, 67, e28499. [Google Scholar] [CrossRef]
- Yahaya, I.A.; Isah, H.S.; Anaja, P.O. Serum Fructosamine in the Assessment of Glycaemic Status in Patients with Sickle Cell Anaemia. Niger. Postgrad. Med. J. 2006, 13, 95–98. [Google Scholar] [CrossRef]
- Kosecki, S.M.; Rodgers, P.T.; Adams, M.B. Glycemic Monitoring in Diabetics with Sickle Cell Plus β-Thalassemia Hemoglobinopathy. Ann. Pharmacother. 2005, 39, 1557–1560. [Google Scholar] [CrossRef] [PubMed]
- Badawy, S.M.; Payne, A.B. Association between Clinical Outcomes and Metformin Use in Adults with Sickle Cell Disease and Diabetes Mellitus. Blood Adv. 2019, 3, 3297–3306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Paikari, A.; Sumazin, P.; Ginter Summarell, C.C.; Crosby, J.R.; Boerwinkle, E.; Weiss, M.J.; Sheehan, V.A. Metformin Induces FOXO3-Dependent Fetal Hemoglobin Production in Human Primary Erythroid Cells. Blood 2018, 132, 321–333. [Google Scholar] [CrossRef]
- Maertens, J.A.; Raad, I.I.; Marr, K.A.; Patterson, T.F.; Kontoyiannis, D.P.; Cornely, O.A.; Bow, E.J.; Rahav, G.; Neofytos, D.; Aoun, M.; et al. Isavuconazole versus Voriconazole for Primary Treatment of Invasive Mould Disease Caused by Aspergillus and Other Filamentous Fungi (SECURE): A Phase 3, Randomised-Controlled, Non-Inferiority Trial. Lancet 2016, 387, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Gathwala, G.; Das, K.; Agrawal, N. Thyroid Hormone Profile in Beta-Thalassemia Major Children. Bangladesh Med. Res. Counc. Bull. 2009, 35, 71–72. [Google Scholar] [CrossRef][Green Version]
- Abdel-Razek, A.-R.A.; Abdel-Salam, A.; El-Sonbaty, M.M.; Youness, E.R. Study of Thyroid Function in Egyptian Children with β-Thalassemia Major and β-Thalassemia Intermedia. J. Egypt. Public Health Assoc. 2013, 88, 148–152. [Google Scholar] [CrossRef]
- Haghpanah, S.; Jelodari, S.; Karamifar, H.; Saki, F.; Rahimi, R.; De Sanctis, V.; Dehbozorgian, J.; Karimi, M. The Frequency of Hypothyroidism and Its Relationship with HCV Positivity in Patients with Thalassemia Major in Southern Iran. Acta Bio-Medica Atenei Parm. 2018, 89, 55–60. [Google Scholar] [CrossRef]
- Magro, S.; Puzzonia, P.; Consarino, C.; Galati, M.C.; Morgione, S.; Porcelli, D.; Grimaldi, S.; Tancrè, D.; Arcuri, V.; De Santis, V.; et al. Hypothyroidism in Patients with Thalassemia Syndromes. Acta Haematol. 1990, 84, 72–76. [Google Scholar] [CrossRef]
- Zervas, A.; Katopodi, A.; Protonotariou, A.; Livadas, S.; Karagiorga, M.; Politis, C.; Tolis, G. Assessment of Thyroid Function in Two Hundred Patients with β -Thalassemia Major. Thyroid 2002, 12, 151–154. [Google Scholar] [CrossRef]
- Landau, H.; Matoth, I.; Landau-Cordova, Z.; Goldfarbs, A.; Rachmilewitz, E.A.; Glaser, B. Cross-Sectional and Longitudinal Study of the Pituitary-Thyroid Axis in Patients with Thalassaemia Major. Clin. Endocrinol. 1993, 38, 55–61. [Google Scholar] [CrossRef]
- Bazi, A.; Harati, H.; Khosravi-Bonjar, A.; Rakhshani, E.; Delaramnasab, M. Hypothyroidism and Hypoparathyroidism in Thalassemia Major Patients: A Study in Sistan and Baluchestan Province, Iran. Int. J. Endocrinol. Metab. 2018, 16, e13228. [Google Scholar] [CrossRef]
- Filosa, A.; Di Maio, S.; Aloj, G.; Acampora, C. Longitudinal Study on Thyroid Function in Patients with Thalassemia Major. J. Pediatr. Endocrinol. Metab. 2006, 19, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Baghersalimi, A.; Rad, A.H.; Koohmanaee, S.; Darbandi, B.; Mirzaee, M.M.; Aminzadeh, V.; Medghalchi, A.; Dalili, S. The Cutoff of Ferritin for Evaluation of Hypothyroidism in Patients With Thalassemia. J. Pediatr. Hematol. Oncol. 2019, 41, 515–518. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.T.; Canatan, D.; Elsedfy, H.; Karimi, M.; Daar, S.; Rimawi, H.; Christou, S.; Skordis, N.; Tzoulis, P.; et al. An ICET- A Survey on Hypoparathyroidism in Patients with Thalassaemia Major and Intermedia: A Preliminary Report. Acta Bio-Medica Atenei Parm. 2018, 88, 435–444. [Google Scholar] [CrossRef]
- Angelopoulos, N.G.; Goula, A.; Rombopoulos, G.; Kaltzidou, V.; Katounda, E.; Kaltsas, D.; Tolis, G. Hypoparathyroidism in Transfusion-Dependent Patients with β-Thalassemia. J. Bone Miner. Metab. 2006, 24, 138–145. [Google Scholar] [CrossRef]
- Tangngam, H.; Mahachoklertwattana, P.; Poomthavorn, P.; Chuansumrit, A.; Sirachainan, N.; Chailurkit, L.; Khlairit, P. Under-Recognized Hypoparathyroidism in Thalassemia. J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 324. [Google Scholar] [CrossRef]
- Gaudio, A.; Morabito, N.; Catalano, A.; Rapisarda, R.; Xourafa, A.; Lasco, A. Pathogenesis of Thalassemia Major-Associated Osteoporosis: A Review with Insights from Clinical Experience. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 110–117. [Google Scholar] [CrossRef]
- Bozdağ, M.; Bayraktaroğlu, S.; Aydınok, Y.; Çallı, M.C. MRI Assessment of Pituitary Iron Accumulation by Using Pituitary-R2 in β-Thalassemia Patients. Acta Radiol. 2018, 59, 732–739. [Google Scholar] [CrossRef]
- Albu, A.; Barbu, C.G.; Antonie, L.; Vladareanu, F.; Fica, S. Risk Factors Associated with Hypogonadism in β–Thalassemia Major Patients: Predictors for a Frequent Complication of a Rare Disease. Postgrad. Med. 2014, 126, 121–127. [Google Scholar] [CrossRef]
- Abdelrazik, N.; Ghanem, H. Failure of Puberty in Egyptian Beta Thalassemic Patients: Experience in North East Region—Dakahlia Province. Hematology 2007, 12, 449–456. [Google Scholar] [CrossRef]
- Al-Rimawi, H.S.; Jallad, M.F.; Amarin, Z.O.; Obeidat, B.R. Hypothalamic-Pituitary-Gonadal Function in Adolescent Females with Beta-Thalassemia Major. Int. J. Gynecol. Obstet. 2005, 90, 44–47. [Google Scholar] [CrossRef]
- Arab-Zozani, M.; Kheyrandish, S.; Rastgar, A.; Miri-Moghadam, E. A Systematic Review and Meta-Analysis of Stature Growth Complications in β-Thalassemia Major Patients. Ann. Glob. Health 2021, 87, 48. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.E.; Mittelman, S.D.; Coates, T.D.; Geffner, M.E.; Wood, J.C. A Significant Proportion of Thalassemia Major Patients Have Adrenal Insufficiency Detectable on Provocative Testing. J. Pediatr. Hematol. Oncol. 2015, 37, 54–59. [Google Scholar] [CrossRef]
- Poomthavorn, P.; Isaradisaikul, B.; Chuansumrit, A.; Khlairit, P.; Sriphrapradang, A.; Mahachoklertwattana, P. High Prevalence of “Biochemical” Adrenal Insufficiency in Thalassemics: Is It a Matter of Different Testings or Decreased Cortisol Binding Globulin? J. Clin. Endocrinol. Metab. 2010, 95, 4609–4615. [Google Scholar] [CrossRef] [PubMed]
- ElAlfy, M.S.; El-Sherif, N.H.; Sakr, H.M.; El Ashkar, M.N.M. Thyroid Hemodynamic Alterations in Egyptian Patients with Sickle Cell Disease: Relation to Disease Severity, Total Body Iron and Thyroid Function. Expert Rev. Hematol. 2019, 12, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Karazincir, S.; Balci, A.; Yonden, Z.; Gali, E.; Daplan, T.; Beyoglu, Y.; Kaya, H.; Egilmez, E. Thyroid Doppler Indices in Patients with Sickle Cell Disease. Clin. Imaging 2013, 37, 852–855. [Google Scholar] [CrossRef]
- Garadah, T.; Jaradat, A.; Al Alalawi, M.; Hassan, A.B. Hormonal and Echocardiographic Abnormalities in Adult Patients with Sickle-Cell Anemia in Bahrain. J. Blood Med. 2016, 7, 283–289. [Google Scholar] [CrossRef]
- Evliyaoǧlu, N.; Kilinç, Y.; Sargin, Ö. Thyroid Functions in Mild and Severe Forms of Sickle Cell Anemia. Pediatr. Int. 1996, 38, 460–463. [Google Scholar] [CrossRef]
- Kaudha, G.; Piloya, T.; Musiime, V.; Kuteesa, M.G.; Namugerwa, S.; Owomugisha, G.; Wachepa, S.A.; Lubwama, S.K.; Kiguli, S.; Tumwine, J.K. Prevalence and Factors Associated with Hypothyroidism in Children with Sickle Cell Anemia Aged 6 Months—17 Years Attending the Sickle Cell Clinic, Mulago Hospital, Uganda; a Cross-Sectional Study. BMC Endocr. Disord. 2023, 23, 60. [Google Scholar] [CrossRef]
- Özen, S.; Ünal, S.; Erçetin, N.; Taşdelen, B. Frequency and Risk Factors of Endocrine Complications in Turkish Children and Adolescents with Sickle Cell Anemia. Turk. J. Hematol. 2013, 30, 25–31. [Google Scholar] [CrossRef]
- Mandese, V.; Bigi, E.; Bruzzi, P.; Palazzi, G.; Predieri, B.; Lucaccioni, L.; Cellini, M.; Iughetti, L. Endocrine and Metabolic Complications in Children and Adolescents with Sickle Cell Disease: An Italian Cohort Study. BMC Pediatr. 2019, 19, 56. [Google Scholar] [CrossRef] [PubMed]
- Arlet, J.-B.; Courbebaisse, M.; Chatellier, G.; Eladari, D.; Souberbielle, J.-C.; Friedlander, G.; de Montalembert, M.; Prié, D.; Pouchot, J.; Ribeil, J.-A. Relationship between Vitamin D Deficiency and Bone Fragility in Sickle Cell Disease: A Cohort Study of 56 Adults. Bone 2013, 52, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Chapelon, E.; Garabedian, M.; Brousse, V.; Souberbielle, J.C.; Bresson, J.L.; de Montalembert, M. Osteopenia and Vitamin D Deficiency in Children with Sickle Cell Disease. Eur. J. Haematol. 2009, 83, 572–578. [Google Scholar] [CrossRef]
- Garrido, C.; Cela, E.; Beléndez, C.; Mata, C.; Huerta, J. Status of Vitamin D in Children with Sickle Cell Disease Living in Madrid, Spain. Eur. J. Pediatr. 2012, 171, 1793–1798. [Google Scholar] [CrossRef]
- Batte, A.; Kasirye, P.; Baluku, R.; Kiguli, S.; Kalyesubula, R.; John, C.C.; Schwaderer, A.L.; Imel, E.A.; Conroy, A.L. Mineral Bone Disorders and Kidney Disease in Hospitalized Children with Sickle Cell Anemia. Front. Pediatr. 2022, 10, 1078853. [Google Scholar] [CrossRef]
- Eskiocak, Ö.; Yılmaz, M.Ö.; İlhan, G. Metabolic Bone Diseases in Sickle Cell Anemia Patients and Evaluation of Associated Factors. Am. J. Med. Sci. 2022, 363, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Fung, E.B.; Kawchak, D.A.; Zemel, B.S.; Rovner, A.J.; Ohene-Frempong, K.; Stallings, V.A. Markers of Bone Turnover Are Associated with Growth and Development in Young Subjects with Sickle Cell Anemia. Pediatr. Blood Cancer 2008, 50, 620–623. [Google Scholar] [CrossRef]
- Adams-Graves, P.; Daniels, A.B.; Womack, C.R.; Freire, A.X. Bone Mineral Density Patterns in Vitamin D Deficient African American Men With Sickle Cell Disease. Am. J. Med. Sci. 2014, 347, 262–266. [Google Scholar] [CrossRef]
- Garadah, T.S.; Hassan, A.B.; Jaradat, A.A.; Diab, D.E.; Kalafalla, H.O.; Kalifa, A.K.; Sequeira, R.P.; Alawadi, A.H.A. Predictors of Abnormal Bone Mass Density in Adult Patients with Homozygous Sickle-Cell Disease. Clin. Med. Insights. Endocrinol. Diabetes 2015, 8, 35–40. [Google Scholar] [CrossRef]
- Miller, R.G.; Segal, J.B.; Ashar, B.H.; Leung, S.; Ahmed, S.; Siddique, S.; Rice, T.; Lanzkron, S. High Prevalence and Correlates of Low Bone Mineral Density in Young Adults with Sickle Cell Disease. Am. J. Hematol. 2006, 81, 236–241. [Google Scholar] [CrossRef]
- De Franceschi, L.; Gabbiani, D.; Giusti, A.; Forni, G.; Stefanoni, F.; Pinto, V.M.; Sartori, G.; Balocco, M.; Dal Zotto, C.; Valenti, M.T.; et al. Development of Algorithm for Clinical Management of Sickle Cell Bone Disease: Evidence for a Role of Vertebral Fractures in Patient Follow-Up. J. Clin. Med. 2020, 9, 1601. [Google Scholar] [CrossRef] [PubMed]
- Taddesse, A.; Woldie, I.L.; Khana, P.; Swerdlow, P.S.; Chu, J.-W.; Abrams, J.; Abou-Samra, A.-B. Hypogonadism in Patients with Sickle Cell Disease: Central or Peripheral? Acta Haematol. 2012, 128, 65–68. [Google Scholar] [CrossRef]
- Makino, J.; Ndzengue, A.; Adekolujo, S.; Tipu, A.; Dogar, U.; Mezher, H.; Sivasambu, B.; Trauber, D.; Guillaume, E.; Jaffe, E.; et al. High Prevalence of Adrenal Insufficiency in Patients with Sickle Cell Disease: Results from a Community Hospital in the U.S. Exp. Clin. Endocrinol. Diabetes 2013, 121, 32–36. [Google Scholar] [CrossRef][Green Version]
- Taher, A.T.; Musallam, K.M.; Karimi, M.; El-Beshlawy, A.; Belhoul, K.; Daar, S.; Saned, M.-S.; El-Chafic, A.-H.; Fasulo, M.R.; Cappellini, M.D. Overview on Practices in Thalassemia Intermedia Management Aiming for Lowering Complication Rates across a Region of Endemicity: The OPTIMAL CARE Study. Blood 2010, 115, 1886–1892. [Google Scholar] [CrossRef] [PubMed]
- Hussein, S.Z. Evaluation of Thyroid Hormones and Ferritin Level in Patients with β- Thalassemia. Med. Pharm. Rep. 2022, 95, 152. [Google Scholar] [CrossRef]
- Sabato, A.R.; De Sanctis, V.; Atti, G.; Capra, L.; Bagni, B.; Vullo, C. Primary Hypothyroidism and the Low T3 Syndrome in Thalassaemia Major. Arch. Dis. Child. 1983, 58, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Chirico, V.; Antonio, L.; Vincenzo, S.; Luca, N.; Valeria, F.; Basilia, P.; Luciana, R.; Carmelo, S.; Teresa, A. Thyroid Dysfunction in Thalassaemic Patients: Ferritin as a Prognostic Marker and Combined Iron Chelators as an Ideal Therapy. Eur. J. Endocrinol. 2013, 169, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, D.; Tandon, N. Overt and Subclinical Hypothyroidism. Drugs 2012, 72, 17–33. [Google Scholar] [CrossRef]
- Phillips, G.; Becker, B.; Keller, V.A.; Hartman, J. Hypothyroidism in Adults with Sickle Cell Anemia. Am. J. Med. 1992, 92, 567–570. [Google Scholar] [CrossRef]
- El-Hazmi, M.A.F.; Bahakim, H.M.; Al-Fawaz, I. Endocrine Functions in Sickle Cell Anaemia Patients. J. Trop. Pediatr. 1992, 38, 307–313. [Google Scholar] [CrossRef]
- Lukanmbi, F.A.; Adeyokunnu, A.A.; Osifo, B.O.; Bolodeoku, J.O.; Dada, O.A. Endocrine Function and Haemoglobinopathies: Biochemical Assessment of Thyroid Function in Children with Sickle-Cell Disease. Afr. J. Med. Med. Sci. 1986, 15, 25–28. [Google Scholar] [PubMed]
- Hagag, A.A.; El-Asy, H.M.; Badraia, I.M.; Hablas, N.M.; El-Latif, A.E.A. Thyroid Function in Egyptian Children with Sickle Cell Anemia in Correlation with Iron Load. Endocr. Metab. Immune Disord.-Drug Targets 2019, 19, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Parshad, O.; Stevens, M.C.G.; Hudson, C.; Rosenthal, J.; Melville, G.N.; Dunn, D.T.; Serjeant, G.R. Abnormal Thyroid Hormone and Thyrotropin Levels in Homozygous Sickle Cell Disease. Clin. Lab. Haematol. 1989, 11, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Yassin, M.A.; Soliman, A.T.; De Sanctis, V.; Abdula, M.A.J.; Riaz, L.M.; Ghori, F.F.; Yousaf, A.; Nashwan, A.J.; Abusamaan, S.; Moustafa, A.; et al. Statural Growth and Prevalence of Endocrinopathies in Relation to Liver Iron Content (LIC) in Adult Patients with Beta Thalassemia Major (BTM) and Sickle Cell Disease (SCD). Acta Bio-Medica Atenei Parm. 2018, 89, 33–40. [Google Scholar] [CrossRef]
- Saki, F.; Salehifar, A.; Kassaee, S.R.; Omrani, G.R. Association of Vitamin D and FGF23 with Serum Ferritin in Hypoparathyroid Thalassemia: A Case Control Study. BMC Nephrol. 2020, 21, 482. [Google Scholar] [CrossRef]
- Zafeiriou, D.I.; Athanasiou, M.; Katzos, G.; Economou, M.; Kontopoulos, E. Hypoparathyroidism and Intracranial Calcifications in β-Thalassemia Major. J. Pediatr. 2001, 138, 411. [Google Scholar] [CrossRef]
- Karimi, M.; Rasekhi, A.R.; Rasekh, M.; Nabavizadeh, S.A.; Assadsangabi, R.; Amirhakimi, G.H. Hypoparathyroidism and Intracerebral Calcification in Patients with Beta-Thalassemia Major. Eur. J. Radiol. 2009, 70, 481–484. [Google Scholar] [CrossRef]
- Mohammed, S.; Addae, S.; Suleiman, S.; Adzaku, F.; Annobil, S.; Kaddoumi, O.; Richards, J. Serum Calcium, Parathyroid Hormone, and Vitamin D Status in Children and Young Adults with Sickle Cell Disease. Ann. Clin. Biochem. 1993, 30 Pt 1, 45–51. [Google Scholar] [CrossRef]
- Nolan, V.G.; Nottage, K.A.; Cole, E.W.; Hankins, J.S.; Gurney, J.G. Prevalence of Vitamin D Deficiency in Sickle Cell Disease: A Systematic Review. PLoS ONE 2015, 10, e0119908. [Google Scholar] [CrossRef]
- Denoix, E.; Bomahou, C.; Clavier, L.; Ribeil, J.-A.; Lionnet, F.; Bartolucci, P.; Courbebaisse, M.; Pouchot, J.; Arlet, J.-B. Primary Hyperparathyroidism in Sickle Cell Disease: An Unknown Complication of the Disease in Adulthood. J. Clin. Med. 2020, 9, 308. [Google Scholar] [CrossRef]
- Caocci, G.; Orofino, M.G.; Vacca, A.; Piroddi, A.; Piras, E.; Addari, M.C.; Caria, R.; Pilia, M.P.; Origa, R.; Moi, P.; et al. Long-Term Survival of Beta Thalassemia Major Patients Treated with Hematopoietic Stem Cell Transplantation Compared with Survival with Conventional Treatment. Am. J. Hematol. 2017, 92, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Elshal, M.F.; Bernawi, A.E.; Al-Ghamdy, M.A.; Jalal, J.A. The Association of Bone Mineral Density and Parathyroid Hormone with Serum Magnesium in Adult Patients with Sickle-Cell Anaemia. Arch. Med. Sci. AMS 2012, 8, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Balducci, R.; Toscano, V.; Finocchi, G.; Municchi, G.; Mangiantini, A.; Boscherini, B. Effect of HCG or HCG + Treatments in Young Thalassemic Patients with Hypogonadotropic Hypogonadism. J. Endocrinol. Investig. 1990, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.T.; Yassin, M.A.; Di Maio, S.; Daar, S.; Elsedfy, H.; Soliman, N.; Kattamis, C. Hypogonadism in Male Thalassemia Major Patients: Pathophysiology, Diagnosis and Treatment. Acta Bio-Medica Atenei Parm. 2018, 89, 6–15. [Google Scholar] [CrossRef]
- Grundy, R.G.; Woods, K.A.; Savage, M.O.; Evans, J.P. Relationship of Endocrinopathy to Iron Chelation Status in Young Patients with Thalassaemia Major. Arch. Dis. Child. 1994, 71, 128–132. [Google Scholar] [CrossRef]
- Singh, P.; Samaddar, S.; Parakh, N.; Chandra, J.; Seth, A. Pubertal Development and Its Determinants in Adolescents With Transfusion-Dependent Thalassemia. Indian Pediatr. 2021, 58, 635–638. [Google Scholar] [CrossRef]
- Shahid, Z.; Hassan, S.; Ghazanfar, S.; Kaneez, M.; Khan, M.S.; Tariq, H.T.; Jawad, A.; Shuaib, A.; Bhatti, A.A.; Razzaq, M.T. Investigating the Role of Ferritin in Determining Sexual Underdevelopment in Beta-Thalassemia Major Patients: A Cross-Sectional Analysis From Pakistan. Cureus 2021, 13, e15572. [Google Scholar] [CrossRef]
- De Sanctis, V.; Soliman, A.T.; Daar, S.; Di Maio, S.; Yassin, M.A.; Canatan, D.; Vives Corrons, J.-L.; Elsedfy, H.; Kattamis, A.; Kattamis, C. The Experience of a Tertiary Unit on the Clinical Phenotype and Management of Hypogonadism in Female Adolescents and Young Adults with Transfusion Dependent Thalassemia. Acta Bio-Medica Atenei Parm. 2019, 90, 158–167. [Google Scholar] [CrossRef]
- Bronspiegel-Weintrob, N.; Olivieri, N.F.; Tyler, B.; Andrews, D.F.; Freedman, M.H.; Holland, F.J. Effect of Age at the Start of Iron Chelation Therapy on Gonadal Function in β-Thalassemia Major. N. Engl. J. Med. 1990, 323, 713–719. [Google Scholar] [CrossRef]
- Argyropoulou, M.; Kiortsis, D.; Metafratzi, Z.; Bitsis, S.; Tsatoulis, A.; Efremidis, S. Pituitary Gland Height Evaluated by MR in Patients with β-Thalassemia Major: A Marker of Pituitary Gland Function. Neuroradiology 2001, 43, 1056–1058. [Google Scholar] [CrossRef]
- De Sanctis, V.; Soliman, A.T.; Elsedfy, H.; Di Maio, S.; Canatan, D.; Soliman, N.; Karimi, M.; Kattamis, C. Gonadal Dysfunction in Adult Male Patients with Thalassemia Major: An Update for Clinicians Caring for Thalassemia. Expert Rev. Hematol. 2017, 10, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Vullo, C.; Katz, M.; Wonke, B.; Tanas, R.; Bagni, B. Gonadal Function in Patients with Beta Thalassaemia Major. J. Clin. Pathol. 1988, 41, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.A.; Prasad, A.S.; Ortega, J.; Congco, E.; Oberleas, D. Gonadal Function Abnormalities in Sickle Cell Anemia. Studies in Adult Male Patients. Ann. Intern. Med. 1976, 85, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Osegbe, D.N.; Akinyanju, O.O. Testicular Dysfunction in Men with Sickle Cell Disease. Postgrad. Med. J. 1987, 63, 95–98. [Google Scholar] [CrossRef]
- Dada, O.A.; Nduka, E.U. Endocrine Function and Haemoglobinopathies: Relation between the Sickle Cell Gene and Circulating Plasma Levels of Testosterone, Luteinising Hormone (LH) and Follicle Stimulating Hormone (FSH) in Adult Males. Clin. Chim. Acta 1980, 105, 269–273. [Google Scholar] [CrossRef]
- Parshad, O.; Stevens, M.C.; Preece, M.A.; Thomas, P.W.; Serjeant, G.R. The Mechanism of Low Testosterone Levels in Homozygous Sickle-Cell Disease. W. Indian Med. J. 1994, 43, 12–14. [Google Scholar]
- Singhal, A.; Gabay, L.; Serjeant, G.R. Testosterone Deficiency and Extreme Retardation of Puberty in Homozygous Sickle-Cell Disease. W. Indian Med. J. 1995, 44, 20–23. [Google Scholar]
- Ribeiro, A.P.M.R.; Silva, C.S.; Zambrano, J.C.C.; de Oliveira Freitas Miranda, J.; Molina, C.A.F.; Gomes, C.M.; de Miranda, E.P.; de Bessa, J. Compensated Hypogonadism in Men with Sickle Cell Disease. Clin. Endocrinol. 2021, 94, 968–972. [Google Scholar] [CrossRef]
- Martins, P.R.J.; De Vito, F.B.; Resende, G.A.D.; Kerbauy, J.; de Araújo Pereira, G.; Moraes-Souza, H.; Figueiredo, M.S.; Verreschi, I.T. Male Sickle Cell Patients, Compensated Transpubertal Hypogonadism and Normal Final Growth. Clin. Endocrinol. 2019, 91, 676–682. [Google Scholar] [CrossRef]
- Musicki, B.; Burnett, A.L. Testosterone Deficiency in Sickle Cell Disease: Recognition and Remediation. Front. Endocrinol. 2022, 13, 892184. [Google Scholar] [CrossRef]
- Claudino, M.A.; Fertrin, K.Y. Sickling Cells, Cyclic Nucleotides, and Protein Kinases: The Pathophysiology of Urogenital Disorders in Sickle Cell Anemia. Anemia 2012, 2012, 723520. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Fogarty, J.; Whitney, K.D.; Stone, P. Repeated Testicular Infarction in a Patient with Sickle Cell Disease: A Possible Mechanism for Testicular Failure. Urology 2003, 62, 551. [Google Scholar] [CrossRef] [PubMed]
- Holmes, N.M.; Kane, C.J. Testicular Infarction Associated with Sickle Cell Disease. J. Urol. 1998, 160, 130. [Google Scholar] [CrossRef] [PubMed]
- Brachet, C.; Heinrichs, C.; Tenoutasse, S.; Devalck, C.; Azzi, N.; Ferster, A. Children With Sickle Cell Disease: Growth and Gonadal Function After Hematopoietic Stem Cell Transplantation. J. Pediatr. Hematol. Oncol. 2007, 29, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Smith-Whitley, K. Reproductive Issues in Sickle Cell Disease. Blood 2014, 124, 3538–3543. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.F.; Reid, M.; Madden, W.; Burnett, A.L. Testosterone Replacement Therapy Does Not Promote Priapism in Hypogonadal Men with Sickle Cell Disease: 12-Month Safety Report. Andrology 2013, 1, 576–582. [Google Scholar] [CrossRef]
- Landefeld, C.S.; Schambelan, M.; Kaplan, S.L.; Embury, S.H. Clomiphene-Responsive Hypogonadism in Sickle Cell Anemia. Ann. Intern. Med. 1983, 99, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Osegbe, D.N.; Akinyanju, O.; Amaku, E.O. Fertility in Males with Sickle Cell Disease. Lancet 1981, 2, 275–276. [Google Scholar] [CrossRef]
- Hagag, A.A.; El-Farargy, M.S.; El-Enein, A.M.A. Study of Adrenal Functions Using ACTH Stimulation Test in Egyptian Children with Sickle Cell Anemia: Correlation with Iron Overload. Int. J. Hematol.-Oncol. Stem Cell Res. 2015, 9, 60–66. [Google Scholar]
- Soliman, A.T.; ElZalabany, M.; Amer, M.; Ansari, B.M. Growth and Pubertal Development in Transfusion-Dependent Children and Adolescents with Thalassaemia Major and Sickle Cell Disease: A Comparative Study. J. Trop. Pediatr. 1999, 45, 23–30. [Google Scholar] [CrossRef][Green Version]
- Barden, E.M.; Kawchak, D.A.; Ohene-Frempong, K.; Stallings, V.A.; Zemel, B.S. Body Composition in Children with Sickle Cell Disease. Am. J. Clin. Nutr. 2002, 76, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Platt, O.S.; Rosenstock, W.; Espeland, M.A. Influence of Sickle Hemoglobinopathies on Growth and Development. N. Engl. J. Med. 1984, 311, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, M.; Akohoue, S.A.; Shankar, S.M.; Fleming, I.; Qi An, A.; Yu, C.; Acra, S.; Buchowski, M.S. Growth Patterns in Children with Sickle Cell Anemia during Puberty. Pediatr. Blood Cancer 2009, 53, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Al-Saqladi, A.-W.M.; Cipolotti, R.; Fijnvandraat, K.; Brabin, B.J. Growth and Nutritional Status of Children with Homozygous Sickle Cell Disease. Ann. Trop. Paediatr. 2008, 28, 165–189. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.T.; El Banna, N.; AlSalmi, I.; De Silva, V.; Craig, A.; Asfour, M. Growth Hormone Secretion and Circulating Insulin-like Growth Factor-I (IGF-I) and IGF Binding Protein-3 Concentrations in Children with Sickle Cell Disease. Metab. Clin. Exp. 1997, 46, 1241–1245. [Google Scholar] [CrossRef]
- Collett-Solberg, P.F.; Fleenor, D.; Schultz, W.H.; Ware, R.E. Short Stature in Children with Sickle Cell Anemia Correlates with Alterations in the IGF-I Axis. J. Pediatr. Endocrinol. Metab. JPEM 2007, 20, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.T.; Alaaraj, N.; Yassin, M. The Effects of Treatment with Blood Transfusion, Iron Chelation and Hydroxyurea on Puberty, Growth and Spermatogenesis in Sickle Cell Disease (SCD): A Short Update. Acta Bio-Medica Atenei Parm. 2021, 92, e2021386. [Google Scholar] [CrossRef]
- Osifo, B.O.A.; Lukanmbi, F.A.; Adekile, A. Plasma Cortisol in Sickle Cell Disease. Acta Haematol. 1988, 79, 44–45. [Google Scholar] [CrossRef]
- Sobngwi, E.; Mbango, N.D.; Balti, E.V.; Ngo Sack, F.; Moor, V.A.; Mbanya, J.-C. Relative Adrenal Insufficiency in Adults with Sickle Cell Disease. Pan Afr. Med. J. 2018, 29, 30. [Google Scholar] [CrossRef]
- Rosenbloom, B.E.; Odell, W.D.; Tanaka, K.R. Pituitary-Adrenal Axis Function in Sickle Cell Anemia and Its Relationship to Leukocyte Alkaline Phosphatase. Am. J. Hematol. 1980, 9, 373–379. [Google Scholar] [CrossRef]
- Saad, S.T.; Saad, M.J. Normal Cortisol Secretion in Sickle Cell Anemia. Trop. Geogr. Med. 1992, 44, 86–88. [Google Scholar] [PubMed]
- Hollister, B.M.; Zilbermint, M.; Minniti, C.P.; Buscetta, A.J.; Abdallah, K.E.; You, S.; Soldin, S.J.; Meyer, J.S.; Stratakis, C.A.; Bonham, V.L. Lower Hair Cortisol among Patients with Sickle Cell Disease May Indicate Decreased Adrenal Reserves. Am. J. Blood Res. 2021, 11, 140–148. [Google Scholar] [PubMed]
- Olivieri, N.F. Progression of Iron Overload in Sickle Cell Disease. Semin. Hematol. 2001, 38, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Kimmelsteil, P. Vascular Occlusion and Ischemic Infarction in Sickle Cell Disease. Am. J. Med. Sci. 1948, 216, 11–19. [Google Scholar] [CrossRef]
- Athanasou, N.A.; Hatton, C.; McGee, J.O.; Weatherall, D.J. Vascular Occlusion and Infarction in Sickle Cell Crisis and the Sickle Chest Syndrome. J. Clin. Pathol. 1985, 38, 659–664. [Google Scholar] [CrossRef]
- Coluzzi, F.; LeQuang, J.A.K.; Sciacchitano, S.; Scerpa, M.S.; Rocco, M.; Pergolizzi, J. A Closer Look at Opioid-Induced Adrenal Insufficiency: A Narrative Review. Int. J. Mol. Sci. 2023, 24, 4575. [Google Scholar] [CrossRef]
- Sahmoud, S.; Ibrahim, M.S.; Toraih, E.A.; Kamel, N.; Fawzy, M.S.; Elfiky, S. Association of VDBP Rs4701 Variant, but Not VDR/RXR-α Over-Expression with Bone Mineral Density in Pediatric Well-Chelated β-Thalassemia Patients. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020037. [Google Scholar] [CrossRef]
- Perrotta, S.; Cappellini, M.D.; Bertoldo, F.; Servedio, V.; Iolascon, G.; D’Agruma, L.; Gasparini, P.; Siciliani, M.C.; Iolascon, A. Osteoporosis in Beta-Thalassaemia Major Patients: Analysis of the Genetic Background. Br. J. Haematol. 2000, 111, 461–466. [Google Scholar] [CrossRef]
- Arisal, O.; Deviren, A.; Fenerci, E.Y.; Hacihanefioglu, S.; Ulutin, T.; Erkmen, S.; Buyru, N. Polymorphism Analysis in the COLIA1 Gene of Patients with Thalassemia Major and Intermedia. Haematologia 2002, 32, 475–482. [Google Scholar]
- Hamed, H.M.; Galal, A.; Ghamrawy, M.E.L.; Abd El Azeem, K.; Hussein, I.R.; Abd-Elgawad, M.F. An SP1-Binding Site Polymorphism in the COLIAI Gene and Osteoporosis in Egyptian Patients with Thalassemia Major. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 2011, 22, 81–85. [Google Scholar] [CrossRef]
- Ferrara, M.; Matarese, S.M.R.; Francese, M.; Borrelli, B.; Coppola, A.; Coppola, L.; Esposito, L. Effect of VDR Polymorphisms on Growth and Bone Mineral Density in Homozygous Beta Thalassaemia. Br. J. Haematol. 2002, 117, 436–440. [Google Scholar] [CrossRef] [PubMed]
- El-Edel, R.H.; Ghonaim, M.M.; Abo-Salem, O.M.; El-Nemr, F.M. Bone Mineral Density and Vitamin D Receptor Polymorphism in Beta-Thalassemia Major. Pak. J. Pharm. Sci. 2010, 23, 89–96. [Google Scholar]
- Abbassy, H.A.; Elwafa, R.A.A.; Omar, O.M. Bone Mineral Density and Vitamin D Receptor Genetic Variants in Egyptian Children with Beta Thalassemia Major on Vitamin D Supplementation. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019013. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Kumar, R.; Shukla, A.; Phadke, S.R.; Agarwal, S. Status of 25-Hydroxyvitamin D Deficiency and Effect of Vitamin D Receptor Gene Polymorphisms on Bone Mineral Density in Thalassemia Patients of North India. Hematology 2012, 17, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Ragab, S.M.; Badr, E.A.; Ibrahim, A.S. Evaluation of Glutathione-S-Transferase P1 Polymorphism and Its Relation to Bone Mineral Density in Egyptian Children and Adolescents with Beta-Thalassemia Major. Mediterr. J. Hematol. Infect. Dis. 2016, 8, e2016004. [Google Scholar] [CrossRef]
- Kountouris, P.; Stephanou, C.; Archer, N.; Bonifazi, F.; Giannuzzi, V.; Kuo, K.H.M.; Maggio, A.; Makani, J.; del Mar Mañú-Pereira, M.; Michailidou, K.; et al. The International Hemoglobinopathy Research Network (INHERENT): An International Initiative to Study the Role of Genetic Modifiers in Hemoglobinopathies. Am. J. Hematol. 2021, 96, 948. [Google Scholar] [CrossRef] [PubMed]

| First Author Publication Year Reference Number | Study Type | Disease | Gene | Trait |
|---|---|---|---|---|
| Zhang 2015 [36] | Meta-analysis of cohorts | SCD | C allele of rs59014890 SNP | DM |
| Sahmoud 2020 [157] | Case–control | β-thalassemia | rs4701 SNP at the VDBP | Osteoporosis |
| Perrotta 2000 [158] | Case–control | β-thalassemia | Sp1 binding site SNP at the COLIA1 gene | Osteoporosis |
| Arisal 2002 [159] | Cohort | β-thalassemia | Sp1 binding site SNP at the COLIA1 gene | Low BMD |
| Hamed 2011 [160] | Case–control | β-thalassemia | Sp1 binding site SNP at the COLIA1 gene | Osteoporosis |
| Ragab 2016 [165] | Case–control | β-thalassemia | Ile105Val polymorphism at GSTP1 gene | Low BMD |
| Singh 2012 [164] | Cohort | β-thalassemia | FokI and BsmI polymorphisms at VDR gene | Low BMD |
| Abbassy 2019 [163] | Case–control | β-thalassemia | FokI and BsmI polymorphisms at VDR gene | Osteoporosis |
| El-Edel 2010 [162] | Cohort | β-thalassemia | BsmI polymorphisms at VDR gene | Osteoporosis |
| Ferrara 2002 [161] | Cohort | β-thalassemia | FokI and BsmI polymorphisms at VDR gene | Low BMD Growth failure |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evangelidis, P.; Venou, T.-M.; Fani, B.; Vlachaki, E.; Gavriilaki, E., on behalf of the International Hemoglobinopathy Research Network (INHERENT). Endocrinopathies in Hemoglobinopathies: What Is the Role of Iron? Int. J. Mol. Sci. 2023, 24, 16263. https://doi.org/10.3390/ijms242216263
Evangelidis P, Venou T-M, Fani B, Vlachaki E, Gavriilaki E on behalf of the International Hemoglobinopathy Research Network (INHERENT). Endocrinopathies in Hemoglobinopathies: What Is the Role of Iron? International Journal of Molecular Sciences. 2023; 24(22):16263. https://doi.org/10.3390/ijms242216263
Chicago/Turabian StyleEvangelidis, Paschalis, Theodora-Maria Venou, Barmpageorgopoulou Fani, Efthymia Vlachaki, and Eleni Gavriilaki on behalf of the International Hemoglobinopathy Research Network (INHERENT). 2023. "Endocrinopathies in Hemoglobinopathies: What Is the Role of Iron?" International Journal of Molecular Sciences 24, no. 22: 16263. https://doi.org/10.3390/ijms242216263
APA StyleEvangelidis, P., Venou, T.-M., Fani, B., Vlachaki, E., & Gavriilaki, E., on behalf of the International Hemoglobinopathy Research Network (INHERENT). (2023). Endocrinopathies in Hemoglobinopathies: What Is the Role of Iron? International Journal of Molecular Sciences, 24(22), 16263. https://doi.org/10.3390/ijms242216263