

Evaluation of Biological Equivalence for Generic Tulathromycin Injections in Cattle


Abstract
:1. Introduction
2. Results
2.1. Potency of Generic Products
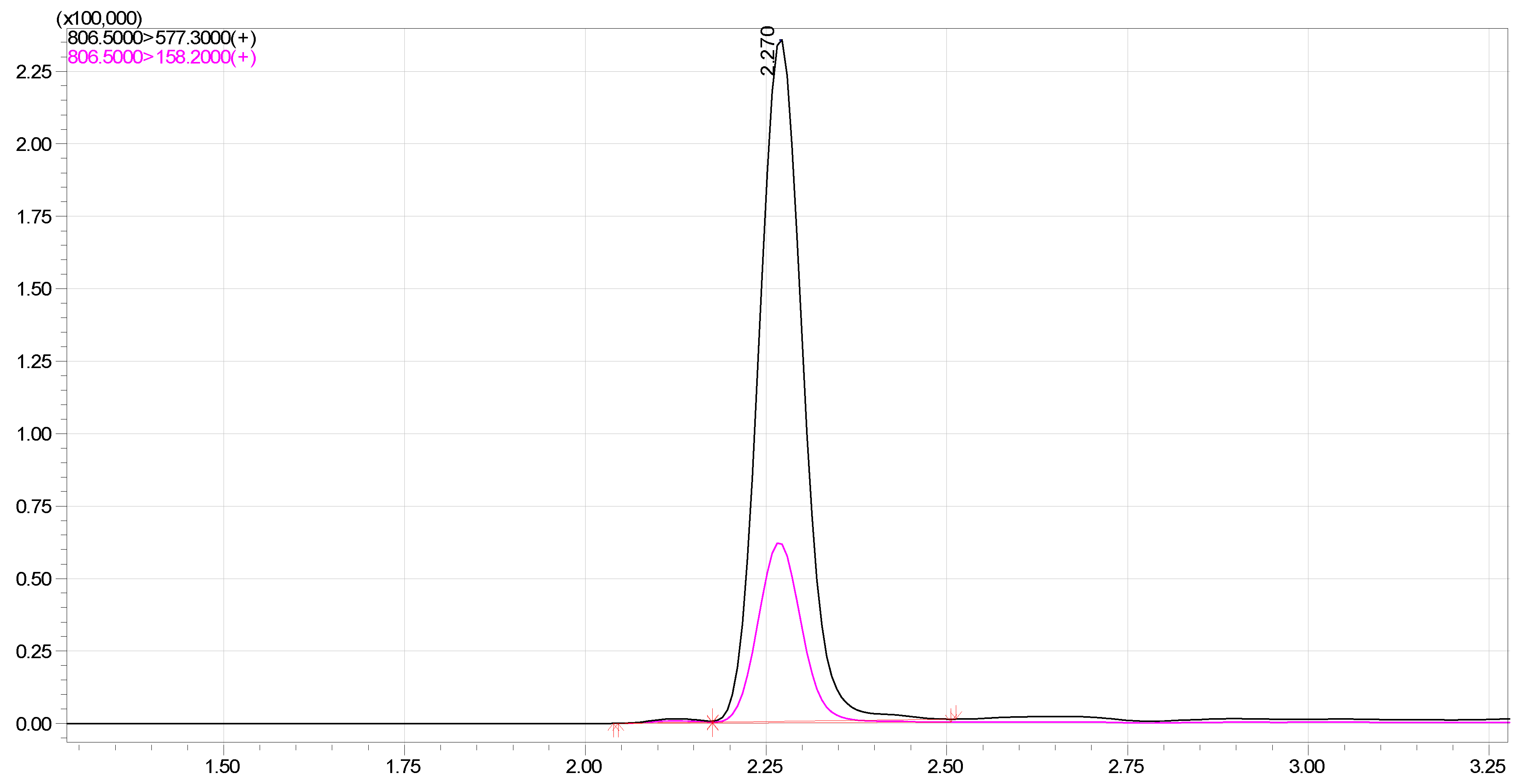
2.2. Validation of LC-MS/MS Method
2.3. Pharmacokinetic Profile
2.4. Bioequivalence Analysis
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. High-Performance Liquid Chromatography (HPLC)
4.3. Animal Experiment
4.4. LC–MS/MS Analysis Conditions
4.5. Validation of LC–MS/MS Method
4.6. Pharmacokinetic and Bioequivalence Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jones, R.N.; Sader, H.S. Antimicrobial activity of tulathromycin against contemporary pathogens isolated from North American and European hospitals. Diagn. Microbiol. Infect. Dis. 2005, 52, 245–250. [Google Scholar]
- Toutain, P.L.; Ferran, A.A.; Bousquet-Mélou, A. Species differences in pharmacokinetics and pharmacodynamics. J. Vet. Pharmacol. Ther. 2002, 25, 455–476. [Google Scholar]
- Sweeney, M.T.; Lubbers, B.V.; Schwarz, S.; Watts, J.L. Antibiotic resistance in animal agriculture: A nexus between the environment, animal health, and human health. Front. Ecol. Environ. 2018, 16, 521–528. [Google Scholar]
- Brown, S.A.; Boucher, J.F. Tulathromycin: A new triamilide antibiotic. J. Vet. Pharmacol. Ther. 2005, 28, 1–12. [Google Scholar]
- Stuart, J.C.; Fales, W.H.; Brown, S.A.; Schultz, L.G. Pharmacokinetics of tulathromycin and its 2-deoxy metabolite in plasma and lung tissue of cattle. J. Vet. Pharmacol. Ther. 2004, 27, 405–412. [Google Scholar]
- Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted under an Abbreviated New Drug Application; Center for Drug Evaluation and Research, US Department of Health and Human Services, Food and Drug Administration: Rockville, MD, USA, 2013.
- Chow, S.C. Bioavailability and bioequivalence in drug development. Wiley Interdiscip. Rev. Comput. Stat. 2014, 6, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.Y.; Lee, J.H.; Shin, M.J.; Kim, M.S.; Song, H.J.; Kim, J.M.; Koo, D.B.; Lee, S.H. Bioequivalence evaluation of generic tulathromycin products available in Korea. J. Vet. Pharmacol. Ther. 2021, 44, 711–717. [Google Scholar]
- Galyean, M.L.; Perino, L.J.; Duff, G.C. Interaction of cattle health/immunity and nutrition. J. Anim. Sci. 1999, 77, 1120–1134. [Google Scholar] [CrossRef] [PubMed]
- Guideline on the Investigation of Bioequivalence; European Medicines Agency: London, UK, 2010.
- Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products-General Considerations; Center for Drug Evaluation and Research, US Department of Health and Human Services, Food and Drug Administration: Rockville, MD, USA, 2019.
- Critically Important Antimicrobials for Human Medicine; World Health Organization: Geneva, Switzerland, 2019.
- Guardabassi, L.; Schwarz, S.; Lloyd, D.H. Pet animals as reservoirs of antimicrobial-resistant bacteria. J. Antimicrob. Chemother. 2004, 54, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Toutain, P.L.; Bousquet-Mélou, A. Bioequivalence and the withdrawal of drugs from the market for safety reasons. J. Vet. Pharmacol. Ther. 2004, 27, 185–193. [Google Scholar]
- Peng, M.; Salaheen, S.; Biswas, D. Animal Health: Global Antibiotic Issues. In Encyclopedia of Agriculture and Food Systems, 2nd ed.; Van Alfen, N.K., Ed.; Elsevier Academic Press: Cambridge, MA, USA, 2014; Volume 1, pp. 346–357. [Google Scholar]
- Gajda, A.; Bladek, T.; Jablonski, A.; Posyniak, A. The influence of Actinobacillus pleuropneumoniae infection on tulathromycin pharmacokinetics and lung tissue disposition in pigs. J. Vet. Pharmacol. Ther. 2016, 39, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tao, Y.F.; Huang, L.L.; Chen, D.M.; Yin, S.Z.; Ihsan, A.; Zhou, W.; Su, S.J.; Liu, Z.L.; Pan, Y.H.; et al. Pharmacokinetics of tulathromycin and its metabolite in swine administered with an intravenous bolus injection and a single gavage. J. Vet. Pharmacol. Ther. 2012, 35, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, S.; Evans, D.R.; Bessias, S.; Haynie, D.G.; Yemeke, T.T.; Laing, S.K.; Herrington, J.E. Prevalence and estimated economic burden of substandard and falsified medicines in low- and middle-income countries: A systematic review and meta-analysis. JAMA Netw. Open 2018, 1, e181662. [Google Scholar] [CrossRef] [PubMed]
- Guideline on the Conduct of Bioequivalence Studies for Veterinary Medicinal Products; European Medicines Agency: London, UK, 2011.
- Yao, L.; Yang, L.; Ling, Y.; Wei, Y.; Shen, X.; Ding, H. Pharmacokinetic/pharmacodynamic relationships of tulathromycin against Actinobacillus pleuropneumoniae in a porcine tissue cage infection model. Front. Vet. Sci. 2022, 9, 822432. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhang, G.; Wang, Q.; Liu, W.; Huang, Y.; Yu, P.; Li, Y.; Ding, H.; Fang, B. Pharmacokinetic/pharmacodynamic modeling of tulathromycin against Pasteurella multocida in a porcine tissue cage model. Front. Pharmacol. 2017, 8, 392. [Google Scholar] [CrossRef] [PubMed]
- Gümüşbaş, A.H.; Bakirel, T. Bioequivalence study of two different formulations of ceftiofur following intramuscular administration in cattle. Acta Vet. Eurasia 2022, 48, 22–29. [Google Scholar]
- Hossain, M.A.; Pervin, R.; Park, N.H.; Kang, J.W.; Lee, K.J.; Suh, J.W.; Park, S.C. Development of a Simple and Sensitive HPLC Method for the Determination of Rifaximin in Serum and its Application. Indian J. Pharm. Sci. 2018, 80, 1108–1114. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Name | Manufacturer | Label Claim (mg/mL) | Found Potency (mg/mL) | Percent of Label Claim (%) |
|---|---|---|---|---|
| Draxxin | Zoetis Inc., Parsippany-Troy Hills, NJ, USA (Pioneer Company) | 100 | 101 | 101 |
| Tulaject | Thumb Vet Co., Ltd., Iksan-si, Korea | 100 | 98 | 98 |
| Tulagen | Hanawin Co., Ltd., Jeonnam, Korea | 100 | 95 | 95 |
| Toulashot | Eagle Vet Co., Ltd., Yesan, Korea | 100 | 100 | 100 |
| T-raxxin | Komipharm International Co., Ltd., Seoul, Korea | 100 | 97 | 97 |
| Parameters (Unit) | Inventor Drug | Tulaject Injection (Thumbvet) | Tulazen Injection (Hanawin) | T-raxxin Injection (Komipharm) | Tulshot Injection (Eaglevet) |
|---|---|---|---|---|---|
| T1/2 (h) | 94.04 ± 36.09 | 102.56 ± 24.31 | 91.40 ± 6.13 | 96.17 ± 14.07 | 96.79 ± 36.43 |
| Tmax (h) | 0.44 ± 0.08 | 0.33 ± 0.00 | 0.50 ± 0.00 | 0.33 ± 0.09 | 0.33 ± 00 |
| Cmax (ng/mL) | 1100.08 ± 102.97 | 1027.97 ± 77.06 | 1110.04 ± 56.78 | 1031.15 ± 80.09 | 1028.99 ± 18.95 |
| AUC (hr.ng/mL) | 27,187.01 ± 4891.20 | 27,452.51 ± 5327.37 | 26,983.37 ± 3146.46 | 26,036.92 ± 3465.66 | 26,962.52 ± 5844.84 |
| Parameter | Inventor Drug | Tulaject Injection (Thumbvet) | Tulazen Injection (Hanawin) | T-raxxin Injection (Komipharm) | Tulshot Injection (Eaglevet) |
|---|---|---|---|---|---|
| Cmax | 1015.00–1185.00 | 964.60–1091.00 | 1063.00–1157.00 | 965.30–1097.00 | 1013.00–1045.00 |
| 92.27–107.72% | 93.84–106.13% | 95.76–104.23% | 93.61–106.39% | 98.45–101.56% | |
| AUC | 23,160.00–31,210.00 | 23,070.00–31,830.00 | 24,400.00–29,570.00 | 23,190.00–28,890.00 | 22,160.00–31,770.00 |
| 85.19–114.80% | 84.04–115.95% | 90.43–109.59% | 89.07–110.96% | 82.19–117.83% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.; Chae, H.; Jeong, S.; Pervin, R.; Hossain, M.A. Evaluation of Biological Equivalence for Generic Tulathromycin Injections in Cattle. Int. J. Mol. Sci. 2023, 24, 16262. https://doi.org/10.3390/ijms242216262
Kang J, Chae H, Jeong S, Pervin R, Hossain MA. Evaluation of Biological Equivalence for Generic Tulathromycin Injections in Cattle. International Journal of Molecular Sciences. 2023; 24(22):16262. https://doi.org/10.3390/ijms242216262
Chicago/Turabian StyleKang, JeongWoo, HyunYoung Chae, SungHoon Jeong, Rokeya Pervin, and Md Akil Hossain. 2023. "Evaluation of Biological Equivalence for Generic Tulathromycin Injections in Cattle" International Journal of Molecular Sciences 24, no. 22: 16262. https://doi.org/10.3390/ijms242216262
APA StyleKang, J., Chae, H., Jeong, S., Pervin, R., & Hossain, M. A. (2023). Evaluation of Biological Equivalence for Generic Tulathromycin Injections in Cattle. International Journal of Molecular Sciences, 24(22), 16262. https://doi.org/10.3390/ijms242216262