
Facilitation of hERG Activation by Its Blocker: A Mechanism to Reduce Drug-Induced Proarrhythmic Risk

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
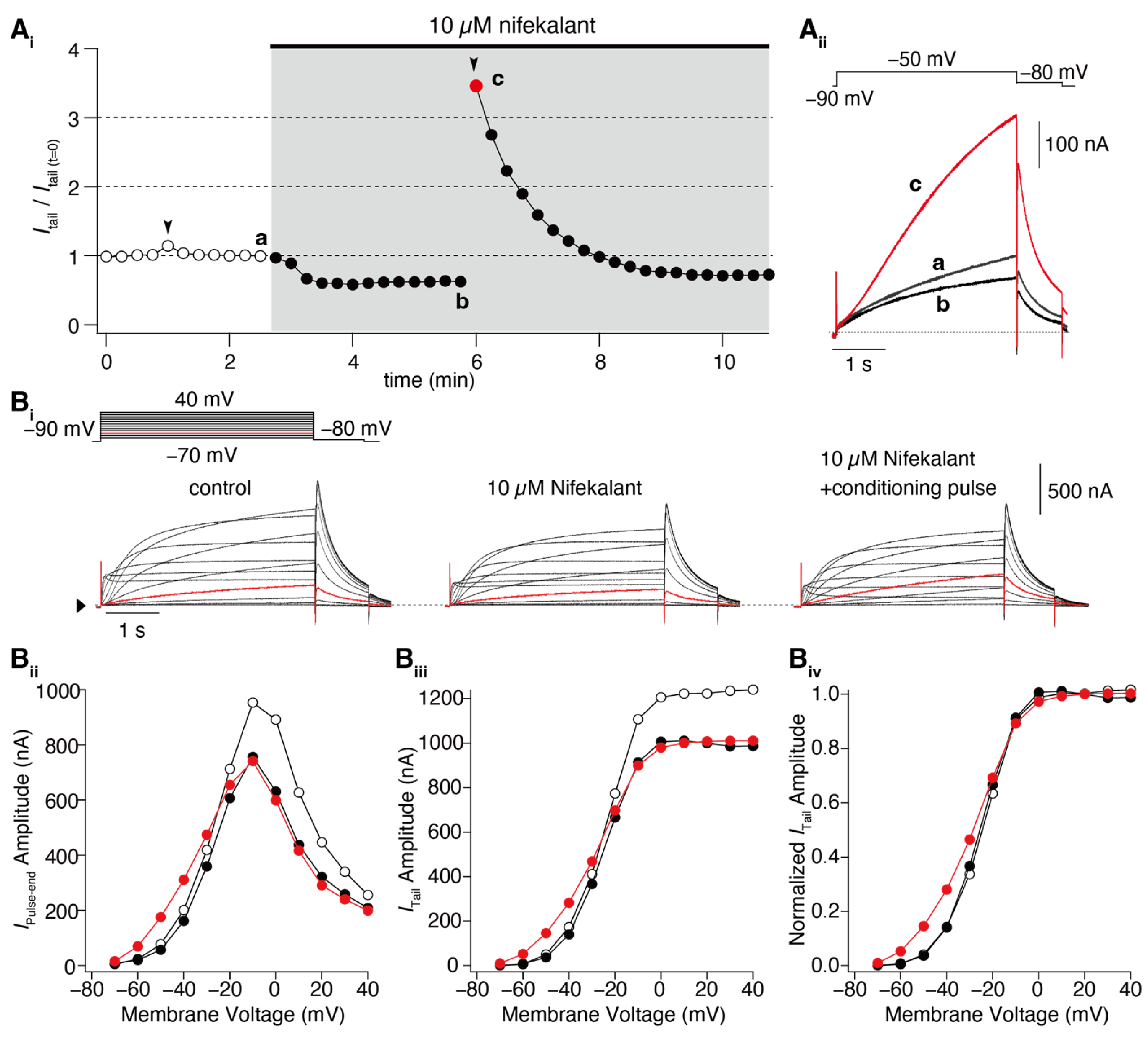
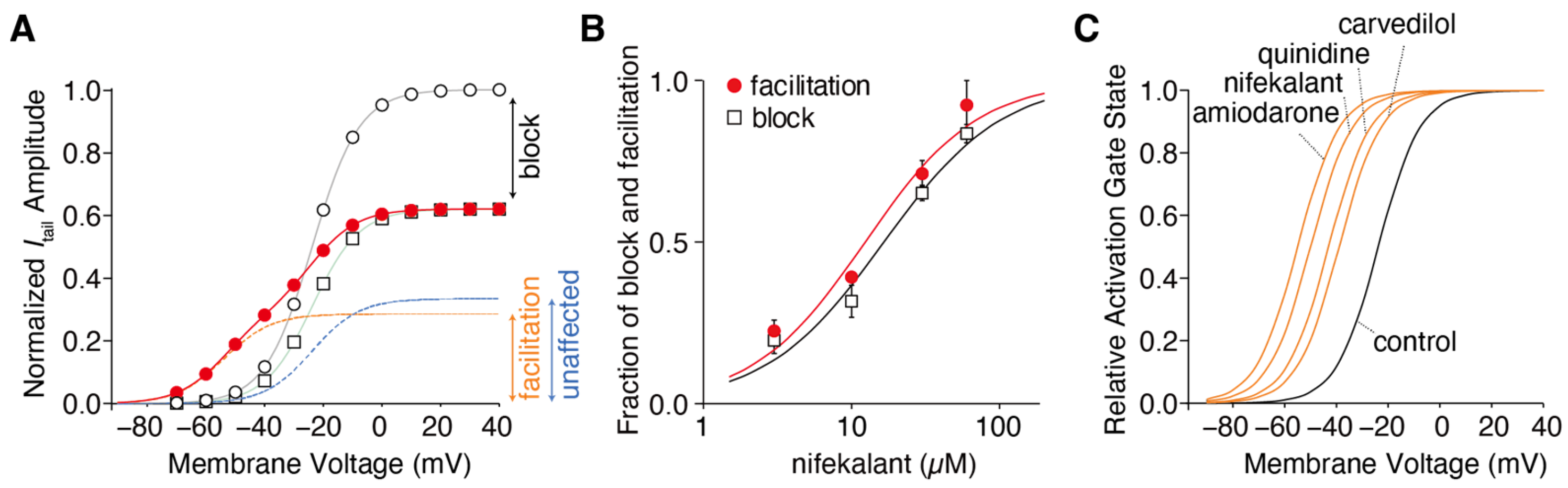
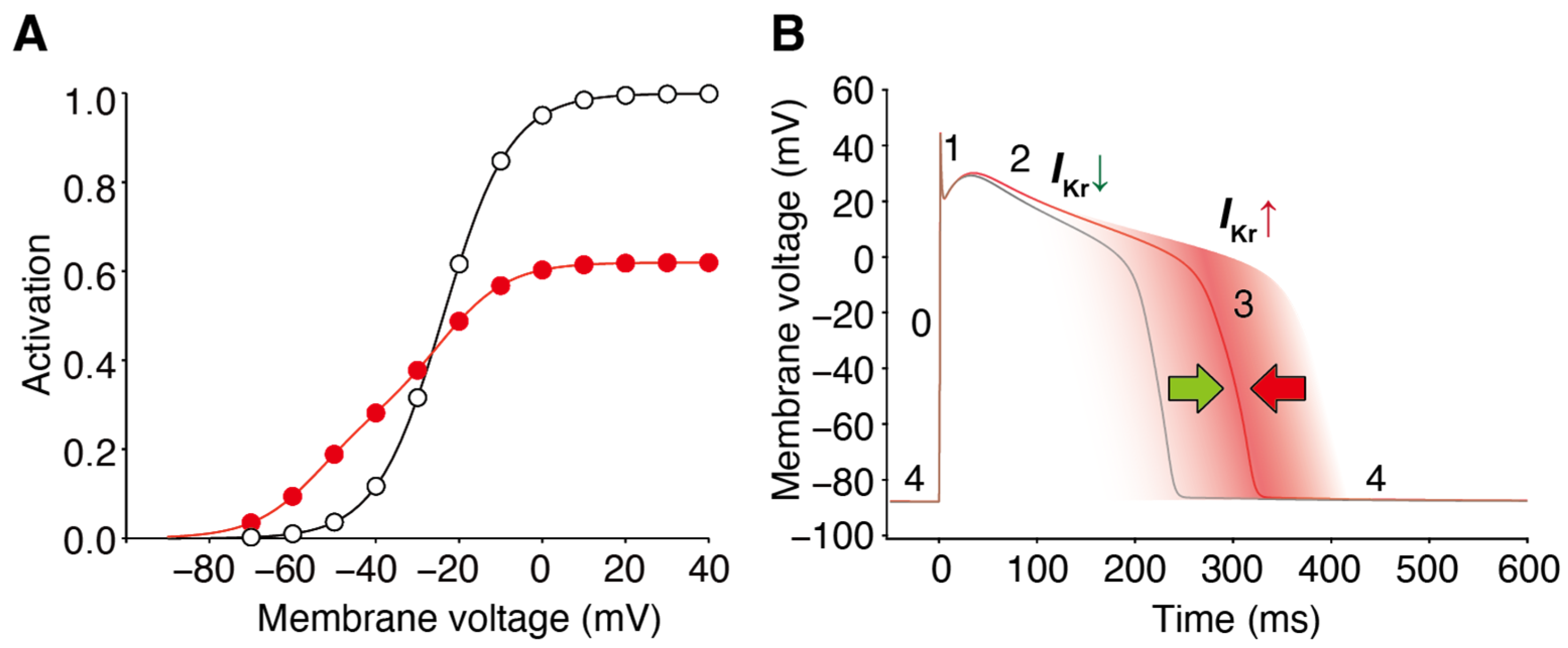
2. Facilitation of hERG Activation by Its Blocker
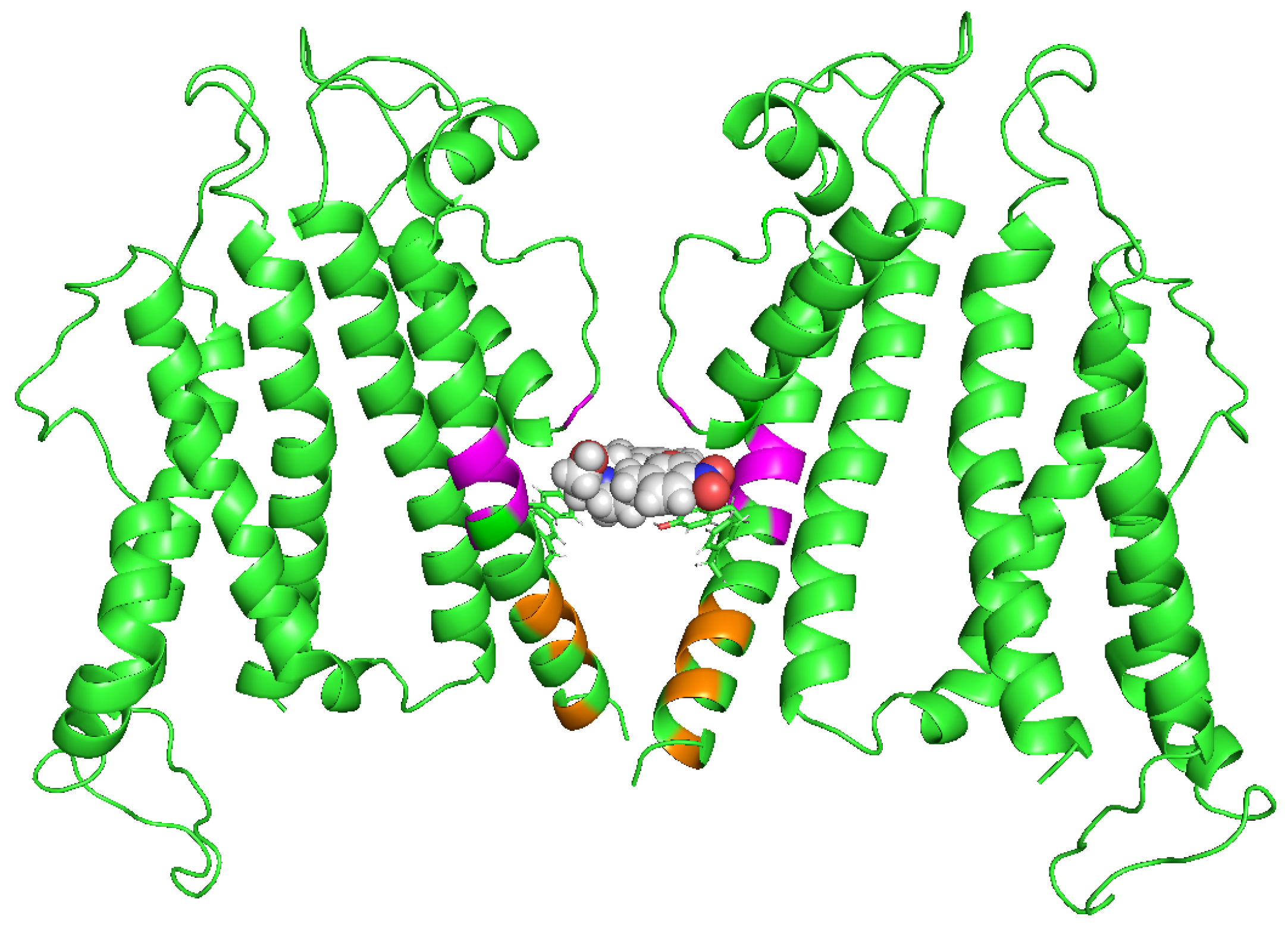
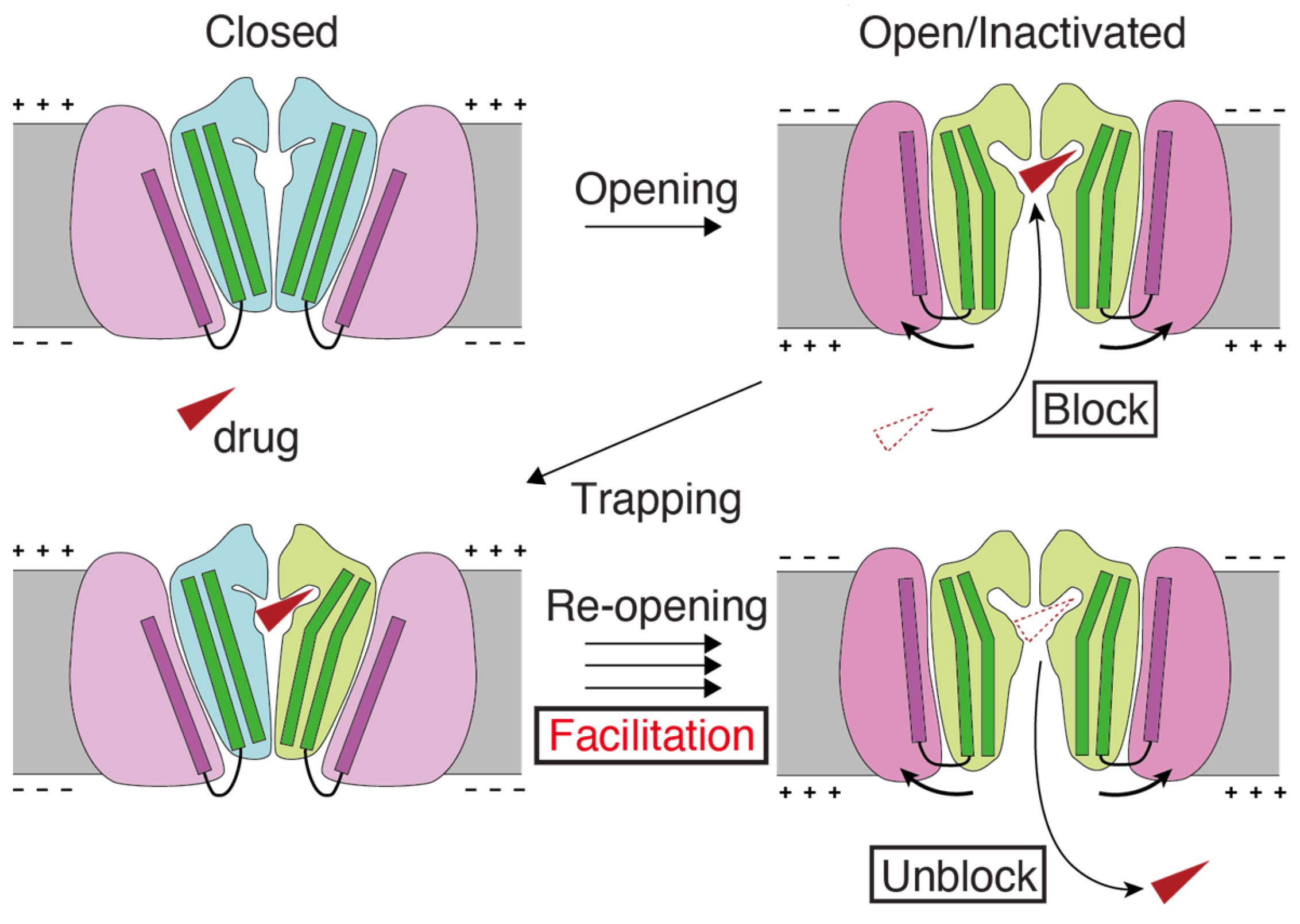
3. Structural Basis of hERG Facilitation
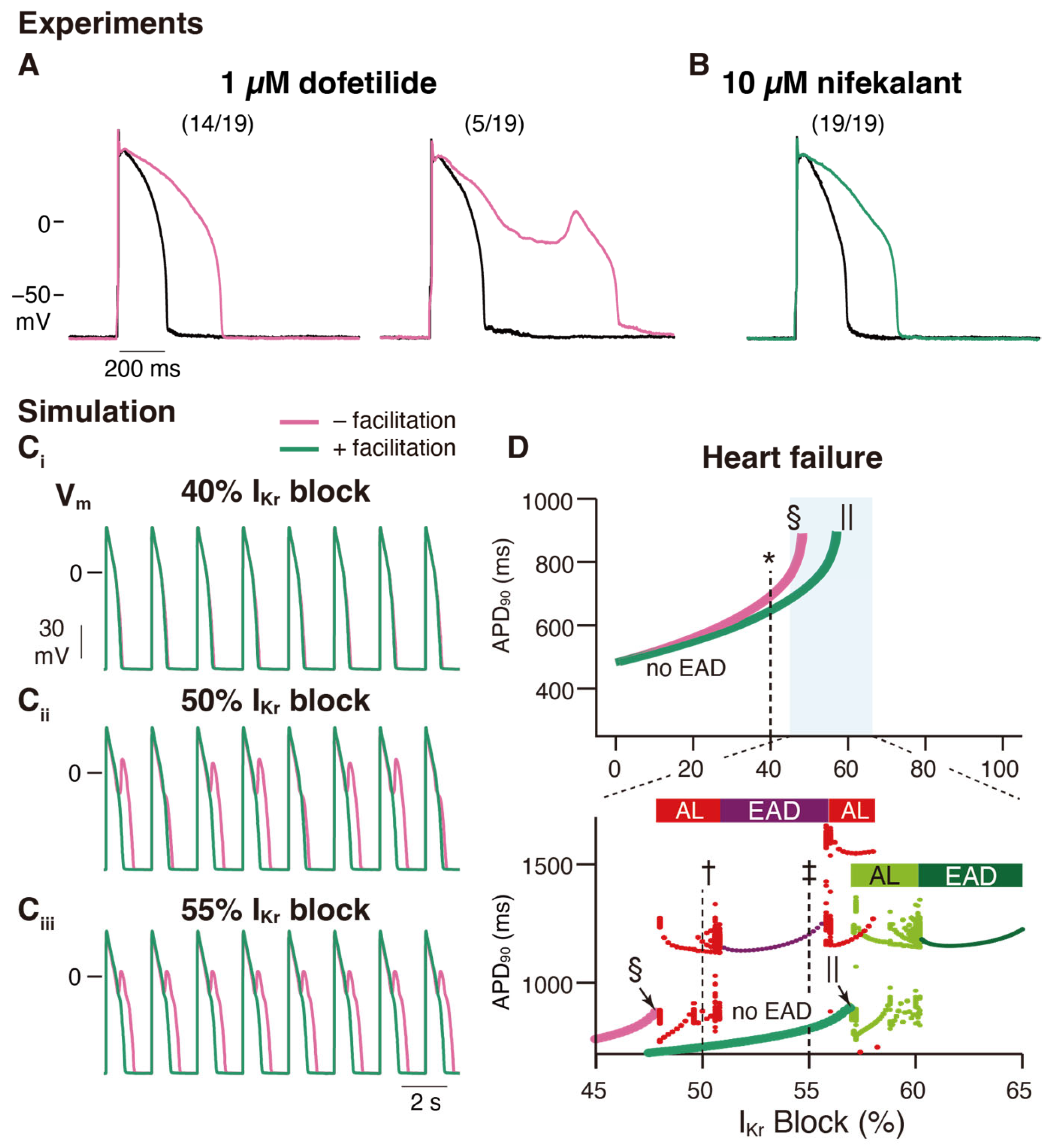
4. A Possible Role of hERG Facilitation in hERG Block-Associated Arrhythmia
5. Conclusions and Future Direction
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef]
- Trudeau, M.C.; Warmke, J.W.; Ganetzky, B.; Robertson, G.A. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 1995, 269, 92–95. [Google Scholar] [CrossRef]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. hERG K(+) channels: Structure, function, and clinical significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef]
- Clancy, C.E.; Kurokawa, J.; Tateyama, M.; Wehrens, X.H.; Kass, R.S. K+ channel structure-activity relationships and mechanisms of drug-induced QT prolongation. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 441–461. [Google Scholar] [CrossRef]
- Nerbonne, J.M.; Kass, R.S. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005, 85, 1205–1253. [Google Scholar] [CrossRef]
- Surawicz, B. Electrophysiologic substrate of torsade de pointes: Dispersion of repolarization or early afterdepolarizations? J. Am. Coll. Cardiol. 1989, 14, 172–184. [Google Scholar] [CrossRef]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincent, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef]
- Roden, D.M. Acquired long QT syndromes and the risk of proarrhythmia. J. Cardiovasc. Electrophysiol. 2000, 11, 938–940. [Google Scholar] [CrossRef]
- Haverkamp, W.; Breithardt, G.; Camm, A.J.; Janse, M.J.; Rosen, M.R.; Antzelevitch, C.; Escande, D.; Franz, M.; Malik, M.; Moss, A.; et al. The potential for QT prolongation and proarrhythmia by non-antiarrhythmic drugs: Clinical and regulatory implications. Report on a policy conference of the European Society of Cardiology. Eur. Heart J. 2000, 21, 1216–1231. [Google Scholar] [CrossRef]
- Roden, D.M. Cellular basis of drug-induced torsades de pointes. Br. J. Pharmacol. 2008, 154, 1502–1507. [Google Scholar] [CrossRef]
- Kannankeril, P.; Roden, D.M.; Darbar, D. Drug-induced long QT syndrome. Pharmacol. Rev. 2010, 62, 760–781. [Google Scholar] [CrossRef] [PubMed]
- Nachimuthu, S.; Assar, M.D.; Schussler, J.M. Drug-induced QT interval prolongation: Mechanisms and clinical management. Ther. Adv. Drug Saf. 2012, 3, 241–253. [Google Scholar] [CrossRef]
- International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH). The Non-Clinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals. Guidance on S7B. 2005. Available online: http://www.ich.org/products/guidelines/safety/safety-single/article/the-non-clinical-evaluation-of-the-potential-for-delayed-ventricular-repolarization-qt-interval-pro.html (accessed on 23 October 2023).
- Sager, P.T.; Gintant, G.; Turner, J.R.; Pettit, S.; Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: A meeting report from the Cardiac Safety Research Consortium. Am. Heart J. 2014, 167, 292–300. [Google Scholar] [CrossRef]
- Gintant, G.; Sager, P.T.; Stockbridge, N. Evolution of strategies to improve preclinical cardiac safety testing. Nat. Rev. Drug Discov. 2016, 15, 457–471. [Google Scholar] [CrossRef]
- Huang, H.; Pugsley, M.K.; Fermini, B.; Curtis, M.J.; Koerner, J.; Accardi, M.; Authier, S. Cardiac voltage-gated ion channels in safety pharmacology: Review of the landscape leading to the CiPA initiative. J. Pharmacol. Toxicol. Methods 2017, 87, 11–23. [Google Scholar] [CrossRef]
- European Medicines Agency. Topics ICH S7B Non-Clinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals—Scientific Guideline; European Medicines Agency: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Redfern, W.S.; Carlsson, L.; Davis, A.S.; Lynch, W.G.; MacKenzie, I.; Palethorpe, S.; Siegl, P.K.; Strang, I.; Sullivan, A.T.; Wallis, R.; et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003, 58, 32–45. [Google Scholar] [CrossRef]
- Mirams, G.R.; Cui, Y.; Sher, A.; Fink, M.; Cooper, J.; Heath, B.M.; McMahon, N.C.; Gavaghan, D.J.; Noble, D. Simulation of multiple ion channel block provides improved early prediction of compounds’ clinical torsadogenic risk. Cardiovasc. Res. 2011, 91, 53–61. [Google Scholar] [CrossRef]
- Martin, R.L.; McDermott, J.S.; Salmen, H.J.; Palmatier, J.; Cox, B.F.; Gintant, G.A. The utility of hERG and repolarization assays in evaluating delayed cardiac repolarization: Influence of multi-channel block. J. Cardiovasc. Pharmacol. 2004, 43, 369–379. [Google Scholar] [CrossRef]
- Finlayson, K.; Witchel, H.J.; McCulloch, J.; Sharkey, J. Acquired QT interval prolongation and HERG: Implications for drug discovery and development. Eur. J. Pharmacol. 2004, 500, 129–142. [Google Scholar] [CrossRef]
- Shah, R.R. Drugs, QTc interval prolongation and final ICH E14 guideline: An important milestone with challenges ahead. Drug Saf. 2005, 28, 1009–1028. [Google Scholar] [CrossRef] [PubMed]
- Raschi, E.; Vasina, V.; Poluzzi, E.; De Ponti, F. The hERG K+ channel: Target and antitarget strategies in drug development. Pharmacol. Res. 2008, 57, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, A. Expression and role of hERG channels in cancer cells. Novartis Found. Symp. 2005, 266, 225–232; discussion 232–234. [Google Scholar] [PubMed]
- Masi, A.; Becchetti, A.; Restano-Cassulini, R.; Polvani, S.; Hofmann, G.; Buccoliero, A.M.; Paglierani, M.; Pollo, B.; Taddei, G.L.; Gallina, P.; et al. hERG1 channels are overexpressed in glioblastoma multiforme and modulate VEGF secretion in glioblastoma cell lines. Br. J. Cancer 2005, 93, 781–792. [Google Scholar] [CrossRef]
- Pardo, L.A.; Contreras-Jurado, C.; Zientkowska, M.; Alves, F.; Stuhmer, W. Role of voltage-gated potassium channels in cancer. J. Membr. Biol. 2005, 205, 115–124. [Google Scholar] [CrossRef]
- Wadhwa, S.; Wadhwa, P.; Dinda, A.K.; Gupta, N.P. Differential expression of potassium ion channels in human renal cell carcinoma. Int. Urol. Nephrol. 2009, 41, 251–257. [Google Scholar] [CrossRef]
- Cicek, M.S.; Koestler, D.C.; Fridley, B.L.; Kalli, K.R.; Armasu, S.M.; Larson, M.C.; Wang, C.; Winham, S.J.; Vierkant, R.A.; Rider, D.N.; et al. Epigenome-wide ovarian cancer analysis identifies a methylation profile differentiating clear-cell histology with epigenetic silencing of the HERG K+ channel. Hum. Mol. Genet. 2013, 22, 3038–3047. [Google Scholar] [CrossRef]
- Calcaterra, N.E.; Hoeppner, D.J.; Wei, H.; Jaffe, A.E.; Maher, B.J.; Barrow, J.C. Schizophrenia-Associated hERG channel Kv11.1-3.1 Exhibits a Unique Trafficking Deficit that is Rescued Through Proteasome Inhibition for High Throughput Screening. Sci. Rep. 2016, 6, 19976. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jurkiewicz, N.K. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 1990, 96, 195–215. [Google Scholar] [CrossRef]
- Vaughan Williams, E.M. Classifying antiarrhythmic actions: By facts or speculation. J. Clin. Pharmacol. 1992, 32, 964–977. [Google Scholar] [CrossRef]
- Nakaya, H.; Tohse, N.; Takeda, Y.; Kanno, M. Effects of MS-551, a new class III antiarrhythmic drug, on action potential and membrane currents in rabbit ventricular myocytes. Br. J. Pharmacol. 1993, 109, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Laurita, K.R.; Rosenbaum, D.S.; Rudy, Y. Two components of the delayed rectifier K+ current in ventricular myocytes of the guinea pig type. Theoretical formulation and their role in repolarization. Circ. Res. 1995, 77, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Kodama, I.; Kamiya, K.; Toyama, J. Cellular electropharmacology of amiodarone. Cardiovasc. Res. 1997, 35, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, Y.; Iwata, M.; Kamiya, N.; Yamada, M.; Kinoshita, K.; Fukunishi, Y.; Tsujimae, K.; Hibino, H.; Aizawa, Y.; Inanobe, A.; et al. Mutational analysis of block and facilitation of HERG current by a class III anti-arrhythmic agent, nifekalant. Channels 2007, 1, 198–208. [Google Scholar] [CrossRef][Green Version]
- Furutani, K.; Yamakawa, Y.; Inanobe, A.; Iwata, M.; Ohno, Y.; Kurachi, Y. A mechanism underlying compound-induced voltage shift in the current activation of hERG by antiarrhythmic agents. Biochem. Biophys. Res. Commun. 2011, 415, 141–146. [Google Scholar] [CrossRef]
- Lo, Y.C.; Kuo, C.C. Temperature Dependence of the Biophysical Mechanisms Underlying the Inhibition and Enhancement Effect of Amiodarone on hERG Channels. Mol. Pharmacol. 2019, 96, 330–344. [Google Scholar] [CrossRef]
- Perry, M.; Sachse, F.B.; Sanguinetti, M.C. Structural basis of action for a human ether-a-go-go-related gene 1 potassium channel activator. Proc. Natl. Acad. Sci. USA 2007, 104, 13827–13832. [Google Scholar] [CrossRef]
- Grunnet, M.; Hansen, R.S.; Olesen, S.P. hERG1 channel activators: A new anti-arrhythmic principle. Prog. Biophys. Mol. Biol. 2008, 98, 347–362. [Google Scholar] [CrossRef]
- Perry, M.; Sachse, F.B.; Abbruzzese, J.; Sanguinetti, M.C. PD-118057 contacts the pore helix of hERG1 channels to attenuate inactivation and enhance K+ conductance. Proc. Natl. Acad. Sci. USA 2009, 106, 20075–20080. [Google Scholar] [CrossRef]
- Perry, M.; Sanguinetti, M.; Mitcheson, J. Revealing the structural basis of action of hERG potassium channel activators and blockers. J. Physiol. 2010, 588, 3157–3167. [Google Scholar] [CrossRef]
- Yamakawa, Y.; Furutani, K.; Inanobe, A.; Ohno, Y.; Kurachi, Y. Pharmacophore modeling for hERG channel facilitation. Biochem. Biophys. Res. Commun. 2012, 418, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Furutani, K.; Tsumoto, K.; Chen, I.S.; Handa, K.; Yamakawa, Y.; Sack, J.T.; Kurachi, Y. Facilitation of I Kr current by some hERG channel blockers suppresses early afterdepolarizations. J. Gen. Physiol. 2019, 151, 214–230. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, E. Use-dependent block and use-dependent unblock of the delayed rectifier K+ current by almokalant in rabbit ventricular myocytes. Circ. Res. 1993, 73, 857–868. [Google Scholar] [CrossRef]
- Jiang, M.; Dun, W.; Fan, J.S.; Tseng, G.N. Use-dependent ‘agonist’ effect of azimilide on the HERG channel. J. Pharmacol. Exp. Ther. 1999, 291, 1324–1336. [Google Scholar] [PubMed]
- Al-Moubarak, E.; Shiels, H.A.; Zhang, Y.; Du, C.; Hanington, O.; Harmer, S.C.; Dempsey, C.E.; Hancox, J.C. Inhibition of the hERG potassium channel by phenanthrene: A polycyclic aromatic hydrocarbon pollutant. Cell. Mol. Life Sci. 2021, 78, 7899–7914. [Google Scholar] [CrossRef]
- Kang, J.; Chen, X.L.; Wang, H.; Ji, J.; Cheng, H.; Incardona, J.; Reynolds, W.; Viviani, F.; Tabart, M.; Rampe, D. Discovery of a small molecule activator of the human ether-a-go-go-related gene (HERG) cardiac K+ channel. Mol. Pharmacol. 2005, 67, 827–836. [Google Scholar] [CrossRef]
- Zhou, J.; Augelli-Szafran, C.E.; Bradley, J.A.; Chen, X.; Koci, B.J.; Volberg, W.A.; Sun, Z.; Cordes, J.S. Novel potent human ether-a-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity. Mol. Pharmacol. 2005, 68, 876–884. [Google Scholar] [CrossRef]
- Hansen, R.S.; Diness, T.G.; Christ, T.; Wettwer, E.; Ravens, U.; Olesen, S.P.; Grunnet, M. Biophysical characterization of the new human ether-a-go-go-related gene channel opener NS3623 [N-(4-bromo-2-(1H-tetrazol-5-yl)-phenyl)-N′-(3′-trifluoromethylphenyl)urea]. Mol. Pharmacol. 2006, 70, 1319–1329. [Google Scholar] [CrossRef]
- Furutani, K.; Kawano, R.; Ichiwara, M.; Adachi, R.; Clancy, C.E.; Sack, J.T.; Kita, S. Pore opening, not voltage sensor movement, underpins the voltage-dependence of facilitation by a hERG blocker. Mol. Pharmacol. 2022, 102, 223–233. [Google Scholar] [CrossRef]
- Abbott, G.W.; Sesti, F.; Splawski, I.; Buck, M.E.; Lehmann, M.H.; Timothy, K.W.; Keating, M.T.; Goldstein, S.A. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999, 97, 175–187. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Xu, Q.P. Mutations of the S4-S5 linker alter activation properties of HERG potassium channels expressed in Xenopus oocytes. J. Physiol. 1999, 514 Pt 3, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Poluzzi, E.; De Ponti, F.; Recanatini, M. Toward a pharmacophore for drugs inducing the long QT syndrome: Insights from a CoMFA study of HERG K(+) channel blockers. J. Med. Chem. 2002, 45, 3844–3853. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; MacKinnon, R. Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell 2017, 169, 422–430. [Google Scholar] [CrossRef]
- Asai, T.; Adachi, N.; Moriya, T.; Oki, H.; Maru, T.; Kawasaki, M.; Suzuki, K.; Chen, S.; Ishii, R.; Yonemori, K.; et al. Cryo-EM Structure of K(+)-Bound hERG Channel Complexed with the Blocker Astemizole. Structure 2021, 29, 203–212.e204. [Google Scholar] [CrossRef] [PubMed]
- Mitcheson, J.S.; Chen, J.; Lin, M.; Culberson, C.; Sanguinetti, M.C. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 12329–12333. [Google Scholar] [CrossRef]
- Lees-Miller, J.P.; Duan, Y.; Teng, G.Q.; Duff, H.J. Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: Involvement of S6 sites. Mol. Pharmacol. 2000, 57, 367–374. [Google Scholar]
- Kamiya, K.; Mitcheson, J.S.; Yasui, K.; Kodama, I.; Sanguinetti, M.C. Open channel block of HERG K(+) channels by vesnarinone. Mol. Pharmacol. 2001, 60, 244–253. [Google Scholar] [CrossRef]
- Chen, J.; Seebohm, G.; Sanguinetti, M.C. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc. Natl. Acad. Sci. USA 2002, 99, 12461–12466. [Google Scholar] [CrossRef]
- Sanchez-Chapula, J.A.; Navarro-Polanco, R.A.; Culberson, C.; Chen, J.; Sanguinetti, M.C. Molecular determinants of voltage-dependent human ether-a-go-go related gene (HERG) K+ channel block. J. Biol. Chem. 2002, 277, 23587–23595. [Google Scholar] [CrossRef]
- Fernandez, D.; Ghanta, A.; Kauffman, G.W.; Sanguinetti, M.C. Physicochemical features of the HERG channel drug binding site. J. Biol. Chem. 2004, 279, 10120–10127. [Google Scholar] [CrossRef]
- Kamiya, K.; Niwa, R.; Mitcheson, J.S.; Sanguinetti, M.C. Molecular determinants of HERG channel block. Mol. Pharmacol. 2006, 69, 1709–1716. [Google Scholar] [CrossRef] [PubMed]
- Milnes, J.T.; Witchel, H.J.; Leaney, J.L.; Leishman, D.J.; Hancox, J.C. hERG K+ channel blockade by the antipsychotic drug thioridazine: An obligatory role for the S6 helix residue F656. Biochem. Biophys. Res. Commun. 2006, 351, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, K.; Niwa, R.; Morishima, M.; Honjo, H.; Sanguinetti, M.C. Molecular determinants of hERG channel block by terfenadine and cisapride. J. Pharmacol. Sci. 2008, 108, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, J.I.; Perozo, E.; Allen, T.W. Towards a Structural View of Drug Binding to hERG K(+) Channels. Trends Pharmacol. Sci. 2017, 38, 899–907. [Google Scholar] [CrossRef]
- Mobley, D.L.; Dill, K.A. Binding of small-molecule ligands to proteins: “what you see” is not always “what you get”. Structure 2009, 17, 489–498. [Google Scholar] [CrossRef]
- Cortez, A.M.E.; DeMarco, K.R.; Furutani, K.; Bekker, S.; Sack, J.T.; Wulff, H.; Clancy, C.E.; Vorobyov, I.; Yarov-Yarovoy, V. Structural Modeling of hERG Channel -Drug Interactions Using Rosetta. Front. Pharmacol. 2023, 14, 1244166. [Google Scholar] [CrossRef]
- Mitcheson, J.S.; Chen, J.; Sanguinetti, M.C. Trapping of a methanesulfonanilide by closure of the HERG potassium channel activation gate. J. Gen. Physiol. 2000, 115, 229–240. [Google Scholar] [CrossRef]
- Windisch, A.; Timin, E.; Schwarz, T.; Stork-Riedler, D.; Erker, T.; Ecker, G.; Hering, S. Trapping and dissociation of propafenone derivatives in HERG channels. Br. J. Pharmacol. 2011, 162, 1542–1552. [Google Scholar] [CrossRef]
- Linder, T.; Bernsteiner, H.; Saxena, P.; Bauer, F.; Erker, T.; Timin, E.; Hering, S.; Stary-Weinzinger, A. Drug trapping in hERG K(+) channels: (not) a matter of drug size? Medchemcomm 2016, 7, 512–518. [Google Scholar] [CrossRef]
- Munawar, S.; Vandenberg, J.I.; Jabeen, I. Molecular Docking Guided Grid-Independent Descriptor Analysis to Probe the Impact of Water Molecules on Conformational Changes of hERG Inhibitors in Drug Trapping Phenomenon. Int. J. Mol. Sci. 2019, 20, 3385. [Google Scholar] [CrossRef]
- O’Hara, T.; Virag, L.; Varro, A.; Rudy, Y. Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Comput. Biol. 2011, 7, e1002061. [Google Scholar] [CrossRef] [PubMed]
- Hondeghem, L.M.; Snyders, D.J. Class III antiarrhythmic agents have a lot of potential but a long way to go. Reduced effectiveness and dangers of reverse use dependence. Circulation 1990, 81, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Jurkiewicz, N.K.; Sanguinetti, M.C. Rate-dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent. Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circ. Res. 1993, 72, 75–83. [Google Scholar] [CrossRef]
- Tande, P.M.; Bjornstad, H.; Yang, T.; Refsum, H. Rate-dependent class III antiarrhythmic action, negative chronotropy, and positive inotropy of a novel Ik blocking drug, UK-68,798: Potent in guinea pig but no effect in rat myocardium. J. Cardiovasc. Pharmacol. 1990, 16, 401–410. [Google Scholar] [CrossRef]
- Hafner, D.; Berger, F.; Borchard, U.; Kullmann, A.; Scherlitz, A. Electrophysiological characterization of the class III activity of sotalol and its enantiomers. New interpretation of use-dependent effects. Arzneimittelforschung 1988, 38, 231–236. [Google Scholar]
- Okada, Y.; Ogawa, S.; Sadanaga, T.; Mitamura, H. Assessment of reverse use-dependent blocking actions of class III antiarrhythmic drugs by 24-hour Holter electrocardiography. J. Am. Coll. Cardiol. 1996, 27, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Elshrif, M.M.; Shi, P.; Cherry, E.M. Representing variability and transmural differences in a model of human heart failure. IEEE J. Biomed. Health Inform. 2015, 19, 1308–1320. [Google Scholar] [CrossRef]
- Dai, J.; Zhou, H.X. An NMDA receptor gating mechanism developed from MD simulations reveals molecular details underlying subunit-specific contributions. Biophys. J. 2013, 104, 2170–2181. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, J.; Perissinotti, L.L.; Lees-Miller, J.; Teng, G.; Durdagi, S.; Duff, H.J.; Noskov, S.Y. Role of the pH in state-dependent blockade of hERG currents. Sci. Rep. 2016, 6, 32536. [Google Scholar] [CrossRef]
- Lev, B.; Murail, S.; Poitevin, F.; Cromer, B.A.; Baaden, M.; Delarue, M.; Allen, T.W. String method solution of the gating pathways for a pentameric ligand-gated ion channel. Proc. Natl. Acad. Sci. USA 2017, 114, E4158–E4167. [Google Scholar] [CrossRef]
- Barbera, N.; Ayee, M.A.A.; Akpa, B.S.; Levitan, I. Molecular Dynamics Simulations of Kir2.2 Interactions with an Ensemble of Cholesterol Molecules. Biophys. J. 2018, 115, 1264–1280. [Google Scholar] [CrossRef] [PubMed]
- Dietzen, N.M.; Arcario, M.J.; Chen, L.J.; Petroff, J.T., 2nd; Moreland, K.T.; Krishnan, K.; Brannigan, G.; Covey, D.F.; Cheng, W.W. Polyunsaturated fatty acids inhibit a pentameric ligand-gated ion channel through one of two binding sites. elife 2022, 11, e74306. [Google Scholar] [CrossRef]
- Zhuang, Y.; Noviello, C.M.; Hibbs, R.E.; Howard, R.J.; Lindahl, E. Differential interactions of resting, activated, and desensitized states of the alpha7 nicotinic acetylcholine receptor with lipidic modulators. Proc. Natl. Acad. Sci. USA 2022, 119, e2208081119. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.; Ocello, R.; Guardiani, C.; Giacomello, A.; Masetti, M. Integrated Approach Including Docking, MD Simulations, and Network Analysis Highlights the Action Mechanism of the Cardiac hERG Activator RPR260243. J. Chem. Inf. Model. 2023, 63, 4888–4899. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furutani, K. Facilitation of hERG Activation by Its Blocker: A Mechanism to Reduce Drug-Induced Proarrhythmic Risk. Int. J. Mol. Sci. 2023, 24, 16261. https://doi.org/10.3390/ijms242216261
Furutani K. Facilitation of hERG Activation by Its Blocker: A Mechanism to Reduce Drug-Induced Proarrhythmic Risk. International Journal of Molecular Sciences. 2023; 24(22):16261. https://doi.org/10.3390/ijms242216261
Chicago/Turabian StyleFurutani, Kazuharu. 2023. "Facilitation of hERG Activation by Its Blocker: A Mechanism to Reduce Drug-Induced Proarrhythmic Risk" International Journal of Molecular Sciences 24, no. 22: 16261. https://doi.org/10.3390/ijms242216261
APA StyleFurutani, K. (2023). Facilitation of hERG Activation by Its Blocker: A Mechanism to Reduce Drug-Induced Proarrhythmic Risk. International Journal of Molecular Sciences, 24(22), 16261. https://doi.org/10.3390/ijms242216261