1. Introduction
As the world population ages, neurodegenerative diseases are increasing. The outcome of these diagnoses varies but can lead to fatality 50% of the time within 15–20 months [
1]. Treatments for neurodegeneration, in particular motor neuron (MN) diseases, are restricted due to the limitations of motor neuron repair and regeneration. Induced pluripotent stem cells (iPSCs) have been proposed as a therapeutic agent due to their ability to successfully differentiate into motor neurons [
2,
3]. However, standard protocols for iPSC differentiation into motor neurons are laborious and can result in mixed populations of motor neuron subtypes [
2,
3,
4,
5]. Therefore, further optimization is needed to leverage iPSC’s therapeutic potential.
iPSC can be programmed into MN lineages using media supplemented with growth factors, pluripotency inhibitors, and other neuron patterning factors [
2]. Alternatively, iPSCs can be genetically reprogrammed to express master regulators of MN development for controlled and rapid progress via neuronal differentiation by reducing or eliminating the intermediate progenitor states [
6,
7]. It has been reported that the expression of three key transcription factors, i.e., NGN2, ISL1, and LHX3, in mouse embryonic stem cells (mESC) is sufficient for the establishment of motor neuron identity [
6,
7,
8]. Neurogenin 2 (NGN2) is normally expressed in neuronal progenitor cells (NPC) to commence neural differentiation and survival programs in the central and peripheral nervous systems, while the LIM Homeobox proteins LHX3 and ISL1 form a hetero-dimer, transcription regulator complex to activate genes that lead to specification of postmitotic neurons [
9,
10]. Previously implemented direct programming strategies have been conducted using lentiviral transduction [
6,
8,
11,
12]. This approach has limited therapeutic application as viral vectors integrate randomly in the genome of iPSCs [
5,
13]. These random integrations can lead to mutagenesis and interference with gene transcription causing genome instability and cancer [
5]. For genetically reprogrammed iPSCs to be considered for therapeutic applications, target specificity must be optimized while off-target effects must be minimized.
CRISPR-Cas9 (clustered regularly interspersed short palindromic repeats) gene editing allows for the necessary high specificity with low rates of off-target effects [
14,
15]. With CRISPR-Cas9-guided events, genes can be inserted, removed, activated, or suppressed at highly specific locations in the genome [
15,
16,
17,
18]. The insertion of exogenous genes into safe harbor sites (SHS) can help prevent genotoxic effects or oncogenic transformation, thereby allowing stable expression of transgenes [
16,
17,
19]. SHS are determined by their location—how far they are positioned from known oncogenes; their functionality—involvement with regulating genes or other important functions; and their accessibility—transcriptional access for editing [
20]. For example, the Rosa26 locus on chromosome 3 is the most widely validated and targeted SHS for genetic editing due to its ubiquitous expression in various tissues and no known function of the expressed sequence tags [
21]. The H11 SHS has no promoter elements and is far from known oncogenes while AAVS1 SHS has an open chromatin structure allowing for the transcription of inserted transgenes, although it was reported to be silenced by DNA methylation in certain cell types [
17,
22,
23].
In this study, we present our findings on CRISPR/Cas9-assisted integration in SHS within the human iPSC genome of ISL1 and LHX3 transgenes under an inducible synthetic promoter. With these modifications, we aimed for rapid and unidirectional MN development from iPSC. Contrary to our expectations, we observed the inhibition of the endogenous transcription factors shaping MN identity and dysregulation of neuronal patterning that impacted MN cholinergic synapse activity.
3. Discussion
A major challenge in the direct application of ESC or iPSC for biomedical regenerative purposes is the limited ability to repair damaged tissue in vivo via targeted differentiation into mature and functional cells. This problem is particularly relevant to regenerative efforts directed at the recovery of damaged motor neurons (MNs). Functional MNs have been derived from human ESC and iPSC in vitro via exposure to complex formulations consisting of neuron patterning molecules, activators of Wnt and Shh pathways, and inhibitors of Notch pathway, supplied at strictly defined concentrations and at narrow time window for the treatment [
2,
3,
4]. To address the challenges of in vivo neuronal differentiation, a revolutionary approach has been proposed by several research groups based on the direct reprogramming of stem cells into MNs via the introduction of recombinant genes encoding transcription master regulators of the MN fate [
6,
7,
9,
11,
12]. In these studies, the transgene cassettes were delivered into ESC or iPSC via retroviral vectors, which creates safety issues for potential clinical use due to the random nature of transgene integration within the recipient genome when using such methods. Here, we mirrored previous methods for a unidirectional transition of iPSC into spinal motor neurons (MNs) via the delivery of
ISL1 and
LHX3 transgenes into well-defined safe harbor sites (H11, ROSA26, and AAVS1) in the human iPSC genome with CRISPR-assisted gene editing techniques. With this method, we attempted to avoid poor outcomes from the interference of the local chromatin structure with the transgene expression and versa vice, and possible interruption of host gene expression by the transgene insertion [
29,
30]. CRISPR gene editing technology has gained popularity for the high precision rates of gene insertion into desired chromosome locus and has found a wide range of applications, including the generation of recombinant cell lines, transgenic animal models, and applications for in vivo gene therapies [
30]. Contemporary CRISPR technology has undergone significant evolution to overcome the natural limitations of first-generation gene editing tools regarding Cas enzyme tolerance to mismatches between the guide RNA and the DNA target. In this study, we have generated recombinant iPSC for direct reprogramming into skeletal MNs by applying high-fidelity recombinant SpCas9 enzyme delivered into target cells as a ribonucleoprotein complex with synthetic single guide RNAs (ssgRNA). This method significantly reduces off-target binding of the Cas9/gRNA complex due to its transiency and significantly lower ribonucleoprotein complex levels compared to methods based on transfection of plasmid DNA encoding for Cas9 and sgRNA. We co-transfected the Cas9/ssgRNA complex with a homology-directed repair (HDR) DNA template (carrier of the transgene cassette), thereby further reducing the probability of introducing deletions and non-specific insertions into the iPSC genome that would normally occur when a non-homologous end joining pathway (NHEJ) is triggered in the absence of HDR. We performed whole genome shotgun sequencing of the CRISPR-modified transgenic iPSC clonal cell lines and found no indels or SNPs in proximity to protospacer adjacent motifs (PAMs) recognized by Cas9 enzyme, indicating that our gene editing approach is safe from introducing cytotoxic off-target mutations or chromosomal rearrangements.
To ensure conditional activation of the transgene master regulators, we inserted
ISL1 and
LHX3 coding sequences downstream of an inducible tetracycline response element (TRE), which is traditionally used for the on-demand activation of gene function [
24]. Here, we report on a phenomenon, previously overlooked by studies applying direct stem cell reprogramming to MNs via transgene expression. Specifically, we demonstrate that the transgene transcription factors (TF) become activated in the modified iPSC prior to the addition of a TRE-inducing agent (doxycycline) and thus established a genetic program committed to the motor neuron fate, although incapable of executing the unidirectional transition into MNs in the absence of extrinsic neuron patterning factors supplied in the differentiation media.
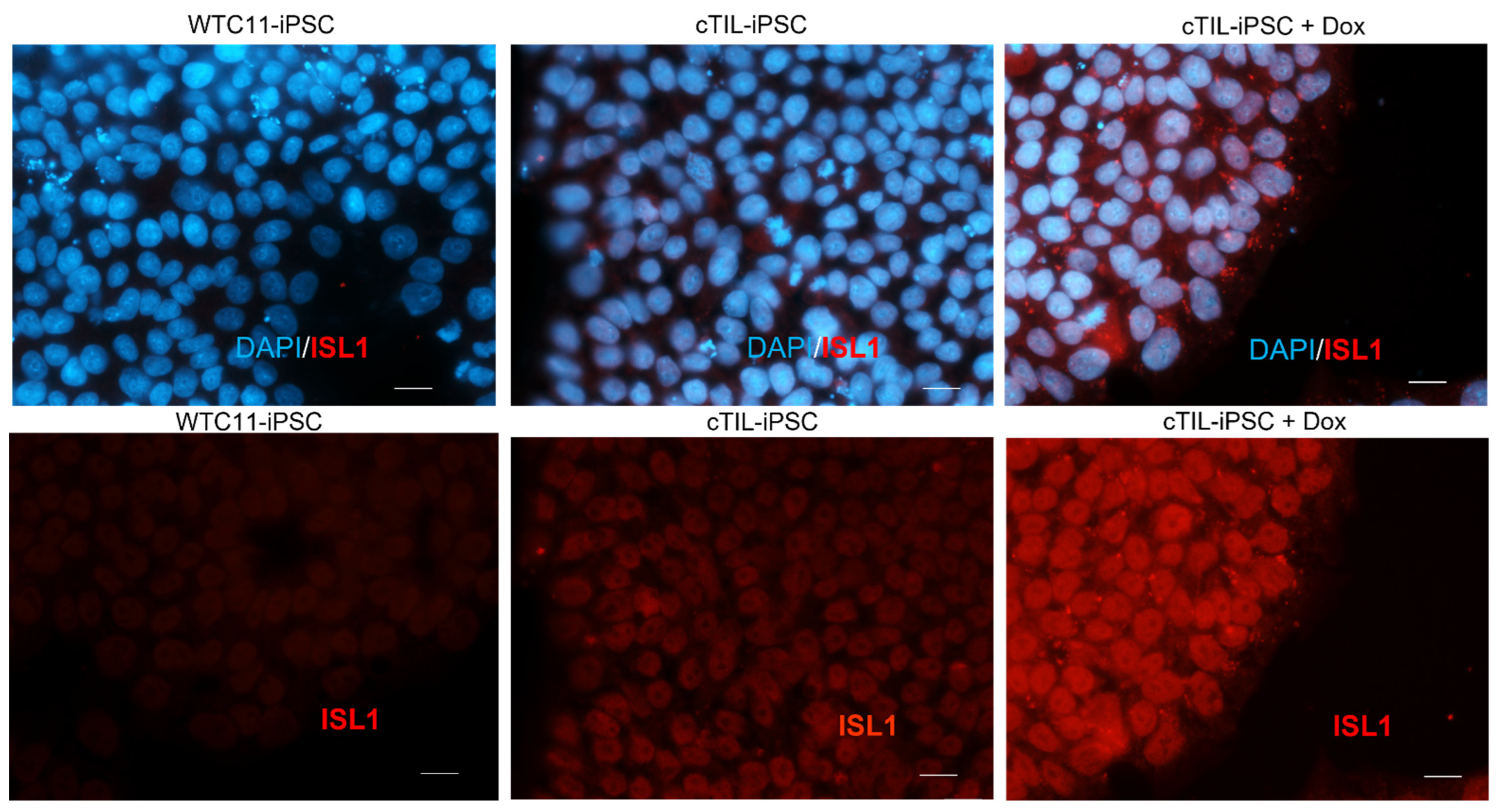
The leaky expression of the TET-On systems has been previously identified as a hindrance to endogenous gene regulations [
31]. Here, we observed the activation of the endogenous
NGN2 gene and accumulation of NGN2 TF into the nucleus of
ISL1-
LHX3 transgene-modified iPSC in the absence of neuronal differentiation stimuli. Normally,
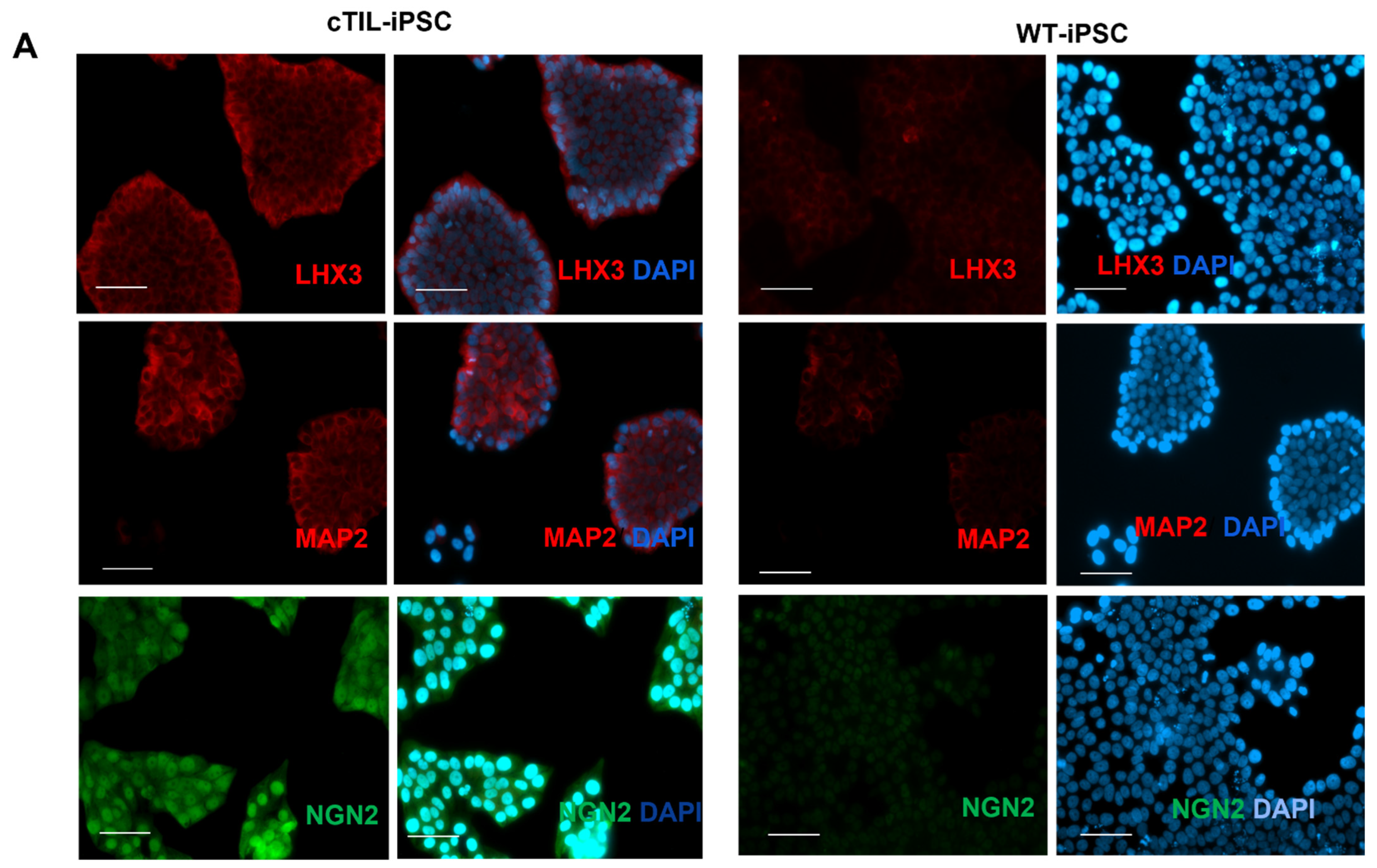
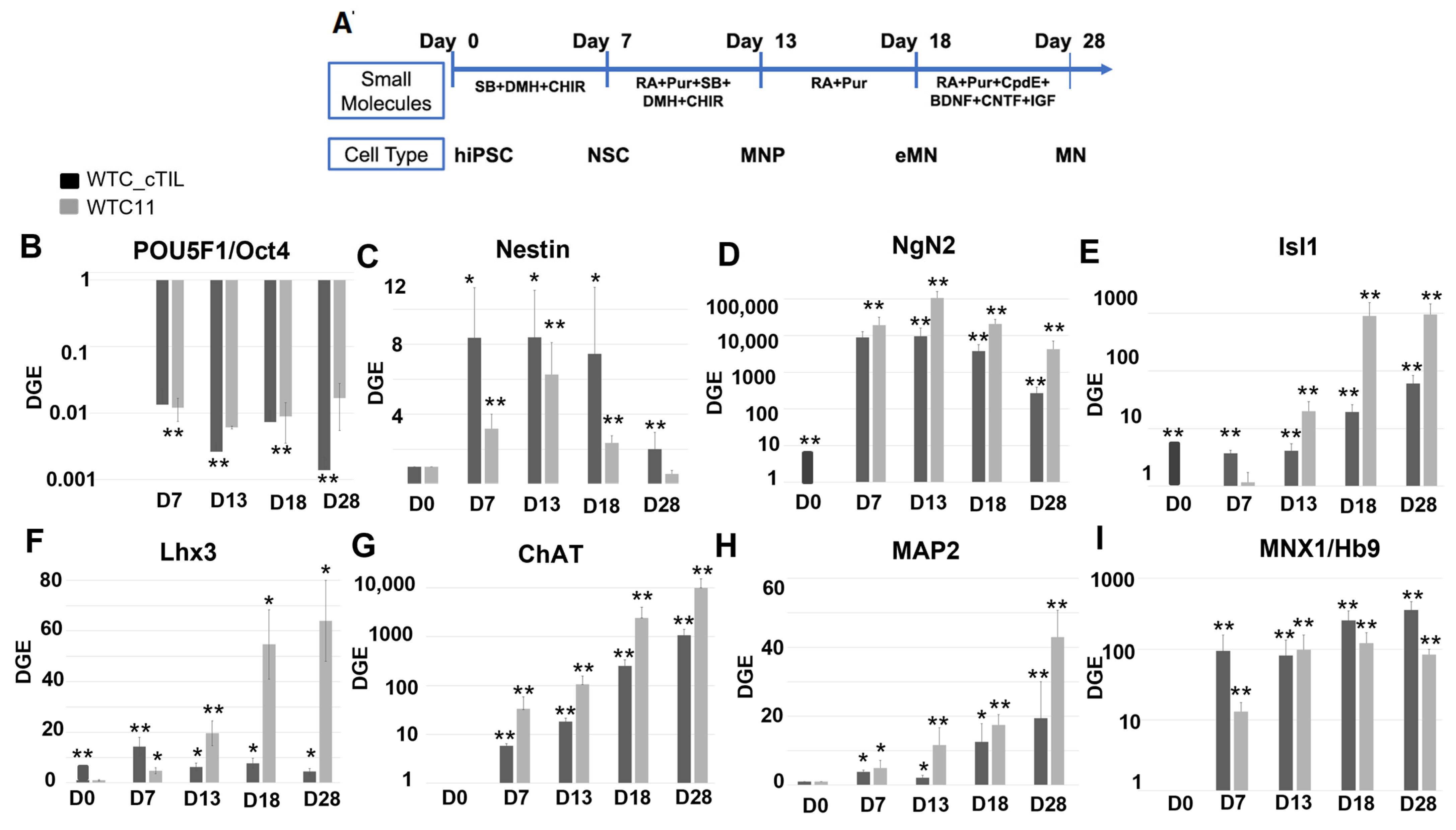
NGN2 is activated in the early stages of neuronal development and downregulated in mature neurons under the program of endogenous ISL1 and LHX3 activation, as shown in
Figure 4 and cited in previous studies [
2,
3]. Consistent with the activation of endogenous
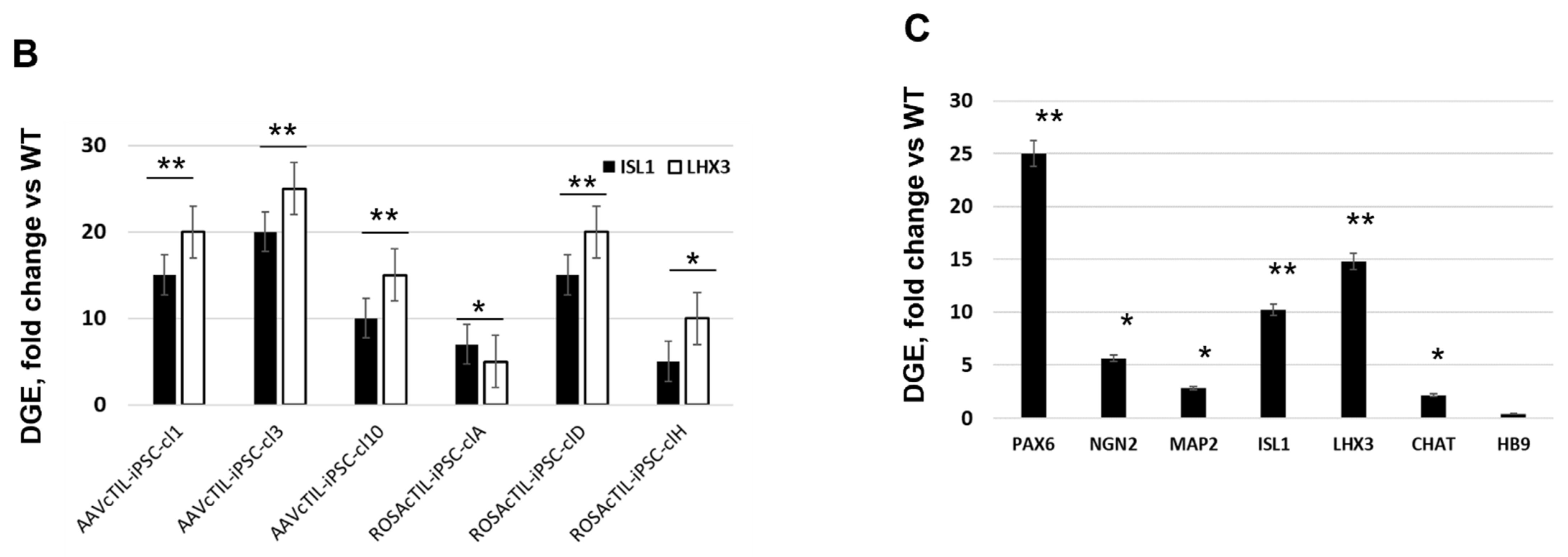
NGN2 (10-fold higher in the
ISL1-
LHX3 modified versus parental iPSC), we registered an increase of the PAX6 transcripts (
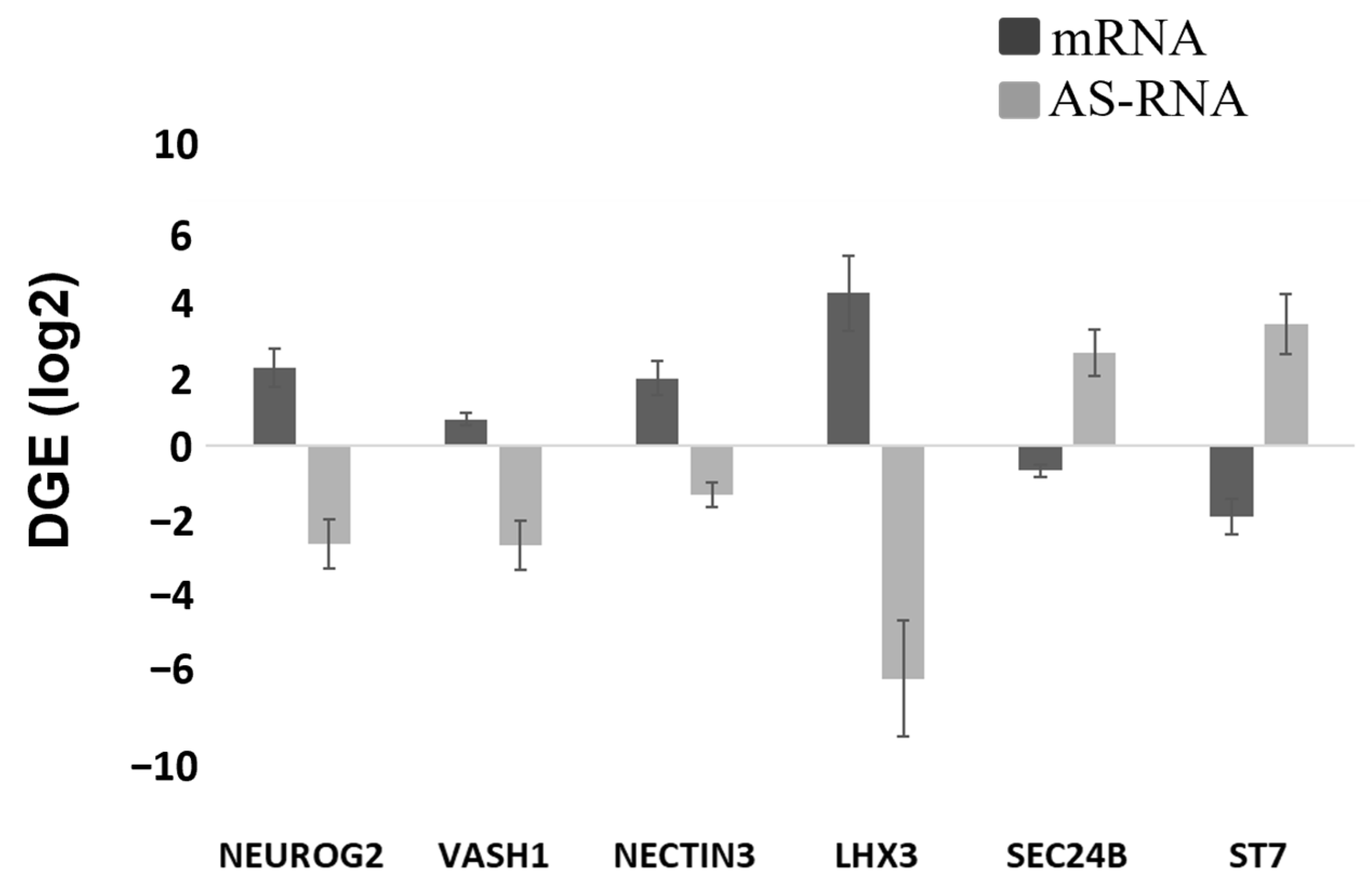
Figure 3C) and a reciprocal decrease in the AS-RNA_Neurog2 levels (
Figure 7), the function of which is to destabilize the NGN2 transcripts. PAX6 is a transcription factor that directly binds to the
NGN2 enhancer element to stimulate NGN2 gene expression in neuronal progenitor cells [
32]. NGN2 inhibits PAX6 expression to allow for transitioning to the next stage of neuronal development [
33]. In this study, we found that endogenous regulatory and functional genes were inhibited throughout the neuron developmental stages of transgene-modified cells (
Figure 4) probably by negative feedback loops (like the NGN2-PAX6 axis), aimed at sustaining the proper function of regulatory circuits in the neuronal cell developmental process. As a result, we observed that the populations of motor neuron progenitors (MNP), expressing key genes which role is to shape the morphology and function of mature skeletal MNs, have substantially declined. As demonstrated in
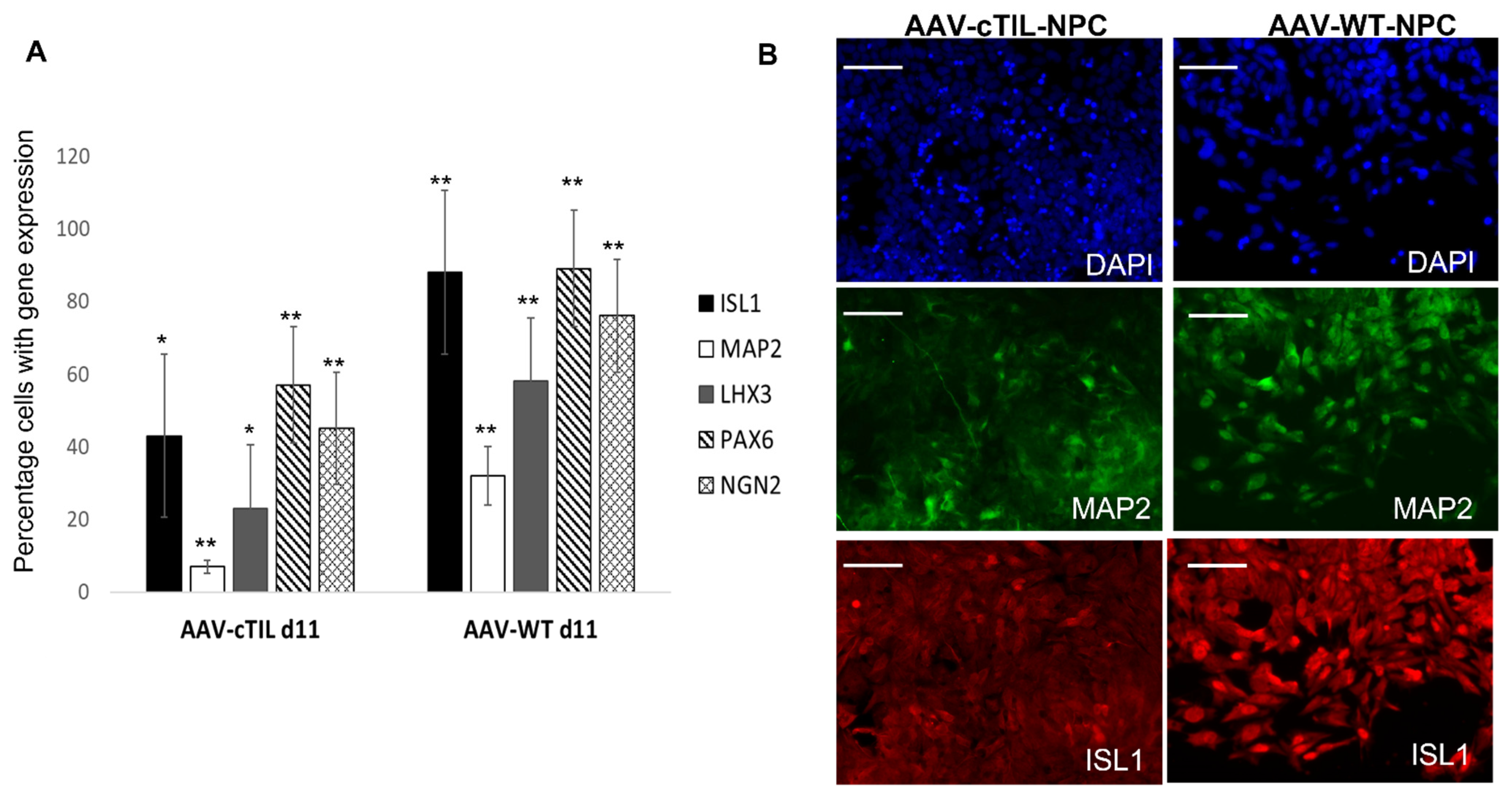
Figure 5, a lesser percentage of cTIL-modified MNP expressed MAP2 compared to their unmodified counterparts, and the ISL1 TF was rarely found in the nucleus of these cells in sharp contrast to the unmodified MNP where on average 90% of the MNP contained activated ISL1 within their nuclei. We attribute this differentiation failure to the premature activation of the
ISL1-
LHX3 transgenes in iPSC causing the dysregulation of endogenous genes responsible for the shaping of neuronal fate.
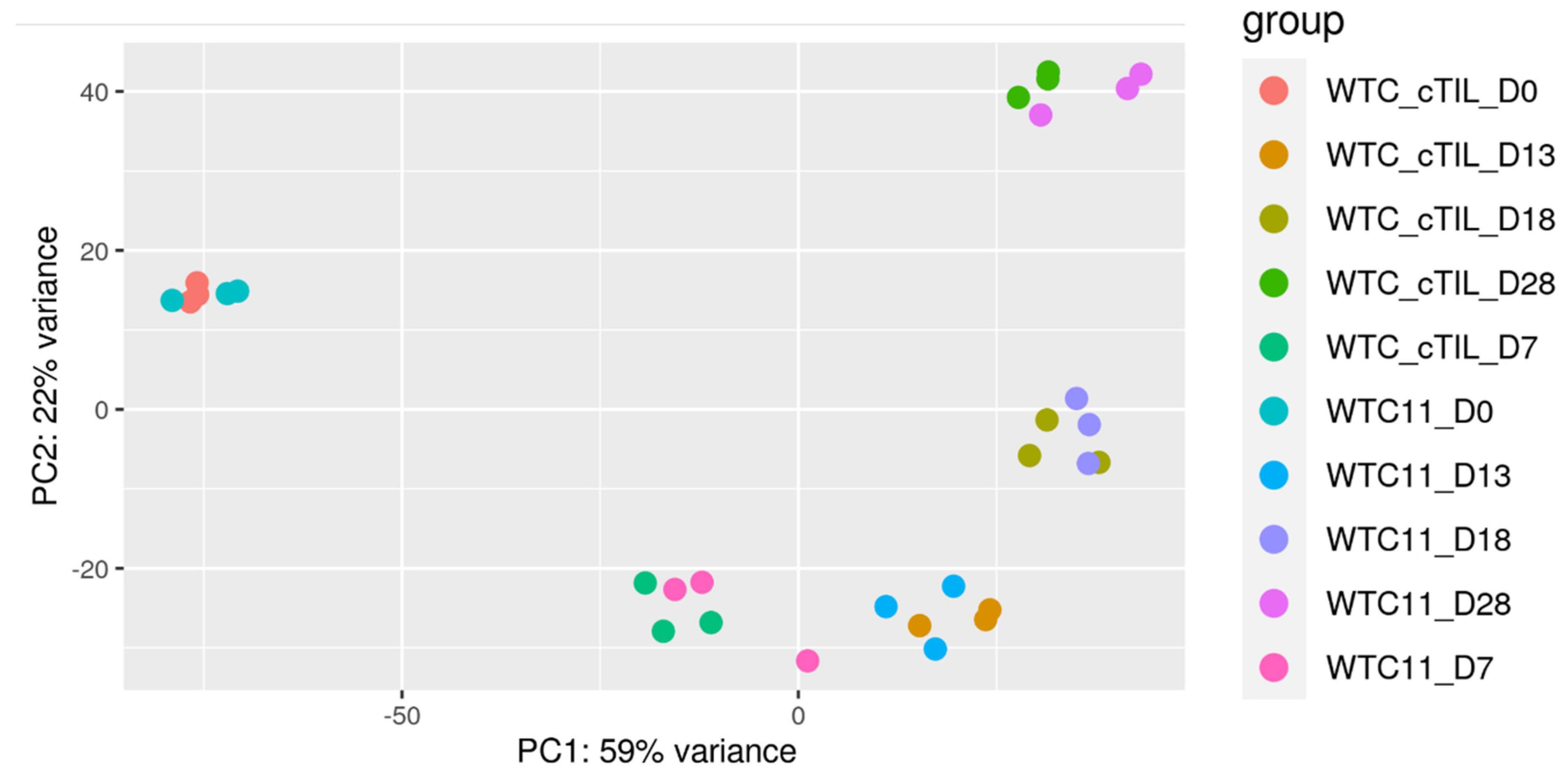
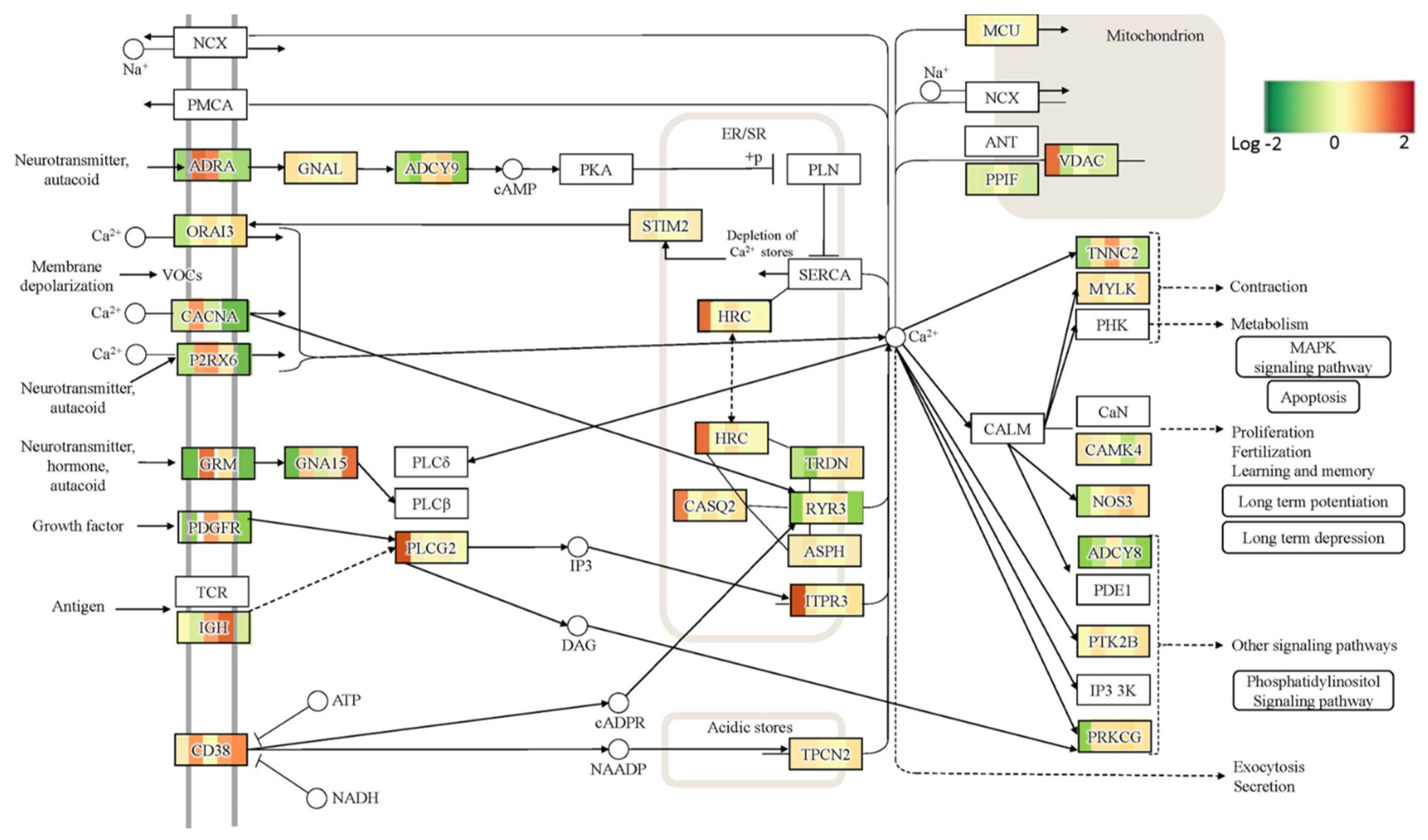
To explore this phenomenon further, we performed a global transcriptome analysis of cTIL-modified iPSCs and populations of NSC, MNP, and early and mature MN derived from these iPSCs. We found that premature activation of ISL1 and LHX3 transgenes in iPSCs altered the gene expression profiles throughout all differentiation stages and affected neuronal development, cell proliferation, and Ca
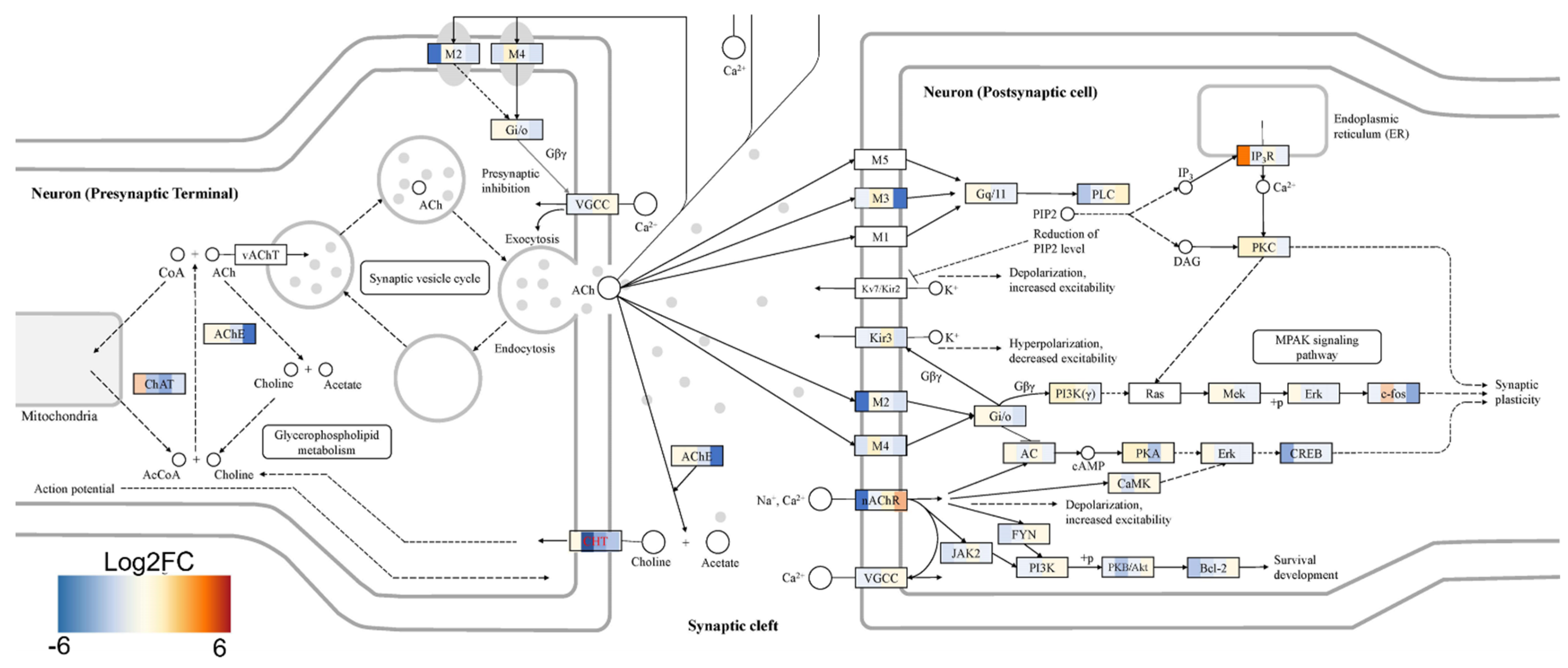
2+ signaling pathways in mature MNs. Specifically, the abnormal global gene expression profiles in the cTIL-modified cells affected the expression of key functional genes in the cholinergic synapse activity responsible for neuronal excitability. Cholinergic synapses utilize acetylcholine (Ach) molecules as the neurotransmitter to trigger electrical signals across the synaptic membranes upon interaction with the Ach receptors (AChR). To prevent overstimulation and fatigue of the cells, Ach is hydrolyzed by Ach esterase enzyme (AChE). Simultaneously, new Ach molecules are synthesized by Choline O-Acetyltransferase (CHAT in the neuronal cells and are released into the synaptic cleft to continue the electro-chemical communication between the neuronal cells and between the neurons and their target organ where the nerve terminals generate cholinergic synapse. Differential gene expression analysis in this study revealed lower transcript levels of AChE, CHAT, muscarinic AChR, Adrenoceptor alpha (ADRA), and high-voltage activated Ca
2+ channels (CACNA) in cTIL-modified compared to wild-type MN populations (
Figure 8 and
Figure 9). We validated these findings using physiological assays evaluating transient calcium currents in mature MNs and electrophysiological activity of spontaneous action potentials across neuronal membranes. In neuronal cells, Ca
2+ flux is fine-tuned by voltage-gated calcium channels, as well as ligand-operated channels, including NMDA, AMPA, and nicotinic acetylcholine receptors (nACHR). Intracellular calcium ions serve as triggers of a multitude of neuronal functions, including metabolic and cell proliferation pathways, induction of activity-dependent synaptic plasticity, and exocytosis of synaptic vesicles loaded with neurotransmitters (summarized in [
34] and
Figure 7). We found that mature MNs, derived from cTIL-modified iPSC, have lower sensitivity to cholinergic neurotransmitter stimulation, more rapid store-operated Ca
2+ entry, and lower retention of intracellular Ca
2+ current compared to unmodified skeletal motor neuron populations (
Figure 10 and
Figure 11). In vivo, such a decrease in MN cholinergic synapse transmissions would translate into muscle weakness or paralysis.
In conclusion, our study demonstrates that in the absence of neuron patterning factors, the overexpression of key transcription factors in human iPSCs using CRISPR technology does not yield the expected unidirectional transition to motor neurons, as previously reported for mouse ESCs. The premature activation of motor neuron genes in iPSCs disrupts the proper neuronal differentiation process, leading to gene dysregulation and lower sensitivity to cholinergic neurotransmitter stimulation. This research sheds light on the complexities of gene regulation and cellular programming, particularly in the context of human iPSCs and their potential for directed differentiation into skeletal motor neurons.
4. Materials and Methods
4.1. Construction of Recombinant Gene Cassettes
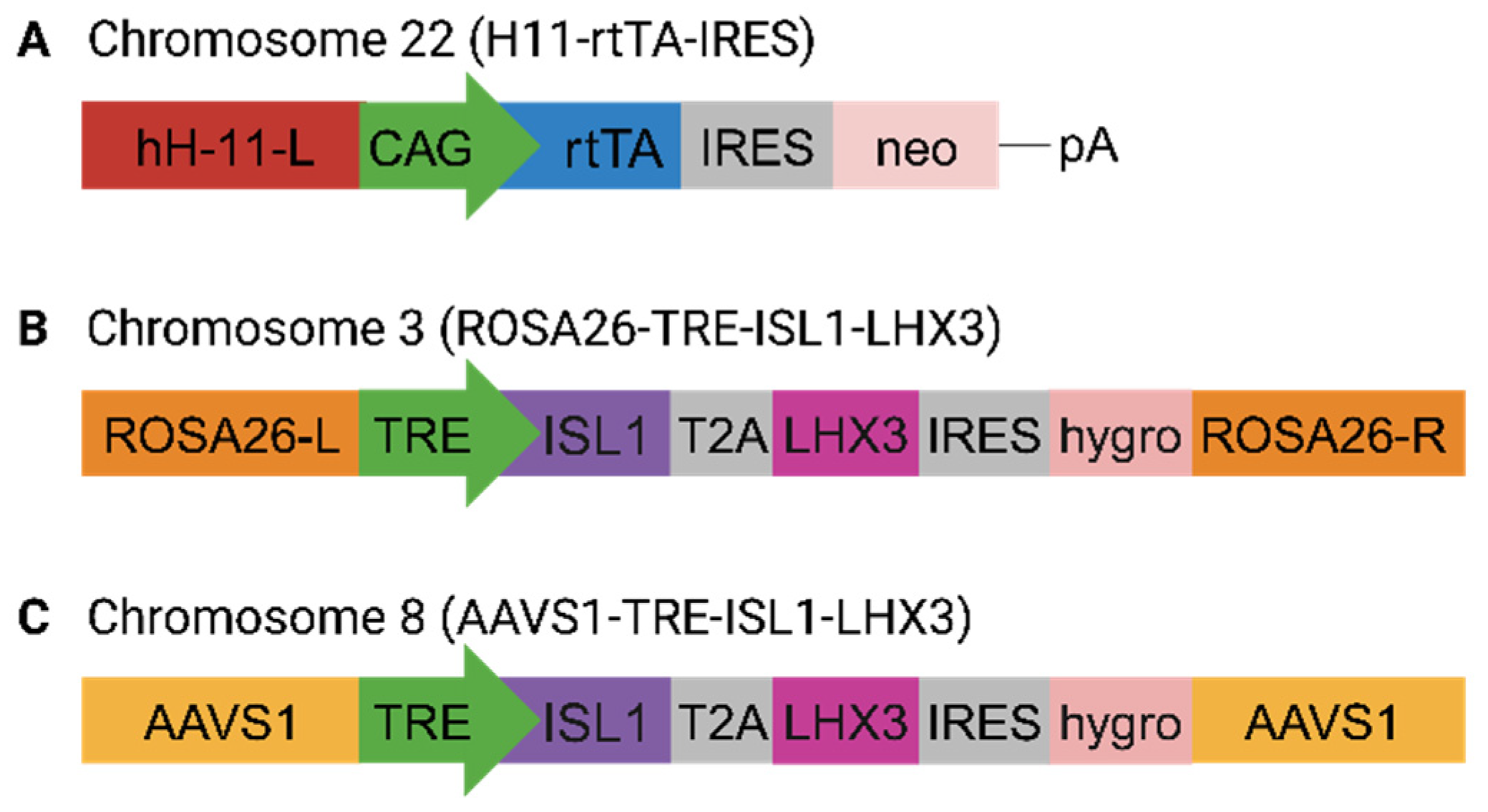
The H11-rtTA-IRES targeting expression was constructed by cloning the coding sequence of rtTA (PCR amplified from Lenti-iCas9-neo, a gift from Qin Yan (Addgene (Watertown, MA, USA) plasmid # 85400;
http://n2t.net/addgene:85400, accessed on 6 November 2023; RRID: Addgene_85400)) into p2attNG-H11-long, a gift from Michele Calos (Addgene plasmid # 51545;
http://n2t.net/addgene:51545, accessed on 6 November 2023; RRID:Addgene_51545). The hROSA26-ISL1-T2A-LHX3 targeting vector was constructed by cloning excised regions of ISL1-T2A-LHX3 from pCSC-ISL1-T2A-LHX3, which was a gift from Chun-Li Zhang (Addgene plasmid # 90215;
http://n2t.net/addgene:90215, accessed on 6 November 2023; RRID: Addgene_90215),and inserted along with a TRE promoter into pMK247 (hROSA26 CMV-MCS-Hygro), which was a gift from Masato Kanemaki (Addgene plasmid # 105926). PCR fragments of the gene templates were linked together by Gibson Assembly Cloning kit (New England Biolabs (NEB), Ipswich, MA, USA). The AAVS1-ISL-T2A-LHX3 targeting vector was constructed by cloning PCR amplified sequences of ISL1-T2A-LHX3 from the pCSC-ISL1-T2A-LHX3 vector, a gift from Chun-Li Zhang (Addgene plasmid # 90215;
http://n2t.net/addgene:90215, accessed on 6 November 2023; RRID: Addgene_90215) The PCR product was inserted along with a TRE promoter into pAAVS1-TLR targeting vector—a gift from Ralf Kuehn (Addgene plasmid # 64215). These fragments were assembled by Gibson Assembly Cloning kit (ThermoFischer Scientific, Waltham, MA, USA).
After each cloning step, the sequences of DNA fragments amplified by PCR were confirmed by Sanger Sequencing on commercial platforms (Eurofins Genomics, Louisville, KY, USA).
4.2. CRISPR/Cas9 Assisted Targeting of Recombinant Cassettes into SHS of iPSC Genome
Lipofectamine™ CRISPRMAX™ Cas9 Transfection Reagent (ThermoFischer Scientific, Waltham, MA, USA) was used according to the manufacturer’s instructions to deliver the targeting vector, Cas9 protein (TrueCut HiFi Cas9, Invitrogen/ThermoFisher), and SHS-specific synthetic guide RNA (sgRNA, Synthego Co., Redwood, CA, USA) to iPSCs cultures. The transfection reaction for 106 iPSC cells in 6-well plate format (70% confluency) consisted of 4μg of TrueCut HiFi Cas9, 1 μg of sgRNA, and 2μg of plasmid DNA of the recombinant plasmid vector. Transfected cells were expanded into a new culture dish (1:6 ratio) 2 days after transfection, and antibiotic selection was applied 4 days after vector delivery to ensure the isolation of clones with an integrated transgene cassette. iPSC colonies surviving antibiotic selection were isolated and expanded, and genomic DNA was isolated to perform PCR confirmation of vector integration. The PCR products were subjected to Sanger Sequencing to confirm transgene integration into the SHS.
4.3. Culturing iPSCs
Human iPSCs (WTC-11, Coriell Institute, Camden, NJ, USA) were cultured and maintained on vitronectin (ThermoFisher Scientific) treated culture plates in mTesR medium (StemCell Technologies, Vancouver, BC, Canada). Differentiation of iPSCs into MNs was directed as previously described by Solomon, 2021. Briefly, iPSCs were cultured in neural media, which consisted of 1:1 DMEM/F12 and Neurobasal medium supplemented with N2, B27, 1× Glutamax and 1×penicillin/streptomycin (all from ThermoFisher Scientific), and 0.1 mM ascorbic acid (StemCell Technology). To maintain the survival of iPSCs during initial seeding, Y-27632 dihydrochloride (Torcis Bioscience, Bristol, UK) was supplemented into media for ~24 h at 1:1000 dilution factor. On days 0–6 3μM of CHIR99021 (StemCell Technology), 2 μM of DMH-1 (Tocris), and 2 μM of SB431542 (StemCell Technology) were added to the neural medium (referred to as NM1); days 6–12 the same media was used with the addition of 0.1 μM of RA (StemCell Technology) and 0.5 μM of Pur (Sigma, St. Louis, MO, USA) referred to as NM2; days 12–18 cells were maintained with 0.1 μM of RA and 0.5 μM of Pur added to the neural media (referred to as NM2); finally, from day 18 onwards, cells were cultured with 0.5 μM of RA, 0.1 μM of Pur, and 0.1 μM of CpdE (StemCell Technology), IGF-1, BDNF, and CNTF (all from R&D Systems, Minneapolis, MN, USA, 10 ng/mL each) referred to as NM4. Accelerated differentiation experiments shortened the exposure time to certain media compositions, as described in the figure legends.
4.4. RNA Extractions
Cells were lifted with accutase, pelleted by centrifugation, and stored at −20 °C. Total RNA was extracted from cell pellets using RNeasy Mini Kit (Qiagen, Hilden, Germany), following the recommendations of the manufacturer. After DNase digestion using a Turbo DNA-free kit (ThermoFisher Scientific), samples were quantified and divided for qPCR and transcriptomic analyses.
4.4.1. RNA-Sequencing (RNA-seq) Analysis
Extracted and DNase-treated RNA was quantified using the Qubit 4 Fluorometer (ThermoFisher) with the High Sensitivity RNA reagents and Bioanalyzer (Agilent, Santa Clara, CA, USA) with RNA 6000 Pico reagents. Ribosomal depletion, DNA conversion, and library preparation were performed on all samples using the Illumina TruSeq Stranded Total RNA kit. A total of 151 base pair reads were sequenced on the Illumina NextSeq. Across fifteen samples (three independent experiments at five time points), the total number of reads generated for each sample ranged from approximately 26 million to 40 million reads. Sequencing data were quality trimmed using FaQCs and a quality score cutoff of Q20. Differential expression analysis was performed using PiReT V 0.3.2 utilizing DEseq2 default parameters and setting a q-value of 0.05 (false discovery rate metric). Human genome version hg38 was used as the reference genome. KEGG pathway mapping was performed using Omics Pathway Viewer—‘OPaver’.
4.4.2. Transcriptome Analysis
DAVID Functional Annotation Bioinformatics Microarray Analysis can be accessed at
https://david.ncifcrf.gov (accessed on 6 November 2023). The raw transcriptomic data of D0-D28 significant DEGs (
p < 0.05) included 2242 upregulated and 1438 downregulated terms, available in the
Supplemental Materials. Each list of Homo sapiens genes was independently analyzed by DAVID to generate an analysis of associated gene ontology (GO) terms. OPaver (Li, unpublished) is a web-based tool to integrate multiple types (e.g., transcriptomics, proteomics, and metabolomics) and time series of data to KEGG biochemical pathways maps. This software analysis tool was developed at Los Alamos National Laboratory. In this case, OPaver was utilized to map significantly differentially expressed genes (
p < 0.05) identified in the DEseq2 analysis performed by PiRet. Differential expression calculation from DEseq2 in Log2 fold change and associated genes (provided in the
Supplementary Materials) were used as input for the OPaver software (
https://gitlab.com/poeli/opaver, accessed on 1 November 2023).
4.4.3. Quantitative Reverse Transcription PCR (RT-qPCR)
Three independent experiments were run in duplicate using a 7500 Fast Real-Time PCR System (Applied Biosciences, Beverly Hill, CA, USA) equipped with QuantStudio Design and Analysis Software v 1.5.2. Fifty ng of each RNA sample were probed for motor neuron differentiation markers using Taqman RNA-to-CT 1-Step Kit (Applied Bioscience) in a 25 μL volume according to the manufacturer’s instructions. Taqman probes included NEUROG2 (Hs00935087_g1), CHAT (Hs00758143_m1), ISL1 (Hs01099686_m1), PAX6 (Hs01088114_m1), MAP2 (Hs00258900_m1), Nestin (Hs04187831_g1), Oct4 (Hs00999632_g1), and HB9 (Hs00907365_m1). Two endogenous control GAPDH (Hs01922876_u1) was used to normalize all qPCR data.
4.5. Immunohistochemistry (IHC) Staining and Single Cell Biomarker Expression Analysis
Immunocytochemistry staining and analysis Nunc Lab-Tek chamber slides (ThermoFisher Scientific, Waltham, MA, USA) were coated with vitronectin, seeded, and subsequently fixed with 4% paraformaldehyde, permeabilized with 0.4% Triton X-100, blocked with 3% BSA in PBS for at least 1 h. Samples were incubated overnight at 4 °C with primary antibody solutions diluted in PBS containing Image-iT FX signal enhancer. Cells were washed with PBS three times prior to incubation with NucBlue Fixed Cell Reagent, Image-iT FX signal enhancer, and secondary Alexa-488-, Alexa 555-, or Alexa 647-conjugated antibodies at 37 °C for 2 h (1:1000 dilution, ThermoFisher Scientific and Jackson ImmunoResearch, West Grove, PA, USA).
Table 1 summarizes the antibodies and their concentrations applied in this study. After three PBS washes, the media chambers were removed from the glass slide, coverslips were mounted using ProLong Diamond Antifade Mountant, and cells were examined using fluorescence microscopy (Zeiss Observer Z.1). For biomarker quantification, images were acquired with an AxioCam camera connected to Axio Observer Z1 microscope, using ZEN 1.1 software. For each culture, images were taken with a 20X objective choosing fields with >100 cells. Images from 5 random fields per culture condition were analyzed with a Cell Profiler pipeline set on nuclei identification with typical diameters of min 8 and max 80-pixel units (manually established to discriminate between single cells and cell clumps). With the Shape function under Cell Profiler, dividing lines between clumped objects were drawn, and an adaptive two-classes threshold strategy was applied. Lower and upper outlier fractions were set at 0.05. The fraction of cells expressing biomarker proteins was calculated as the percent of total cells labeled with DAPI. The following antibodies were applied for biomarker detection: human Islet-1 antibody (PA5-27789, Invitrogen, ThermoFisher Scientific), MAP2 antibody (AP20, Invitrogen, ThermoFisher Scientific), LHX3 antibody (PA5-117410, Invitrogen, ThermoFisher Scientific), PAX6 antibody (D3A9V, Cell Signaling Technology, Danvers, MA, USA), and Neurogenin 2 antibody (D2R3D, Cell Signaling Technology).
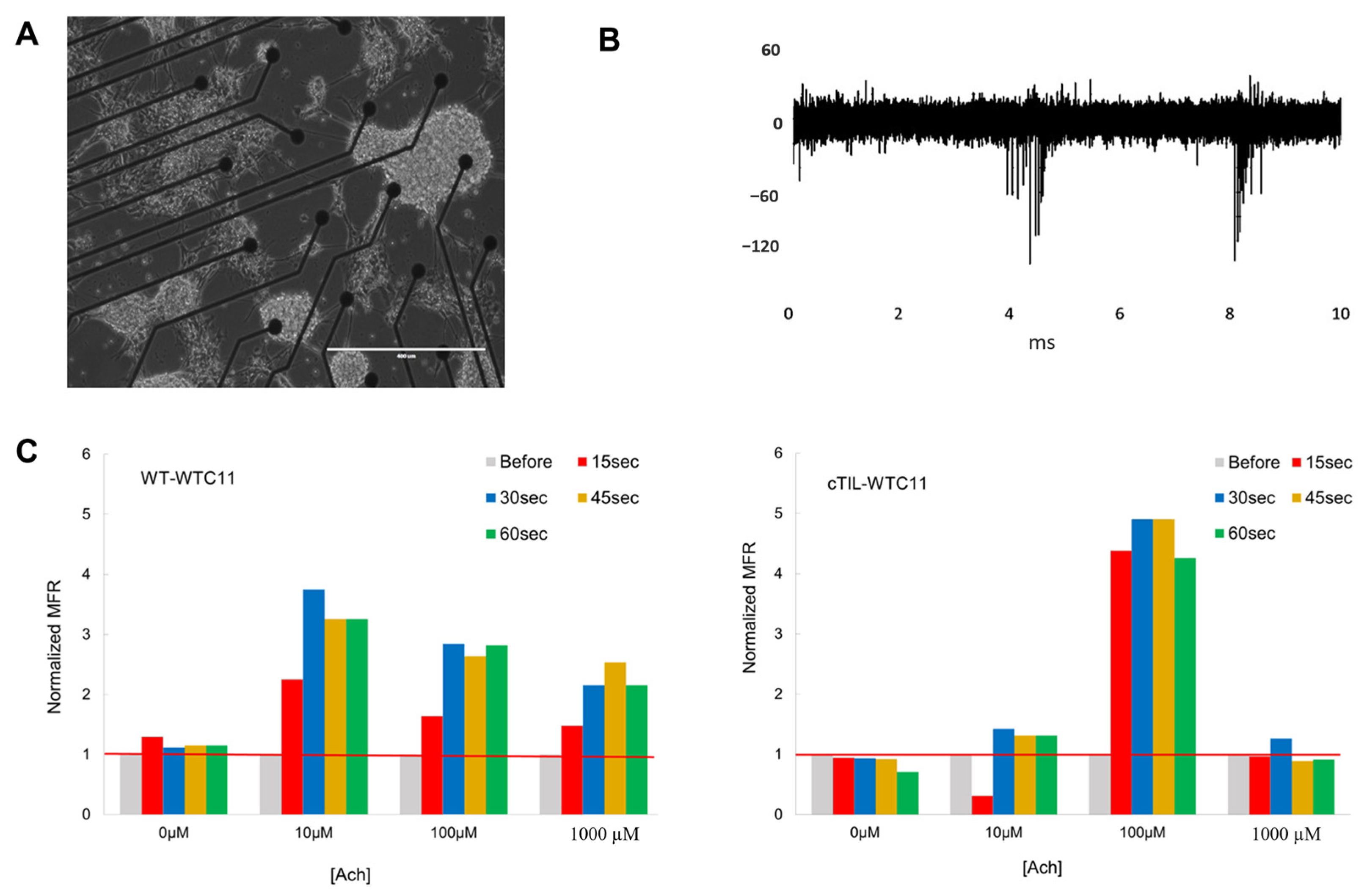
4.6. Functional Analysis of MNs on Microelectrode Array (MEA)
Recording of extracellular action potential from differentiated cTIL-WTC11 and wt-WTC11 using the MEA2100 system (MultiChannel Systems, Kusterdingen, Germany) was performed as previously described [
3]. Briefly, differentiated cells were seeded on poly-D-lysine/vitronectin-coated MEA chips (60MEA200/30iR-Ti arrays or 24 W300/30G-288 (MultiChannel Systems)). The data were collected at a sampling rate of 20 kHz, filtered with a Butterworth band pass, and a threshold was set at 5 × SD. Mean firing rate (MFR) was calculated as the number of spikes per second.
4.7. Calcium Imaging
Cells were seeded in 24-well cell culture plates. They were grown and differentiated as previously described. Intracellular calcium dynamics were visualized using Cal-590 AM dye (AAT Bioquest, Pleasanton, CA, USA). The cells were incubated in 5 µM Cal-590 with 0.04% Pluronic F-127 (AAT Bioquest), 1 mM probenecid (AAT Bioquest) in Hank’s Balanced Salt Solution with 20 mM HEPES Buffer (HHBS, ThermoFisher Scientific, Waltham, MA, USA) for 1 h before they were washed using HHBS buffer supplemented with probenecid. Afterwards, cells were placed on the microscope and fluorescence signals were recorded with a CCD camera at a frame rate of 200 ms/frame before exposure to the neurotransmitter. Acetylcholine was added to the cells at 100 µM, and Cal-590 intensity was immediately recorded for 200 ms. Experiments were performed a minimum of 3 times. Whole imaging region with at least 100 cells was selected for the analysis. Time-lapse imaging data of Cal-590 AM fluorescence intensity (FI), corresponding to intracellular calcium, was recorded using ZEN software 1.1 at 540 nm excitation and 590 nm emission. Data files were exported into Microsoft Excel, where FI was plotted against time. From each graph, calcium flux dynamics post-Ach stimulation were calculated as the slope (5–15 ms of exponentially increasing FI values) or area under the curve (AUC) 5–50 ms of kinetic averages.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}