
Serum-Induced Proliferation of Human Cardiac Stem Cells Is Modulated via TGFβRI/II and SMAD2/3
 ,
, 
Abstract
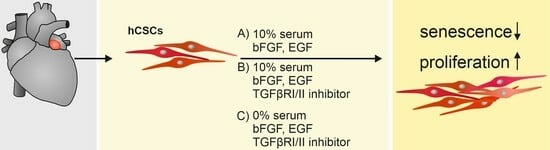
1. Introduction
2. Results
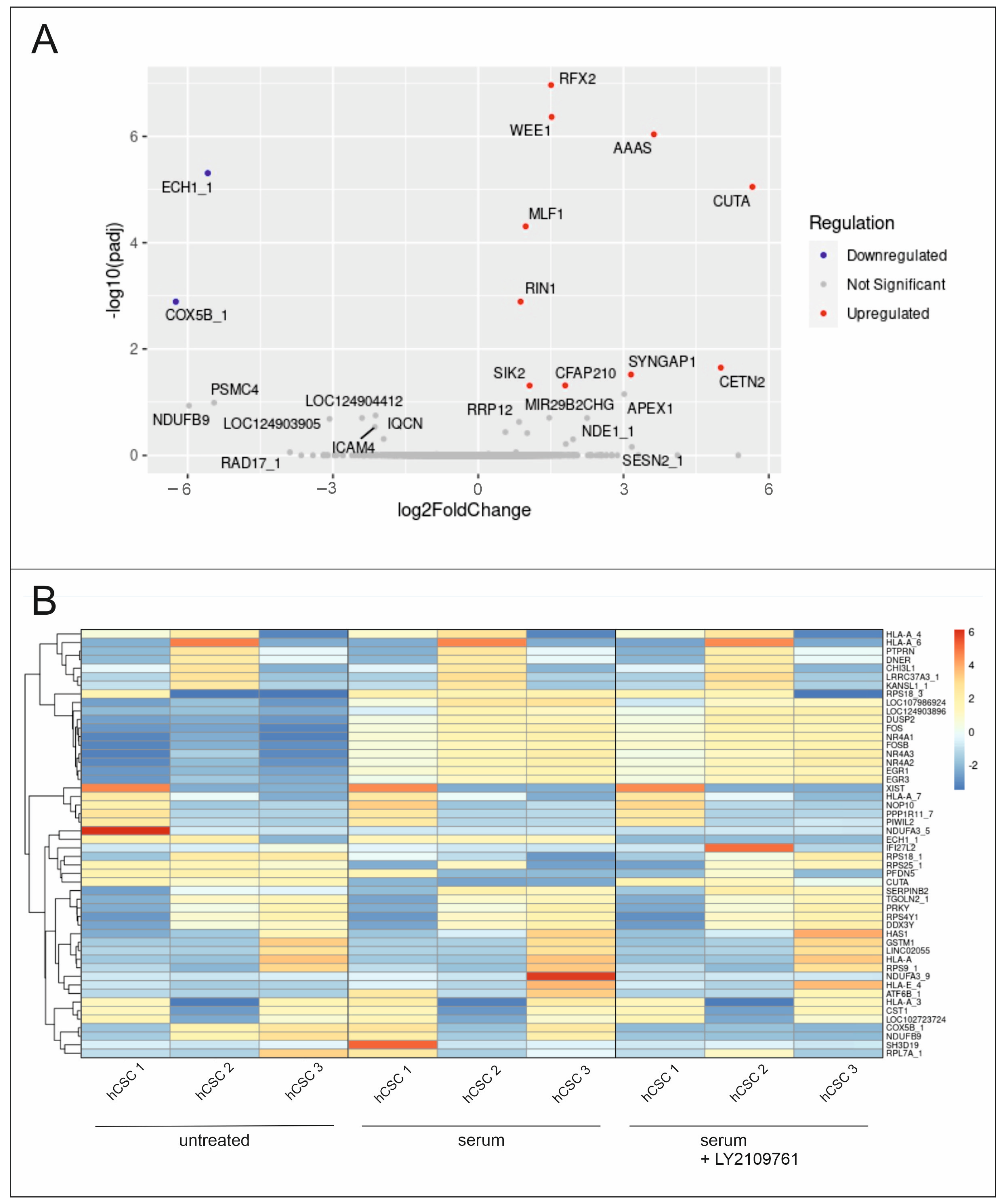
2.1. RNA-Seq Analysis Reveals the Enrichment of Proliferation-Related Processes after Serum Treatment in hCSCs
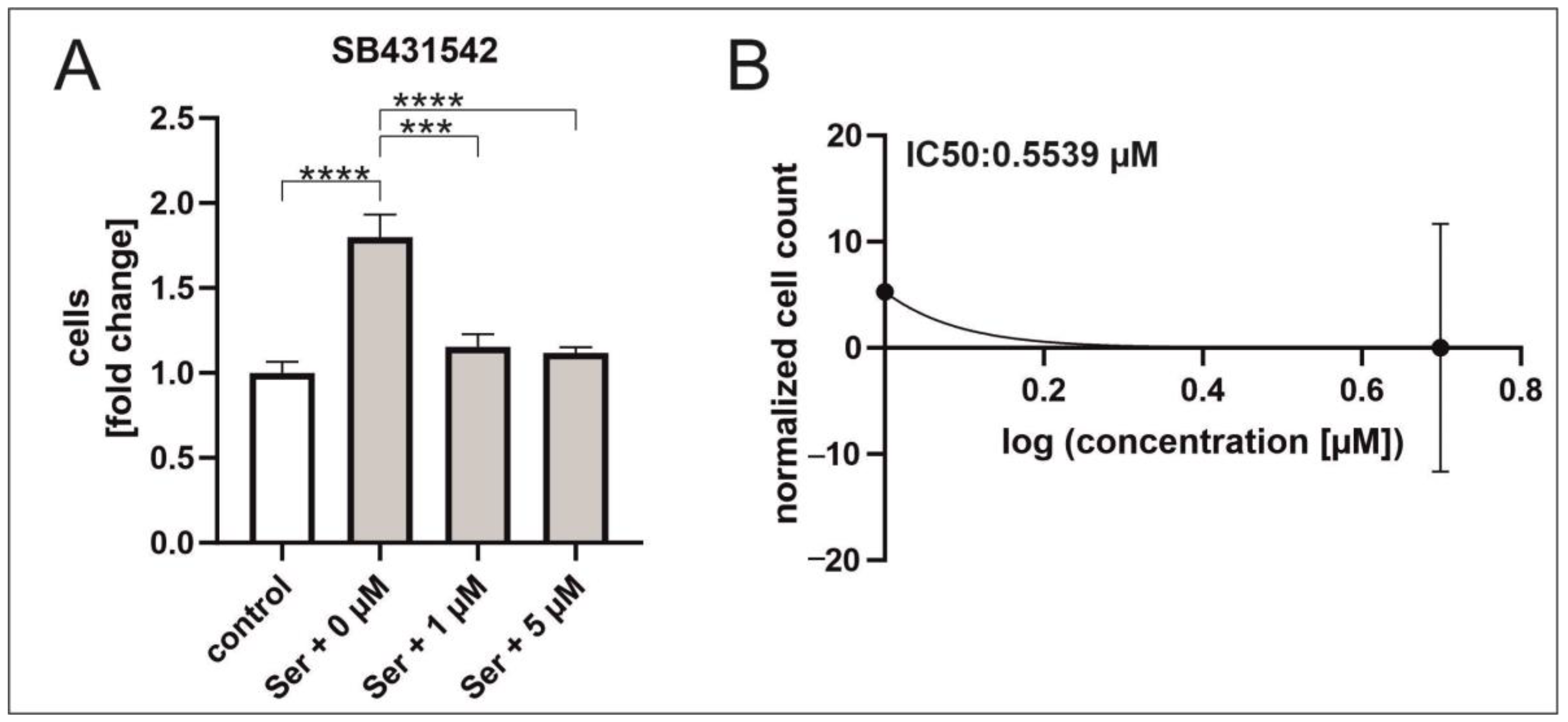
2.2. Inhibition of TGFβRI/II Enhances Proliferation While TGFβ Possesses a Dual Role in Regard to Proliferation and Senescence in hCSCs
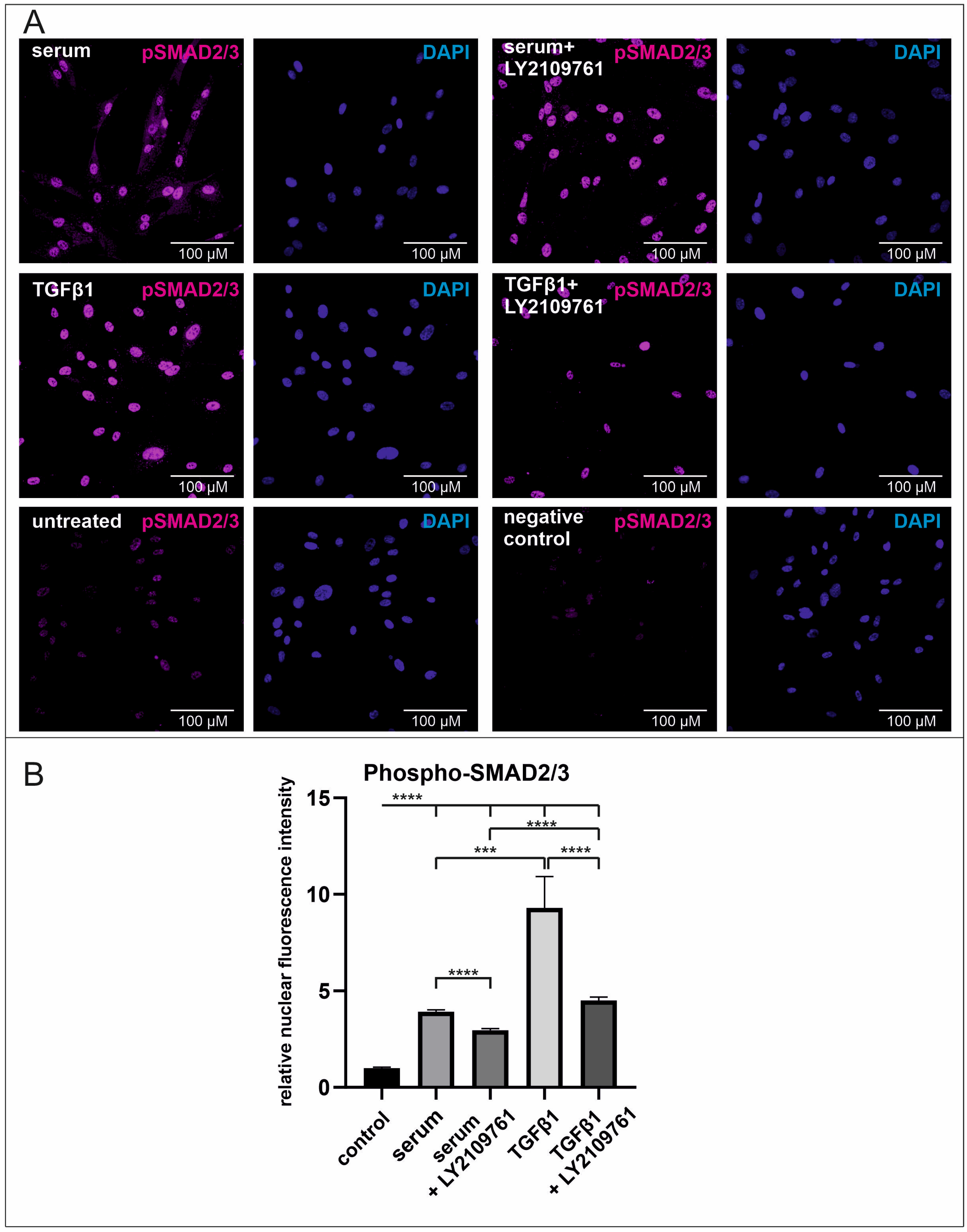
2.3. Human Blood Serum Strongly Induces the Phosphorylation of SMAD2/3 in the Nucleus Which Is Significantly Reduced by the Application of the Selective Inhibitor LY2109761
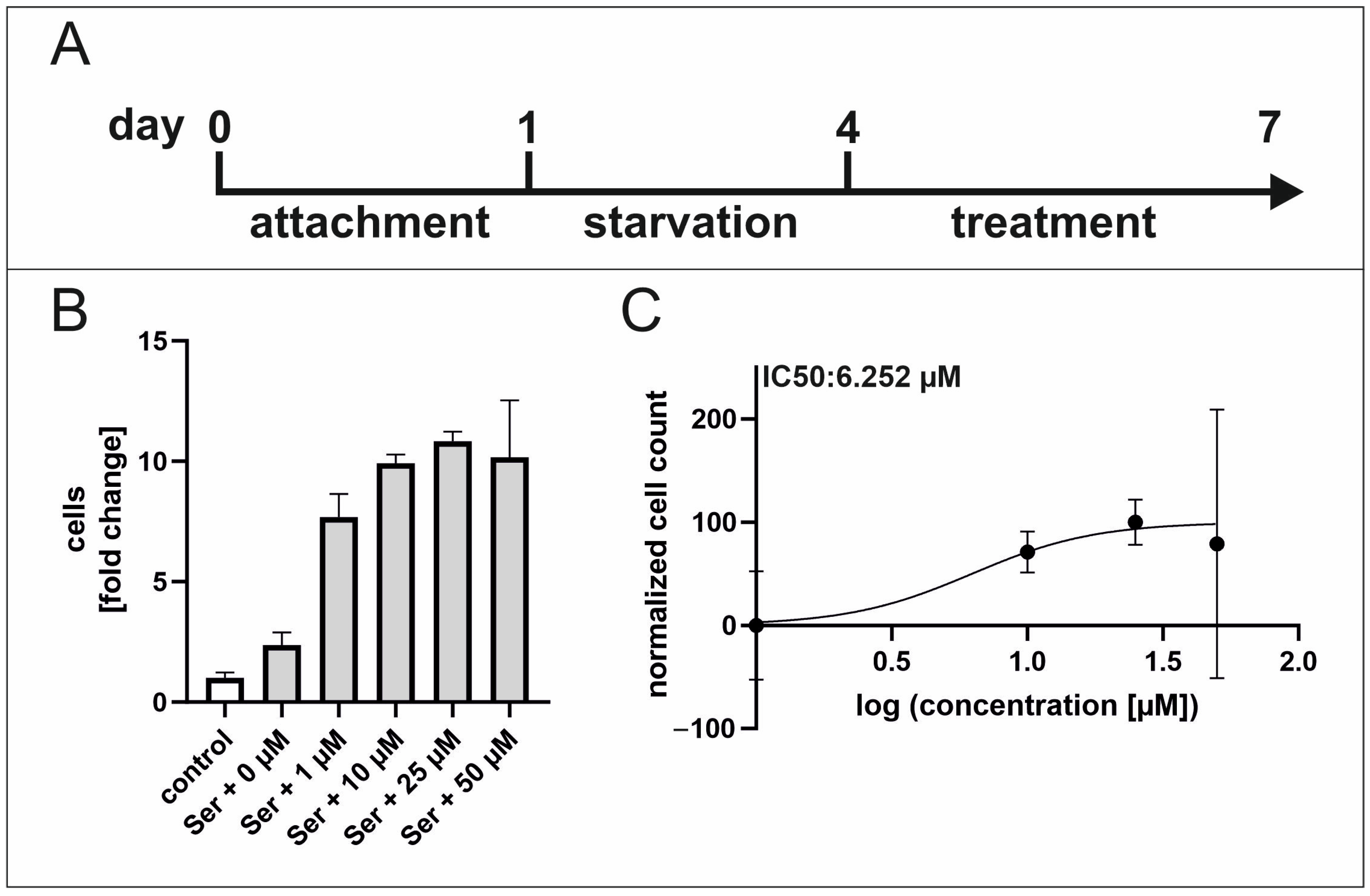
2.4. TGFβ RI Inhibition Reduces Serum-Mediated hCSC Proliferation
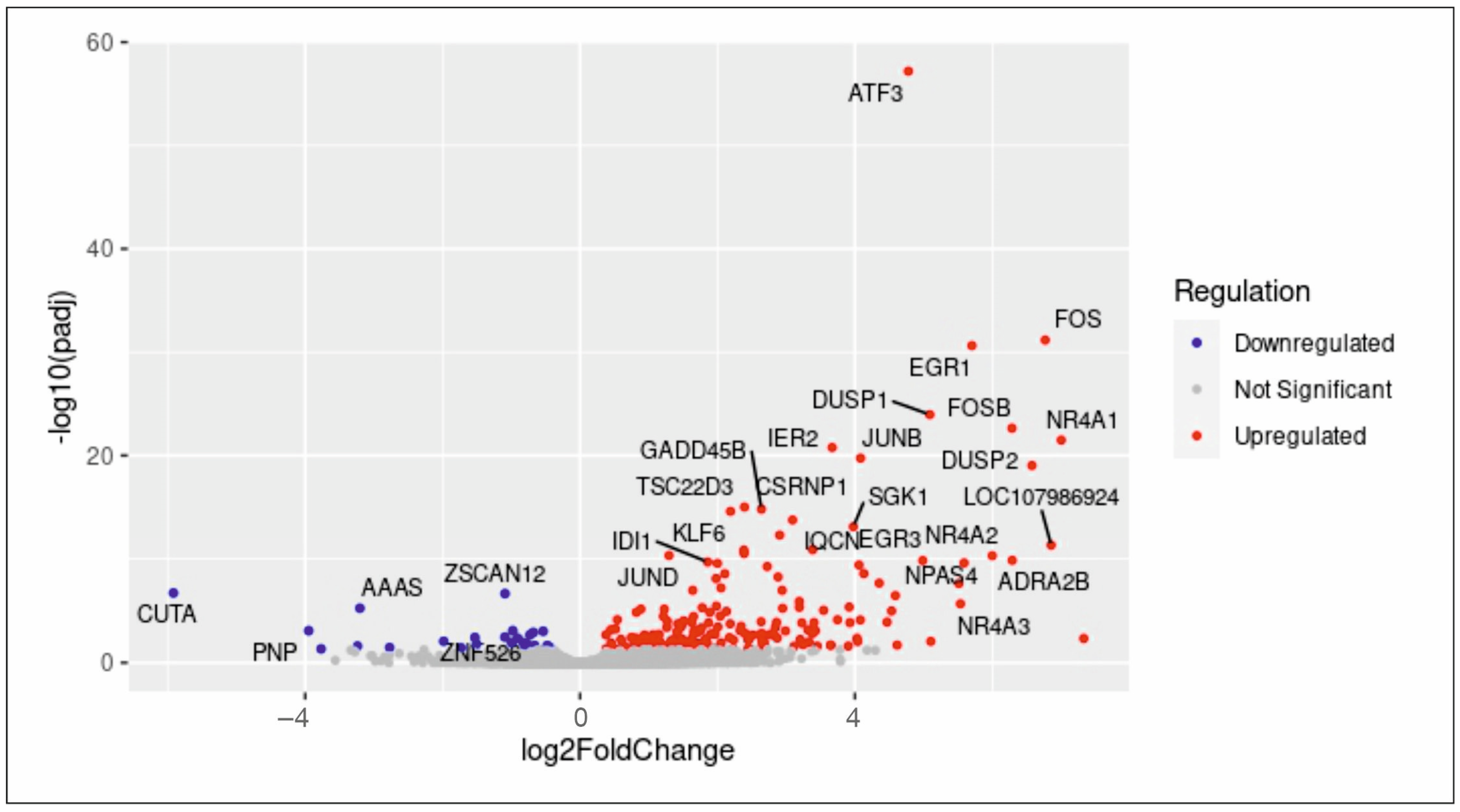
2.5. Differential Global Gene Expression Indicates Upregulation of Proliferation-Related and Ras-Inactivating Genes after TGFβ RI/II Inhibition and Serum Treatment in hCSCs
3. Discussion
4. Materials and Methods
4.1. Isolation and Cultivation of Human Cardiac Stem Cells
4.2. Proliferation Assay
4.3. Senescence-Associated Quantitative β-Galactosidase Assay
4.4. RNA Sequencing and Analysis
4.5. Immunocytochemical Staining
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- He, S.; Nakada, D.; Morrison, S.J. Mechanisms of Stem Cell Self-Renewal. Annu. Rev. Cell Dev. Biol. 2009, 25, 377–406. [Google Scholar] [CrossRef] [PubMed]
- Wagers, A.J.; Weissman, I.L. Plasticity of Adult Stem Cells. Cell 2004, 116, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Cooper, G.M. The Cell, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000; ISBN 978-0-87893-106-4. [Google Scholar]
- Honoki, K. Preventing Aging with Stem Cell Rejuvenation: Feasible or Infeasible? World J. Stem Cells 2017, 9, 1. [Google Scholar] [CrossRef]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult Cardiac Stem Cells Are Multipotent and Support Myocardial Regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Meza-Zepeda, L.A.; Noer, A.; Dahl, J.A.; Micci, F.; Myklebost, O.; Collas, P. High-Resolution Analysis of Genetic Stability of Human Adipose Tissue Stem Cells Cultured to Senescence. J. Cell. Mol. Med. 2008, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Mezey, E.; Key, S.; Vogelsang, G.; Szalayova, I.; Lange, G.D.; Crain, B. Transplanted Bone Marrow Generates New Neurons in Human Brains. Proc. Natl. Acad. Sci. USA 2003, 100, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Murry, C.E. Heart Regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef]
- Aguilar-Sanchez, C.; Michael, M.; Pennings, S. Cardiac Stem Cells in the Postnatal Heart: Lessons from Development. Stem Cells Int. 2018, 2018, 1247857. [Google Scholar] [CrossRef]
- Höving, A.L.; Schmidt, K.E.; Merten, M.; Hamidi, J.; Rott, A.-K.; Faust, I.; Greiner, J.F.W.; Gummert, J.; Kaltschmidt, B.; Kaltschmidt, C.; et al. Blood Serum Stimulates P38-Mediated Proliferation and Changes in Global Gene Expression of Adult Human Cardiac Stem Cells. Cells 2020, 9, 1472. [Google Scholar] [CrossRef]
- Bearzi, C.; Rota, M.; Hosoda, T.; Tillmanns, J.; Nascimbene, A.; De Angelis, A.; Yasuzawa-Amano, S.; Trofimova, I.; Siggins, R.W.; LeCapitaine, N.; et al. Human Cardiac Stem Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 14068–14073. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.M.; van Vliet, P.; Metz, C.H.; Korfage, T.; Sluijter, J.P.; Doevendans, P.A.; Goumans, M.-J. Human Cardiomyocyte Progenitor Cells Differentiate into Functional Mature Cardiomyocytes: An in Vitro Model for Studying Human Cardiac Physiology and Pathophysiology. Nat. Protoc. 2009, 4, 232–243. [Google Scholar] [CrossRef]
- Fuentes, T.; Kearns-Jonker, M. Endogenous Cardiac Stem Cells for the Treatment of Heart Failure. Stem Cells Cloning 2013, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Villeda, S.A.; Plambeck, K.E.; Middeldorp, J.; Castellano, J.M.; Mosher, K.I.; Luo, J.; Smith, L.K.; Bieri, G.; Lin, K.; Berdnik, D.; et al. Young Blood Reverses Age-Related Impairments in Cognitive Function and Synaptic Plasticity in Mice. Nat. Med. 2014, 20, 659–663. [Google Scholar] [CrossRef]
- Conboy, I.M.; Conboy, M.J.; Wagers, A.J.; Girma, E.R.; Weissman, I.L.; Rando, T.A. Rejuvenation of Aged Progenitor Cells by Exposure to a Young Systemic Environment. Nature 2005, 433, 760–764. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Schafer, M.J.; Hooten, N.N.; Atkinson, E.J.; Evans, M.K.; Baker, D.J.; Quarles, E.K.; Robbins, P.D.; Ladiges, W.C.; LeBrasseur, N.K.; et al. Circulating Levels of Monocyte Chemoattractant Protein-1 as a Potential Measure of Biological Age in Mice and Frailty in Humans. Aging Cell 2018, 17, e12706. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, F.S.; Steinhauser, M.L.; Jay, S.M.; Gannon, J.; Pancoast, J.R.; Yalamanchi, P.; Sinha, M.; Dall’Osso, C.; Khong, D.; Shadrach, J.L.; et al. Growth Differentiation Factor 11 Is a Circulating Factor That Reverses Age-Related Cardiac Hypertrophy. Cell 2013, 153, 828–839. [Google Scholar] [CrossRef]
- Höving, A.L.; Schmitz, J.; Schmidt, K.E.; Greiner, J.F.W.; Knabbe, C.; Kaltschmidt, B.; Grünberger, A.; Kaltschmidt, C. Human Blood Serum Induces P38-MAPK- and Hsp27-Dependent Migration Dynamics of Adult Human Cardiac Stem Cells: Single-Cell Analysis via a Microfluidic-Based Cultivation Platform. Biology 2021, 10, 708. [Google Scholar] [CrossRef]
- Roberts, A.B.; Wakefield, L.M. The Two Faces of Transforming Growth Factor β in Carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef]
- Seoane, J. The TGFß Pathway as a Therapeutic Target in Cancer. Clin. Transl. Oncol. 2008, 10, 14–19. [Google Scholar] [CrossRef]
- Moustakas, A.; Pardali, K.; Gaal, A.; Heldin, C.-H. Mechanisms of TGF-β Signaling in Regulation of Cell Growth and Differentiation. Immunol. Lett. 2002, 82, 85–91. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.-F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ Signalling in Context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Suzuki, H.I. TGF-β Signaling in Cellular Senescence and Aging-Related Pathology. Int. J. Mol. Sci. 2019, 20, 5002. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Sheppard, D. TGF-β Signaling in Health and Disease. Cell 2023, 186, 4007–4037. [Google Scholar] [CrossRef]
- Grainger, D.J.; Mosedale, D.E.; Metcalfe, J.C.; Weissberg, P.L.; Kemp, P.R. Active and Acid-Activatable TGF-β in Human Sera, Platelets and Plasma. Clin. Chim. Acta 1995, 235, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β Structure and Activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef]
- Gaussin, V.; Van de Putte, T.; Mishina, Y.; Hanks, M.C.; Zwijsen, A.; Huylebroeck, D.; Behringer, R.R.; Schneider, M.D. Endocardial Cushion and Myocardial Defects after Cardiac Myocyte-Specific Conditional Deletion of the Bone Morphogenetic Protein Receptor ALK3. Proc. Natl. Acad. Sci. USA 2002, 99, 2878–2883. [Google Scholar] [CrossRef]
- Kennedy, K.A.; Porter, T.; Mehta, V.; Ryan, S.D.; Price, F.; Peshdary, V.; Karamboulas, C.; Savage, J.; Drysdale, T.A.; Li, S.-C.; et al. Retinoic Acid Enhances Skeletal Muscle Progenitor Formation and Bypasses Inhibition by Bone Morphogenetic Protein 4 but Not Dominant Negative β-Catenin. BMC Biol. 2009, 7, 67. [Google Scholar] [CrossRef]
- Furtado, M.B.; Solloway, M.J.; Jones, V.J.; Costa, M.W.; Biben, C.; Wolstein, O.; Preis, J.I.; Sparrow, D.B.; Saga, Y.; Dunwoodie, S.L.; et al. BMP/SMAD1 Signaling Sets a Threshold for the Left/Right Pathway in Lateral Plate Mesoderm and Limits Availability of SMAD4. Genes Dev. 2008, 22, 3037–3049. [Google Scholar] [CrossRef]
- Chen, J.-N.; van Eeden, F.J.M.; Warren, K.S.; Chin, A.; Nüsslein-Volhard, C.; Haffter, P.; Fishman, M.C. Left-Right Pattern of Cardiac BMP4 May Drive Asymmetry of the Heart in Zebrafish. Development 1997, 124, 4373–4382. [Google Scholar] [CrossRef]
- Camenisch, T.D.; Molin, D.G.M.; Person, A.; Runyan, R.B.; Gittenberger-de Groot, A.C.; McDonald, J.A.; Klewer, S.E. Temporal and Distinct TGFβ Ligand Requirements during Mouse and Avian Endocardial Cushion Morphogenesis. Dev. Biol. 2002, 248, 170–181. [Google Scholar] [CrossRef]
- Kim, R.Y.; Robertson, E.J.; Solloway, M.J. Bmp6 and Bmp7 Are Required for Cushion Formation and Septation in the Developing Mouse Heart. Dev. Biol. 2001, 235, 449–466. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; D’Amore, P.A. Blood Vessel Formation: What Is Its Molecular Basis? Cell 1996, 87, 1153–1155. [Google Scholar] [CrossRef] [PubMed]
- Pappritz, K.; Savvatis, K.; Koschel, A.; Miteva, K.; Tschöpe, C.; Van Linthout, S. Cardiac (Myo)Fibroblasts Modulate the Migration of Monocyte Subsets. Sci. Rep. 2018, 8, 5575. [Google Scholar] [CrossRef] [PubMed]
- van Nieuwenhoven, F.A.; Turner, N.A. The Role of Cardiac Fibroblasts in the Transition from Inflammation to Fibrosis Following Myocardial Infarction. Vasc. Pharmacol. 2013, 58, 182–188. [Google Scholar] [CrossRef]
- Kang, Y.; Chen, C.-R.; Massagué, J. A Self-Enabling TGFβ Response Coupled to Stress Signaling: Smad Engages Stress Response Factor ATF3 for Id1 Repression in Epithelial Cells. Mol. Cell 2003, 11, 915–926. [Google Scholar] [CrossRef]
- Palumbo-Zerr, K.; Zerr, P.; Distler, A.; Fliehr, J.; Mancuso, R.; Huang, J.; Mielenz, D.; Tomcik, M.; Fürnrohr, B.G.; Scholtysek, C.; et al. Orphan Nuclear Receptor NR4A1 Regulates Transforming Growth Factor-β Signaling and Fibrosis. Nat. Med. 2015, 21, 150–158. [Google Scholar] [CrossRef]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a Novel Transforming Growth Factor Beta Receptor Type I and Type II Dual Inhibitor, as a Therapeutic Approach to Suppressing Pancreatic Cancer Metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef]
- Toma, C.; Pittenger, M.F.; Cahill, K.S.; Byrne, B.J.; Kessler, P.D. Human Mesenchymal Stem Cells Differentiate to a Cardiomyocyte Phenotype in the Adult Murine Heart. Circulation 2002, 105, 93–98. [Google Scholar] [CrossRef]
- Wang, X.-M.; Liu, X.-M.; Wang, Y.; Chen, Z.-Y. Activating Transcription Factor 3 (ATF3) Regulates Cell Growth, Apoptosis, Invasion and Collagen Synthesis in Keloid Fibroblast through Transforming Growth Factor Beta (TGF-Beta)/SMAD Signaling Pathway. Bioengineered 2021, 12, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yue, Z.; Gao, Y.; Jiang, G.; Zeng, F.; Shao, Y.; Huang, J.; Yin, M.; Li, Y. NR4A1 Is Involved in Fibrogenesis in Ovarian Endometriosis. Cell. Physiol. Biochem. 2018, 46, 1078–1090. [Google Scholar] [CrossRef]
- Mihara, K.; Saifeddine, M.; Hollenberg, M.D. Metformin Down-Regulates TGF Beta Signal Transduction and Production of PAR2 N-Terminus Cleaving Protease Activity in an NR4a1 Dependent Manner in a PC3 Prostate Cancer Cell Line. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Arthur, H.M.; Ure, J.; Smith, A.J.H.; Renforth, G.; Wilson, D.I.; Torsney, E.; Charlton, R.; Parums, D.V.; Jowett, T.; Marchuk, D.A.; et al. Endoglin, an Ancillary TGFβ Receptor, Is Required for Extraembryonic Angiogenesis and Plays a Key Role in Heart Development. Dev. Biol. 2000, 217, 42–53. [Google Scholar] [CrossRef]
- Spillmann, F.; Miteva, K.; Pieske, B.; Tschöpe, C.; Van Linthout, S. High-Density Lipoproteins Reduce Endothelial-to-Mesenchymal Transition. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1774–1777. [Google Scholar] [CrossRef]
- Grainger, D.J.; Mosedale, D.E.; Metcalfe, J.C. TGF-β in Blood: A Complex Problem. Cytokine Growth Factor Rev. 2000, 11, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Glick, A.B.; Kulkarni, A.B.; Tennenbaum, T.; Hennings, H.; Flanders, K.C.; O’Reilly, M.; Sporn, M.B.; Karlsson, S.; Yuspa, S.H. Loss of Expression of Transforming Growth Factor Beta in Skin and Skin Tumors Is Associated with Hyperproliferation and a High Risk for Malignant Conversion. Proc. Natl. Acad. Sci. USA 1993, 90, 6076–6080. [Google Scholar] [CrossRef]
- Frippiat, C.; Chen, Q.M.; Zdanov, S.; Magalhaes, J.P.; Remacle, J.; Toussaint, O. Subcytotoxic H2O2 Stress Triggers a Release of Transforming Growth Factor-Beta 1, Which Induces Biomarkers of Cellular Senescence of Human Diploid Fibroblasts. J. Biol. Chem. 2001, 276, 2531–2537. [Google Scholar] [CrossRef]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated Epithelial Cell Senescence in IPF and the Inhibitory Role of SIRT6 in TGF-β-Induced Senescence of Human Bronchial Epithelial Cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef]
- Matsuda, S.; Revandkar, A.; Dubash, T.D.; Ravi, A.; Wittner, B.S.; Lin, M.; Morris, R.; Burr, R.; Guo, H.; Seeger, K.; et al. TGF-β in the Microenvironment Induces a Physiologically Occurring Immune-Suppressive Senescent State. Cell Rep. 2023, 42, 112129. [Google Scholar] [CrossRef]
- Nassar, K.; Grisanti, S.; Tura, A.; Lüke, J.; Lüke, M.; Soliman, M.; Grisanti, S. A TGF-β Receptor 1 Inhibitor for Prevention of Proliferative Vitreoretinopathy. Exp. Eye Res. 2014, 123, 72–86. [Google Scholar] [CrossRef]
- Mu, Y.; Gudey, S.K.; Landström, M. Non-Smad Signaling Pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Pathways in TGF-β Signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Tu, S.; Huang, W.; Huang, C.; Luo, Z.; Yan, X. Contextual Regulation of TGF-β Signaling in Liver Cancer. Cells 2019, 8, 1235. [Google Scholar] [CrossRef]
- Coleman, M.L.; Marshall, C.J.; Olson, M.F. RAS and RHO GTPases in G1-Phase Cell-Cycle Regulation. Nat. Rev. Mol. Cell Biol. 2004, 5, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Keely, P.J. Mechanical Signaling through the Cytoskeleton Regulates Cell Proliferation by Coordinated Focal Adhesion and Rho GTPase Signaling. J. Cell Sci. 2011, 124, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Nicolás, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 Is a Potent and Specific Inhibitor of Transforming Growth Factor-Beta Superfamily Type I Activin Receptor-like Kinase (ALK) Receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Padidam, M.; Cress, D.; Grady, W.M. TGF-β Receptor Levels Regulate the Specificity of Signaling Pathway Activation and Biological Effects of TGF-β. Biochim. Biophys. Acta 2009, 1793, 1165–1173. [Google Scholar] [CrossRef]
- Wagner, K.-D.; Wagner, N. The Senescence Markers p16INK4A, p14ARF/p19ARF, and P21 in Organ Development and Homeostasis. Cells 2022, 11, 1966. [Google Scholar] [CrossRef]
- Grady, W.M.; Willis, J.E.; Trobridge, P.; Romero-Gallo, J.; Munoz, N.; Olechnowicz, J.; Ferguson, K.; Gautam, S.; Markowitz, S.D. Proliferation and Cdk4 Expression in Microsatellite Unstable Colon Cancers with TGFBR2 Mutations. Int. J. Cancer 2006, 118, 600–608. [Google Scholar] [CrossRef]
- Qiu, T.; Wu, X.; Zhang, F.; Clemens, T.L.; Wan, M.; Cao, X. TGF-Beta Type II Receptor Phosphorylates PTH Receptor to Integrate Bone Remodelling Signalling. Nat. Cell Biol. 2010, 12, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Vardouli, L.; Vasilaki, E.; Papadimitriou, E.; Kardassis, D.; Stournaras, C. A Novel Mechanism of TGFbeta-Induced Actin Reorganization Mediated by Smad Proteins and Rho GTPases. FEBS J. 2008, 275, 4074–4087. [Google Scholar] [CrossRef] [PubMed]
- Svensmark, J.H.; Brakebusch, C. Rho GTPases in Cancer: Friend or Foe? Oncogene 2019, 38, 7447–7456. [Google Scholar] [CrossRef]
- Lee, A.J.; Fraser, E.; Flowers, B.; Kim, J.; Wong, K.; Cataisson, C.; Liu, H.; Yang, H.; Lee, M.P.; Yuspa, S.H.; et al. RAS Induced Senescence of Skin Keratinocytes Is Mediated through Rho-Associated Protein Kinase (ROCK). Mol. Carcinog. 2021, 60, 799–812. [Google Scholar] [CrossRef]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-Binding Proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.F.; Marais, R. Ras Protein Signalling. Semin. Immunol. 2000, 12, 63–73. [Google Scholar] [CrossRef]
- Fassl, A.; Geng, Y.; Sicinski, P. CDK4 and CDK6 Kinases: From Basic Science to Cancer Therapy. Science 2022, 375, eabc1495. [Google Scholar] [CrossRef]
- Cobrinik, D. Pocket Proteins and Cell Cycle Control. Oncogene 2005, 24, 2796–2809. [Google Scholar] [CrossRef]
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef]
- Drosten, M.; Dhawahir, A.; Sum, E.Y.M.; Urosevic, J.; Lechuga, C.G.; Esteban, L.M.; Castellano, E.; Guerra, C.; Santos, E.; Barbacid, M. Genetic Analysis of Ras Signalling Pathways in Cell Proliferation, Migration and Survival. EMBO J. 2010, 29, 1091–1104. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS Signalling Pathways in Cancer Therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Bergholz, J.S.; Zhao, J.J. Targeting CDK4 and CDK6 in Cancer. Nat. Rev. Cancer 2022, 22, 356–372. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of P53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Jeyabalan, N.; Clement, J.P. SYNGAP1: Mind the Gap. Front. Cell. Neurosci. 2016, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Funahashi, Y.; Takano, T.; Hossen, E.; Ahammad, R.U.; Tsuboi, D.; Amano, M.; Yamada, K.; Kaibuchi, K. Rho–Rho-Kinase Regulates Ras-ERK Signaling Through SynGAP1 for Dendritic Spine Morphology. Neurochem. Res. 2022, 47, 2757–2772. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, H.; Wang, Q.; Zhou, F.; Liu, Y.; Zhang, Y.; Ding, H.; Yuan, M.; Li, F.; Chen, Y. Involvement of C-Fos in Cell Proliferation, Migration, and Invasion in Osteosarcoma Cells Accompanied by Altered Expression of Wnt2 and Fzd9. PLoS ONE 2017, 12, e0180558. [Google Scholar] [CrossRef] [PubMed]
- McCabe, L.R.; Kockx, M.; Lian, J.; Stein, J.; Stein, G. Selective Expression of Fos- and Jun-Related Genes during Osteoblast Proliferation and Differentiation. Exp. Cell Res. 1995, 218, 255–262. [Google Scholar] [CrossRef]
- Angel, P.; Karin, M. The Role of Jun, Fos and the AP-1 Complex in Cell-Proliferation and Transformation. Biochim. Biophys. Acta (BBA)-Rev. Cancer 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Na, H.-H.; Noh, H.-J.; Cheong, H.-M.; Kang, Y.; Kim, K.-C. SETDB1 Mediated FosB Expression Increases the Cell Proliferation Rate during Anticancer Drug Therapy. BMB Rep. 2016, 49, 238–243. [Google Scholar] [CrossRef]
- Mohan, H.M.; Aherne, C.M.; Rogers, A.C.; Baird, A.W.; Winter, D.C.; Murphy, E.P. Molecular Pathways: The Role of NR4A Orphan Nuclear Receptors in Cancer. Clin. Cancer Res. 2012, 18, 3223–3228. [Google Scholar] [CrossRef]
- Zhao, Y.; Bruemmer, D. NR4A Orphan Nuclear Receptors in Cardiovascular Biology. Drug Discov. Today Dis. Mech. 2009, 6, e43–e48. [Google Scholar] [CrossRef]
- Nomiyama, T.; Nakamachi, T.; Gizard, F.; Heywood, E.B.; Jones, K.L.; Ohkura, N.; Kawamori, R.; Conneely, O.M.; Bruemmer, D. The NR4A Orphan Nuclear Receptor NOR1 Is Induced by Platelet-Derived Growth Factor and Mediates Vascular Smooth Muscle Cell Proliferation. J. Biol. Chem. 2006, 281, 33467–33476. [Google Scholar] [CrossRef]
- Beard, J.A.; Tenga, A.; Chen, T. The Interplay of NR4A Receptors and the Oncogene–Tumor Suppressor Networks in Cancer. Cell Signal. 2015, 27, 257–266. [Google Scholar] [CrossRef]
- Herring, J.A.; Elison, W.S.; Tessem, J.S. Function of Nr4a Orphan Nuclear Receptors in Proliferation, Apoptosis and Fuel Utilization Across Tissues. Cells 2019, 8, 1373. [Google Scholar] [CrossRef]
- Biesiada, E.; Razandi, M.; Levin, E.R. Egr-1 Activates Basic Fibroblast Growth Factor Transcription: Mechanistic Implications for Astrocyte Proliferation. J. Biol. Chem. 1996, 271, 18576–18581. [Google Scholar] [CrossRef]
- Mayer, S.I.; Rössler, O.G.; Endo, T.; Charnay, P.; Thiel, G. Epidermal-Growth-Factor-Induced Proliferation of Astrocytes Requires Egr Transcription Factors. J. Cell Sci. 2009, 122, 3340–3350. [Google Scholar] [CrossRef]
- Sun, T.; Tian, H.; Feng, Y.-G.; Zhu, Y.-Q.; Zhang, W.-Q. Egr-1 Promotes Cell Proliferation and Invasion by Increasing β-Catenin Expression in Gastric Cancer. Dig. Dis. Sci. 2013, 58, 423–430. [Google Scholar] [CrossRef]
- Santiago, F.S.; Atkins, D.G.; Khachigian, L.M. Vascular Smooth Muscle Cell Proliferation and Regrowth after Mechanical Injury in Vitro Are. Egr-1/NGFI-A-Dependent. Am. J. Pathol. 1999, 155, 897–905. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | TGFβ1 [ng/mL] |
|---|---|
| Serum 1 | 0.39 |
| Serum 2 | 0.84 |
| Serum 3 | 1.10 |
| Serum 4 | 1.17 |
| Serum 5 | 0.93 |
| Serum 6 | 1.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, K.E.; Höving, A.L.; Kiani Zahrani, S.; Trevlopoulou, K.; Kaltschmidt, B.; Knabbe, C.; Kaltschmidt, C. Serum-Induced Proliferation of Human Cardiac Stem Cells Is Modulated via TGFβRI/II and SMAD2/3. Int. J. Mol. Sci. 2024, 25, 959. https://doi.org/10.3390/ijms25020959
Schmidt KE, Höving AL, Kiani Zahrani S, Trevlopoulou K, Kaltschmidt B, Knabbe C, Kaltschmidt C. Serum-Induced Proliferation of Human Cardiac Stem Cells Is Modulated via TGFβRI/II and SMAD2/3. International Journal of Molecular Sciences. 2024; 25(2):959. https://doi.org/10.3390/ijms25020959
Chicago/Turabian StyleSchmidt, Kazuko E., Anna L. Höving, Sina Kiani Zahrani, Katerina Trevlopoulou, Barbara Kaltschmidt, Cornelius Knabbe, and Christian Kaltschmidt. 2024. "Serum-Induced Proliferation of Human Cardiac Stem Cells Is Modulated via TGFβRI/II and SMAD2/3" International Journal of Molecular Sciences 25, no. 2: 959. https://doi.org/10.3390/ijms25020959
APA StyleSchmidt, K. E., Höving, A. L., Kiani Zahrani, S., Trevlopoulou, K., Kaltschmidt, B., Knabbe, C., & Kaltschmidt, C. (2024). Serum-Induced Proliferation of Human Cardiac Stem Cells Is Modulated via TGFβRI/II and SMAD2/3. International Journal of Molecular Sciences, 25(2), 959. https://doi.org/10.3390/ijms25020959