
Sphingosine-1-Phosphate Receptor 3 Induces Endothelial Barrier Loss via ADAM10-Mediated Vascular Endothelial-Cadherin Cleavage

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
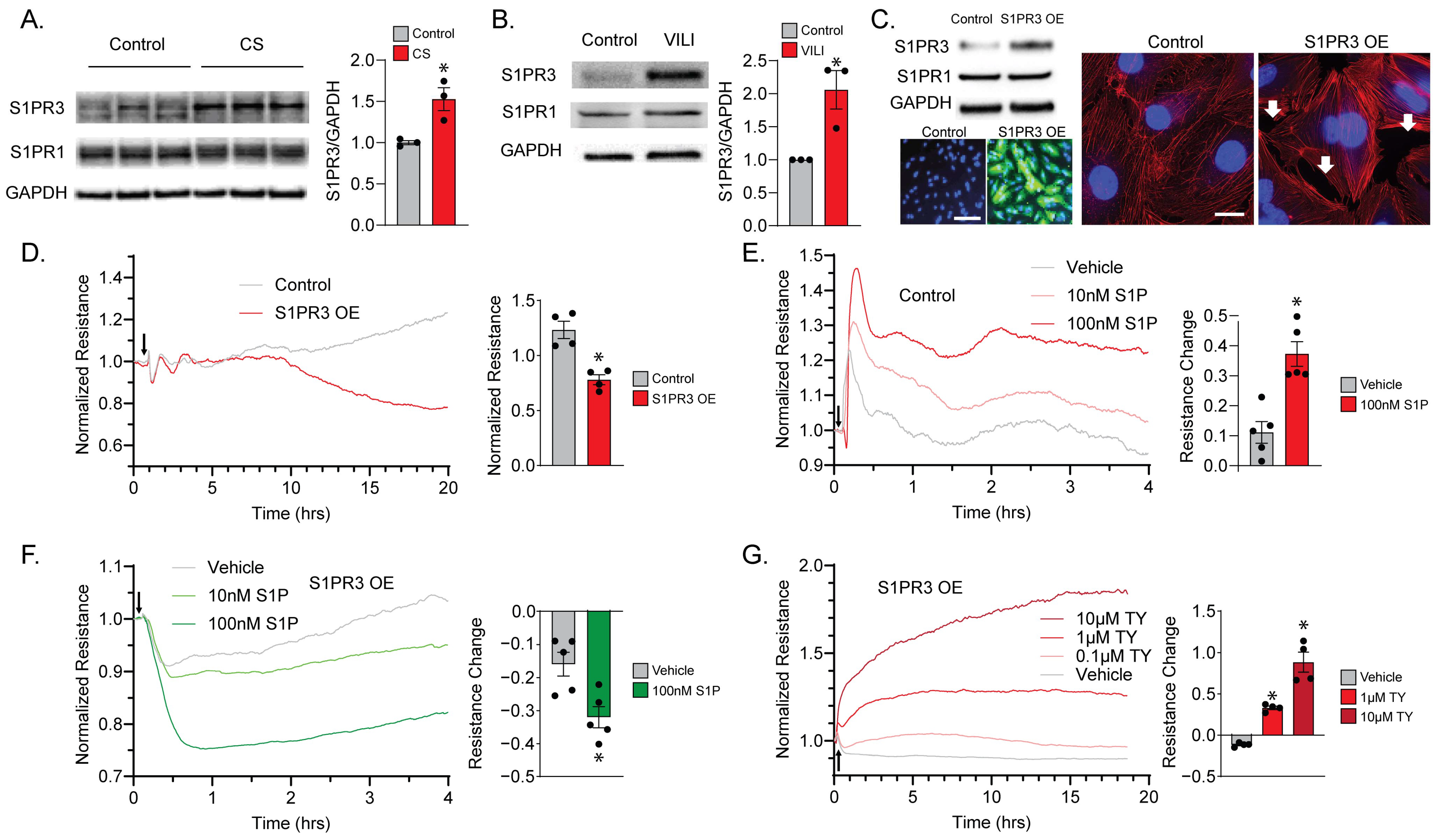
2.1. S1PR3 Overexpression (OE) Remodels Endothelial Barrier Regulation via S1P
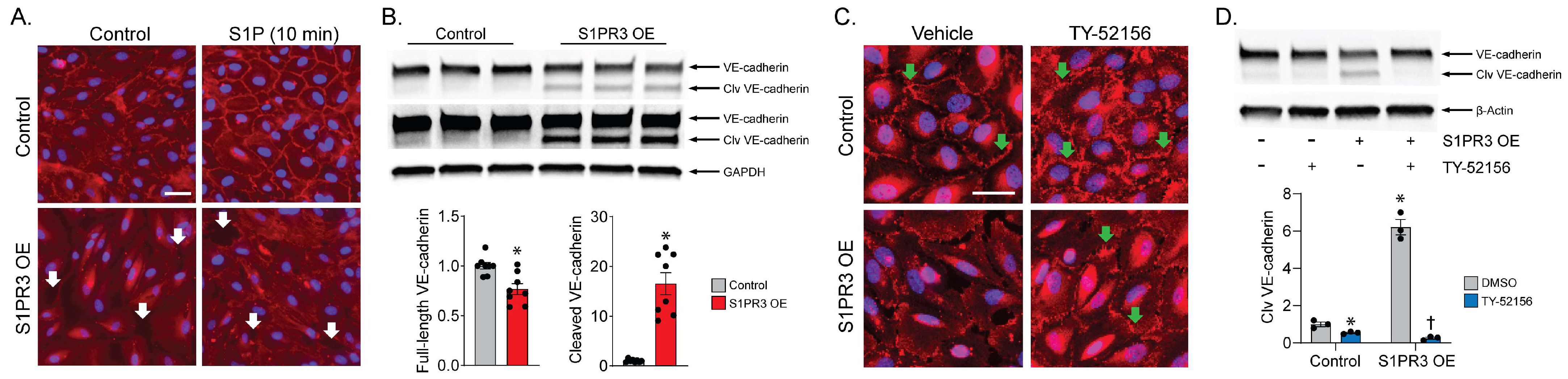
2.2. S1PR3 OE Induces VE-Cadherin Cleavage in PECs
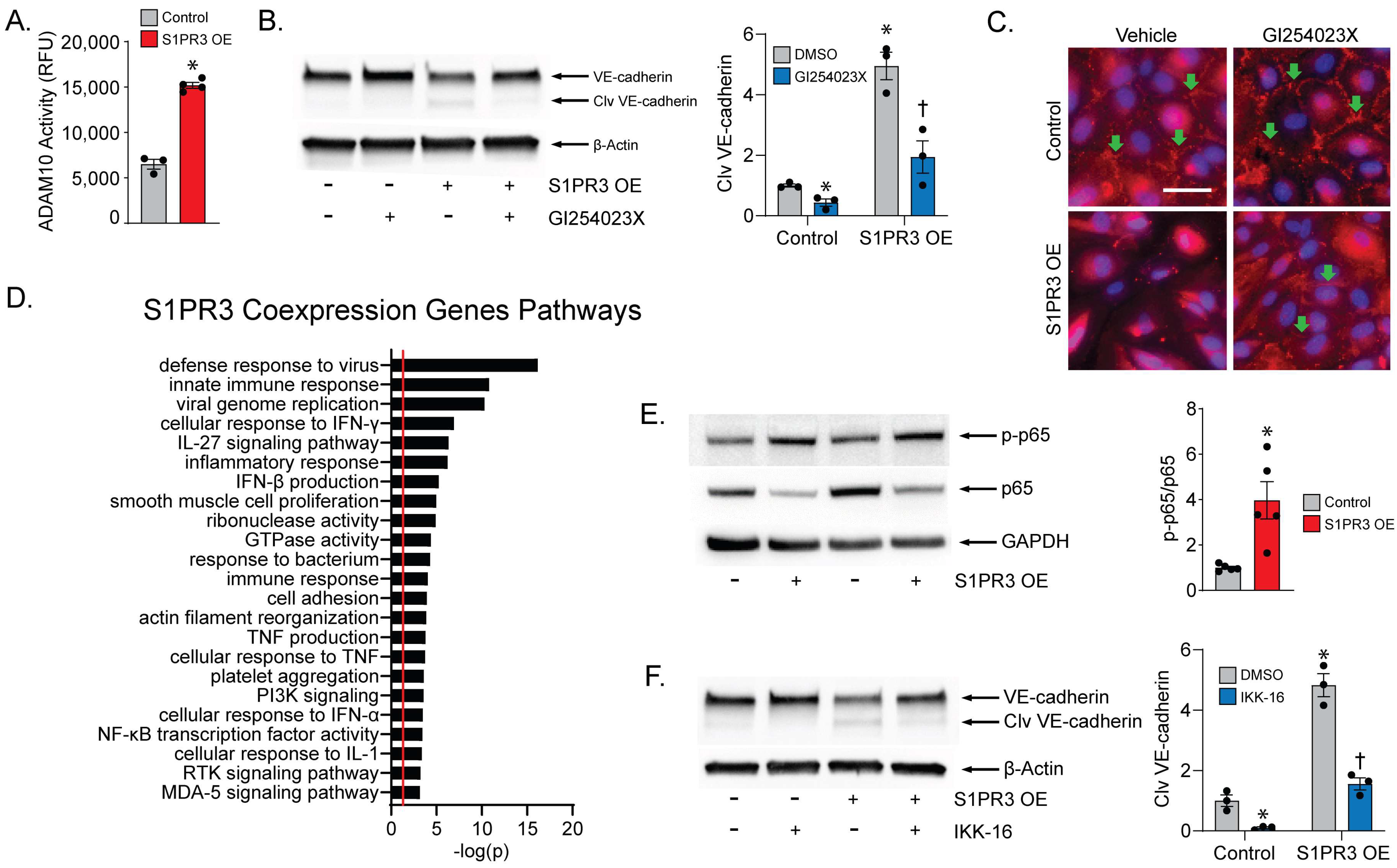
2.3. ADAM10 Mediates VE-Cadherin Cleavage
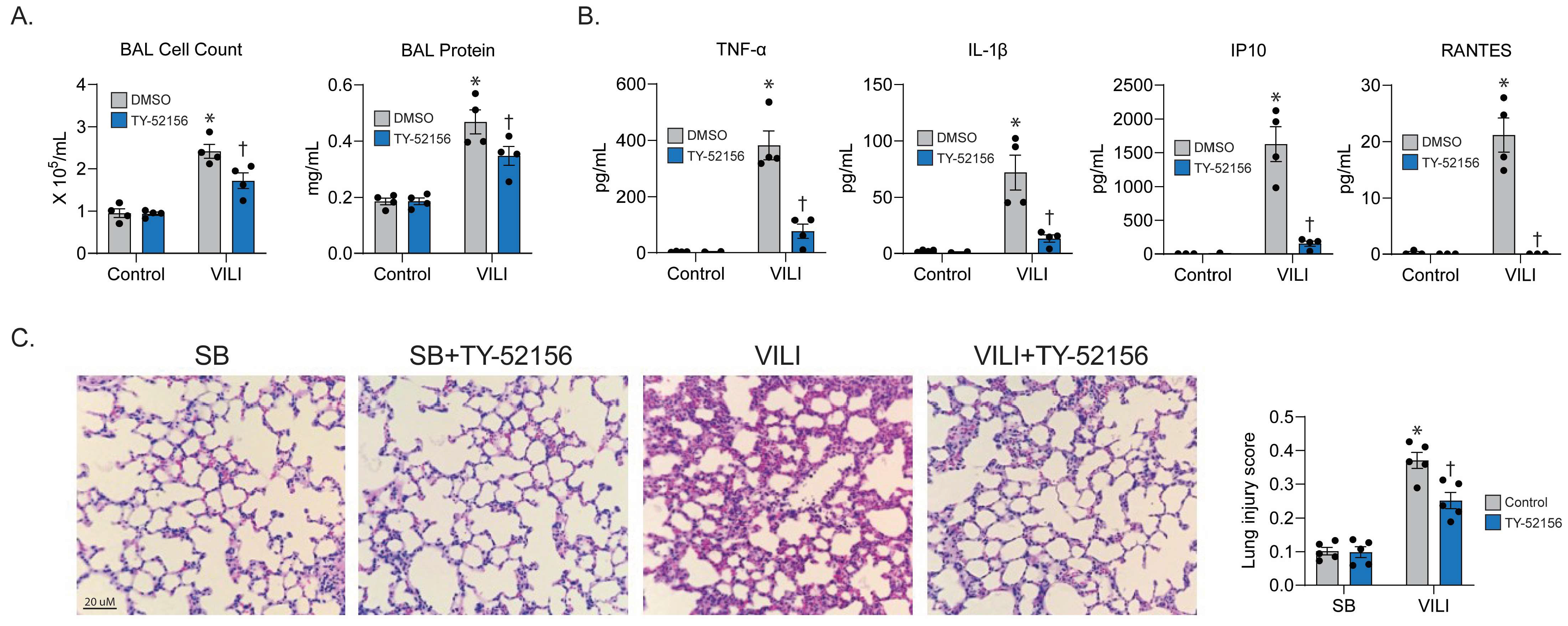
2.4. S1PR3 Inhibitor Prevents Experimental VILI
3. Discussion
3.1. S1P and S1P Receptors in Acute Lung Injury
3.2. S1PR3 and Inflammation
3.3. Therapeutic Potential of TY-52156
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Transendothelial Electrical Resistance (TER)
4.3. Western Blotting
4.4. Immunofluorescence
4.5. Biological Pathway Analysis
4.6. Mouse VILI Model
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abrams, D.; Schmidt, M.; Pham, T.; Beitler, J.R.; Fan, E.; Goligher, E.C.; McNamee, J.J.; Patroniti, N.; Wilcox, M.E.; Combes, A.; et al. Mechanical Ventilation for Acute Respiratory Distress Syndrome during Extracorporeal Life Support. Research and Practice. Am. J. Respir. Crit. Care Med. 2020, 201, 514–525. [Google Scholar] [CrossRef]
- Slutsky, A.S.; Ranieri, V.M. Ventilator-induced lung injury. N. Engl. J. Med. 2013, 369, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gross, C.; Desai, A.A.; Zemskov, E.; Wu, X.; Garcia, A.N.; Jacobson, J.R.; Yuan, J.X.; Garcia, J.G.; Black, S.M. Endothelial cell signaling and ventilator-induced lung injury: Molecular mechanisms, genomic analyses, and therapeutic targets. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L452–L476. [Google Scholar] [CrossRef] [PubMed]
- Pyne, S.; Pyne, N. Sphingosine 1-phosphate signalling via the endothelial differentiation gene family of G-protein-coupled receptors. Pharmacol. Ther. 2000, 88, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; An, S. Diversity of cellular receptors and functions for the lysophospholipid growth factors lysophosphatidic acid and sphingosine 1-phosphate. FASEB J. 1998, 12, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Waeber, C.; Blondeau, N.; Salomone, S. Vascular sphingosine-1-phosphate S1P1 and S1P3 receptors. Drug News Perspect. 2004, 17, 365–382. [Google Scholar] [CrossRef]
- Vogt, W. Pharamacologically active acidic phospholipids and glycolipids. Biochem. Pharmacol. 1963, 12, 415–420. [Google Scholar] [CrossRef]
- Hla, T.; Maciag, T. Isolation of immediate-early differentiation mRNAs by enzymatic amplification of subtracted cDNA from human endothelial cells. Biochem. Biophys. Res. Commun. 1990, 167, 637–643. [Google Scholar] [CrossRef]
- Akhter, M.Z.; Chandra Joshi, J.; Balaji Ragunathrao, V.A.; Maienschein-Cline, M.; Proia, R.L.; Malik, A.B.; Mehta, D. Programming to S1PR1+ Endothelial Cells Promotes Restoration of Vascular Integrity. Circ. Res. 2021, 129, 221–236. [Google Scholar] [CrossRef]
- Anwar, M.; Mehta, D. Post-translational modifications of S1PR1 and endothelial barrier regulation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158760. [Google Scholar] [CrossRef]
- Diab, K.J.; Adamowicz, J.J.; Kamocki, K.; Rush, N.I.; Garrison, J.; Gu, Y.; Schweitzer, K.S.; Skobeleva, A.; Rajashekhar, G.; Hubbard, W.C.; et al. Stimulation of sphingosine 1-phosphate signaling as an alveolar cell survival strategy in emphysema. Am. J. Respir. Crit. Care Med. 2010, 181, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Singleton, P.A.; Letsiou, E.; Zhao, J.; Belvitch, P.; Sammani, S.; Chiang, E.T.; Moreno-Vinasco, L.; Wade, M.S.; Zhou, T.; et al. Sphingosine-1-phosphate receptor-3 is a novel biomarker in acute lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Flemming, S.; Burkard, N.; Renschler, M.; Vielmuth, F.; Meir, M.; Schick, M.A.; Wunder, C.; Germer, C.T.; Spindler, V.; Waschke, J.; et al. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc. Res. 2015, 107, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liang, Y.; Feng, A.; Tieu, K.; Black, S.M.; Wang, T. S1PR3 Plays a Key Role in Ventilator-induced Lung Injury: Promoter Activation, Barrier Disruption, and Therapeutic Evaluation. Free Radic. Biol. Med. 2022, 192, 75–76. [Google Scholar] [CrossRef]
- Wu, J.; Liang, Y.; Sun, X.; Lu, Q.; Tieu, K.; Black, S.M.; Wang, T. Upregulated Sphingosine-1-Phosphate Receptor 3 Disrupts Endothelial Cell Barrier and Mitochondrial Network Dynamics. Circulation 2022, 146, A14973. [Google Scholar] [CrossRef]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef]
- Singleton, P.A.; Dudek, S.M.; Ma, S.F.; Garcia, J.G. Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J. Biol. Chem. 2006, 281, 34381–34393. [Google Scholar] [CrossRef]
- Errasfa, M.; Russo-Marie, F. A purified lipocortin shares the anti-inflammatory effect of glucocorticosteroids in vivo in mice. Br. J. Pharmacol. 1989, 97, 1051–1058. [Google Scholar] [CrossRef]
- Wilkerson, B.A.; Argraves, K.M. The role of sphingosine-1-phosphate in endothelial barrier function. Biochim. Biophys. Acta 2014, 1841, 1403–1412. [Google Scholar] [CrossRef]
- Zeng, Y.; Adamson, R.H.; Curry, F.R.; Tarbell, J.M. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H363–H372. [Google Scholar] [CrossRef]
- Jiang, Y.; Gong, Q.; Huang, J.; Gong, Y.; Tang, Q.; Wei, D.; Tang, Q.; Zhao, J.; Song, J.; Meng, L. ADAM-10 Regulates MMP-12 during Lipopolysaccharide-Induced Inflammatory Response in Macrophages. J. Immunol. Res. 2022, 2022, 3012218. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Marazzi, I.; Beg, A.A.; Natoli, G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor kappaB response. J. Exp. Med. 2004, 200, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.C.; Zhu, R.; Huang, T.; Tomsig, J.L.; Mathews, T.P.; David, M.; Peyruchaud, O.; Macdonald, T.L.; Lynch, K.R. Characterization of a sphingosine 1-phosphate receptor antagonist prodrug. J. Pharmacol. Exp. Ther. 2011, 338, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Punsawad, C.; Viriyavejakul, P. Expression of sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 in malaria-associated acute lung injury/acute respiratory distress syndrome in a mouse model. PLoS ONE 2019, 14, e0222098. [Google Scholar] [CrossRef]
- Hou, J.; Chen, Q.; Wu, X.; Zhao, D.; Reuveni, H.; Licht, T.; Xu, M.; Hu, H.; Hoeft, A.; Ben-Sasson, S.A.; et al. S1PR3 Signaling Drives Bacterial Killing and Is Required for Survival in Bacterial Sepsis. Am. J. Respir. Crit. Care Med. 2017, 196, 1559–1570. [Google Scholar] [CrossRef]
- Heo, J.Y.; Im, D.S. Pro-Inflammatory Role of S1P3 in Macrophages. Biomol. Ther. 2019, 27, 373–380. [Google Scholar] [CrossRef]
- Gong, L.; Wu, X.; Li, X.; Ni, X.; Gu, W.; Wang, X.; Ji, H.; Hu, L.; Zhu, L. S1PR3 deficiency alleviates radiation-induced pulmonary fibrosis through the regulation of epithelial-mesenchymal transition by targeting miR-495-3p. J. Cell. Physiol. 2020, 235, 2310–2324. [Google Scholar] [CrossRef]
- Aguilar, B.J.; Zhu, Y.; Lu, Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 97. [Google Scholar] [CrossRef]
- Clayton, N.S.; Ridley, A.J. Targeting Rho GTPase Signaling Networks in Cancer. Front. Cell Dev. Biol. 2020, 8, 222. [Google Scholar] [CrossRef]
- Kim, S.; Kim, S.A.; Han, J.; Kim, I.S. Rho-Kinase as a Target for Cancer Therapy and Its Immunotherapeutic Potential. Int. J. Mol. Sci. 2021, 22, 12916. [Google Scholar] [CrossRef]
- Sykes, D.A.; Riddy, D.M.; Stamp, C.; Bradley, M.E.; McGuiness, N.; Sattikar, A.; Guerini, D.; Rodrigues, I.; Glaenzel, A.; Dowling, M.R.; et al. Investigating the molecular mechanisms through which FTY720-P causes persistent S1P1 receptor internalization. Br. J. Pharmacol. 2014, 171, 4797–4807. [Google Scholar] [CrossRef] [PubMed]
- Feng, A.; Ma, W.; Faraj, R.; Kelly, G.T.; Black, S.M.; Fallon, M.B.; Wang, T. Identification of S1PR3 gene signature involved in survival of sepsis patients. BMC Med. Genom. 2021, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Ishii, I.; Friedman, B.; Ye, X.; Kawamura, S.; McGiffert, C.; Contos, J.J.; Kingsbury, M.A.; Zhang, G.; Brown, J.H.; Chun, J. Selective loss of sphingosine 1-phosphate signaling with no obvious phenotypic abnormality in mice lacking its G protein-coupled receptor, LP(B3)/EDG-3. J. Biol. Chem. 2001, 276, 33697–33704. [Google Scholar] [CrossRef] [PubMed]
- Perez-Jeldres, T.; Alvarez-Lobos, M.; Rivera-Nieves, J. Targeting Sphingosine-1-Phosphate Signaling in Immune-Mediated Diseases: Beyond Multiple Sclerosis. Drugs 2021, 81, 985–1002. [Google Scholar] [CrossRef] [PubMed]
- Salomone, S.; Waeber, C. Selectivity and specificity of sphingosine-1-phosphate receptor ligands: Caveats and critical thinking in characterizing receptor-mediated effects. Front. Pharmacol. 2011, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, H.S.; Lee, J.S.; Bastarache, J.A.; Kuebler, W.M.; Downey, G.P.; Albaiceta, G.M.; Altemeier, W.A.; Artigas, A.; Bates, J.H.T.; Calfee, C.S.; et al. Update on the Features and Measurements of Experimental Acute Lung Injury in Animals: An Official American Thoracic Society Workshop Report. Am. J. Respir. Cell Mol. Biol. 2022, 66, e1–e14. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Liang, Y.; Fu, P.; Feng, A.; Lu, Q.; Unwalla, H.J.; Marciano, D.P.; Black, S.M.; Wang, T. Sphingosine-1-Phosphate Receptor 3 Induces Endothelial Barrier Loss via ADAM10-Mediated Vascular Endothelial-Cadherin Cleavage. Int. J. Mol. Sci. 2023, 24, 16083. https://doi.org/10.3390/ijms242216083
Wu J, Liang Y, Fu P, Feng A, Lu Q, Unwalla HJ, Marciano DP, Black SM, Wang T. Sphingosine-1-Phosphate Receptor 3 Induces Endothelial Barrier Loss via ADAM10-Mediated Vascular Endothelial-Cadherin Cleavage. International Journal of Molecular Sciences. 2023; 24(22):16083. https://doi.org/10.3390/ijms242216083
Chicago/Turabian StyleWu, Jialin, Ying Liang, Panfeng Fu, Anlin Feng, Qing Lu, Hoshang J. Unwalla, David P. Marciano, Stephen M. Black, and Ting Wang. 2023. "Sphingosine-1-Phosphate Receptor 3 Induces Endothelial Barrier Loss via ADAM10-Mediated Vascular Endothelial-Cadherin Cleavage" International Journal of Molecular Sciences 24, no. 22: 16083. https://doi.org/10.3390/ijms242216083
APA StyleWu, J., Liang, Y., Fu, P., Feng, A., Lu, Q., Unwalla, H. J., Marciano, D. P., Black, S. M., & Wang, T. (2023). Sphingosine-1-Phosphate Receptor 3 Induces Endothelial Barrier Loss via ADAM10-Mediated Vascular Endothelial-Cadherin Cleavage. International Journal of Molecular Sciences, 24(22), 16083. https://doi.org/10.3390/ijms242216083