Comparative Computational Analysis of Spike Protein Structural Stability in SARS-CoV-2 Omicron Subvariants

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. SARS-CoV-2 S Protein Genome/Protein Analysis in COVID-19 Patients
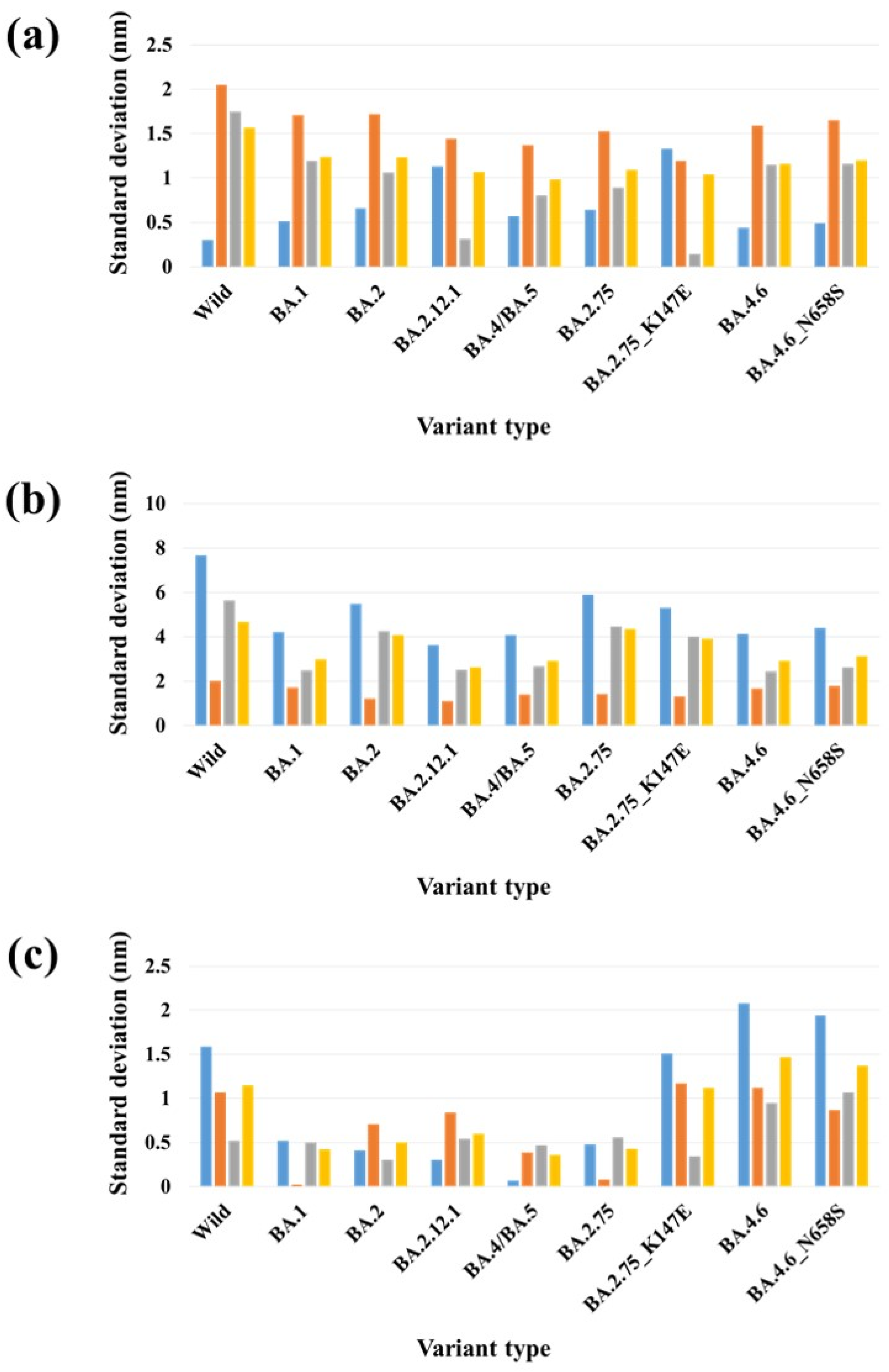
2.2. Binding Free Energy Analysis of Full-Length S Protein–ACE2 Complex
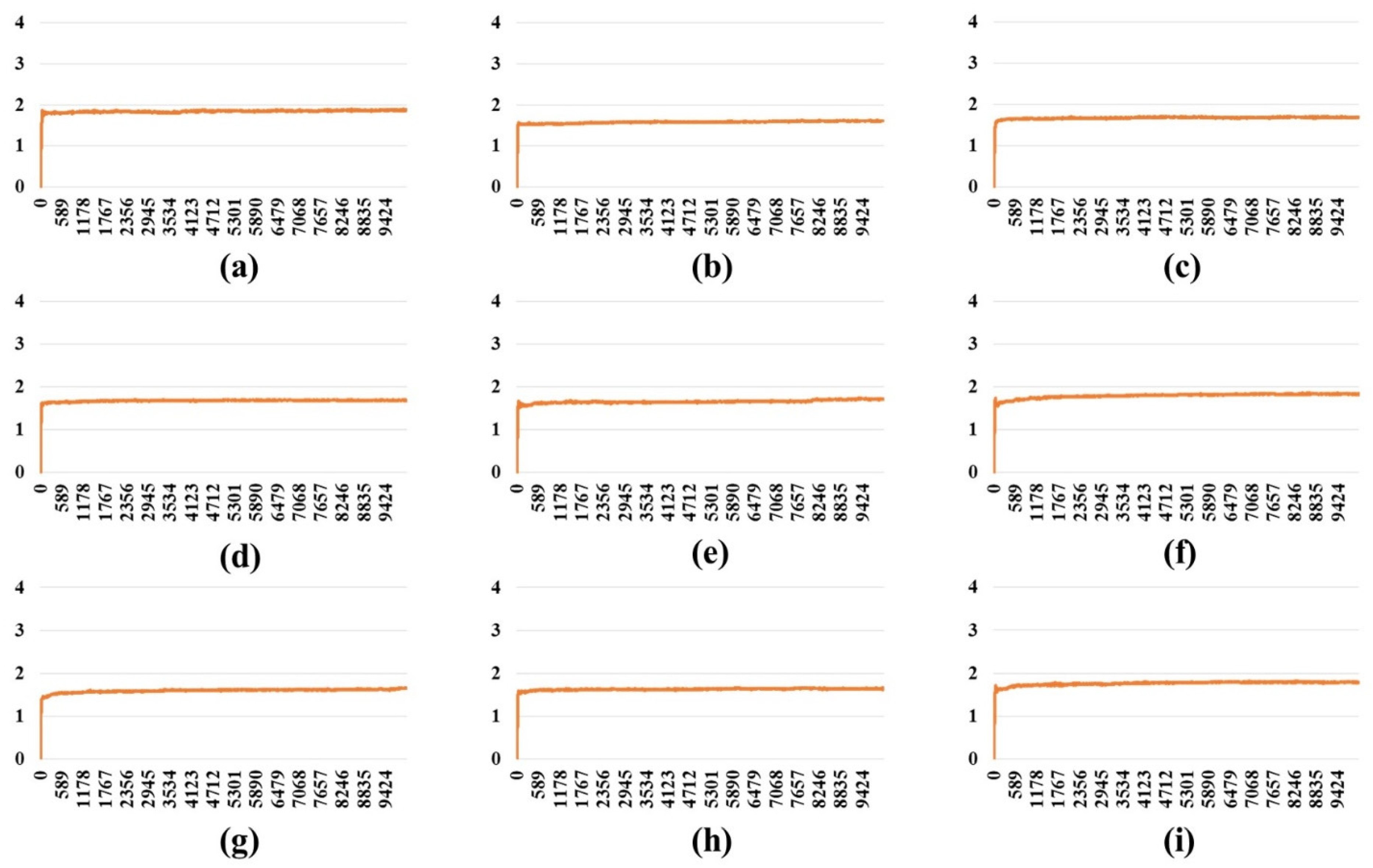
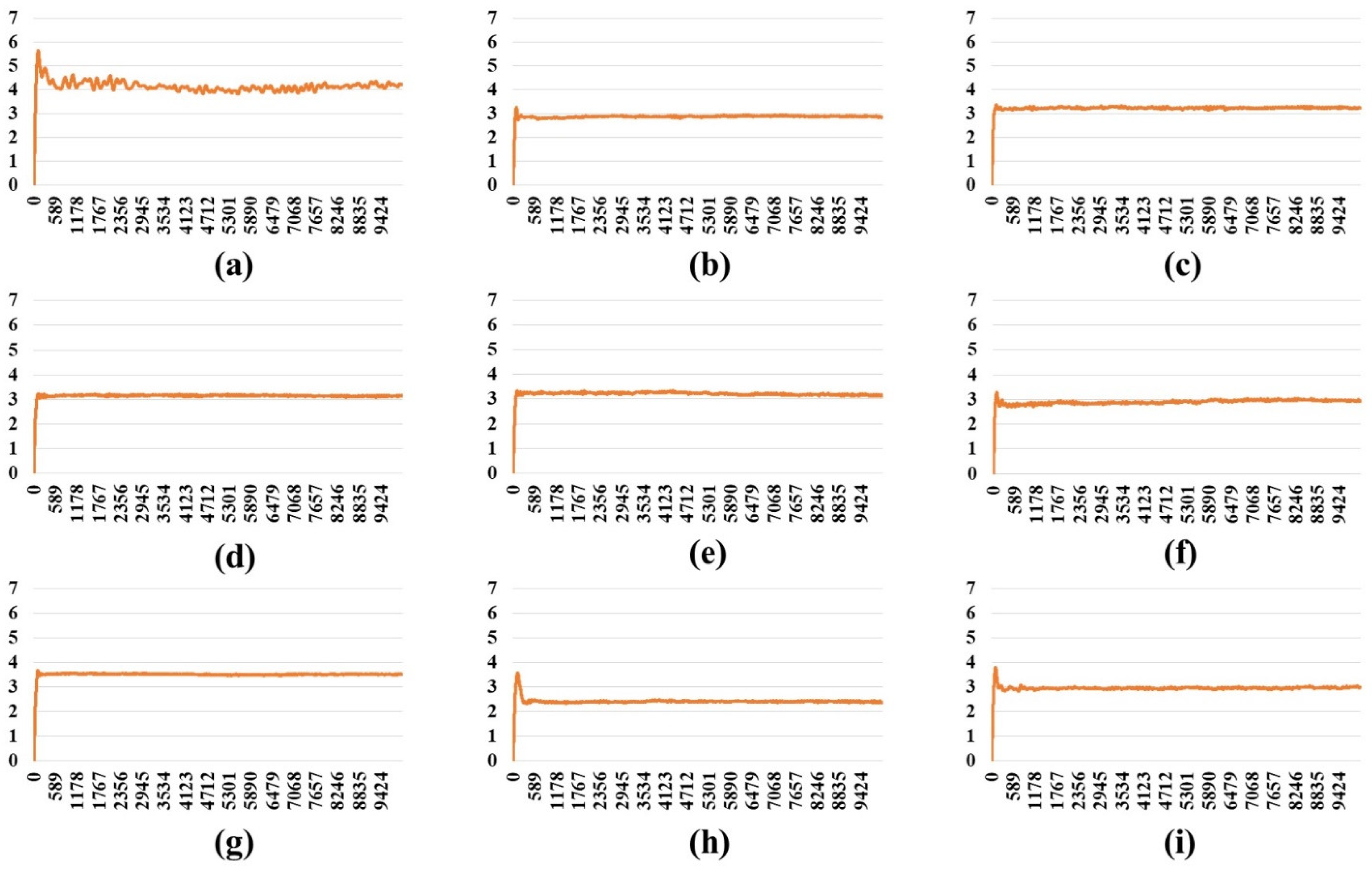
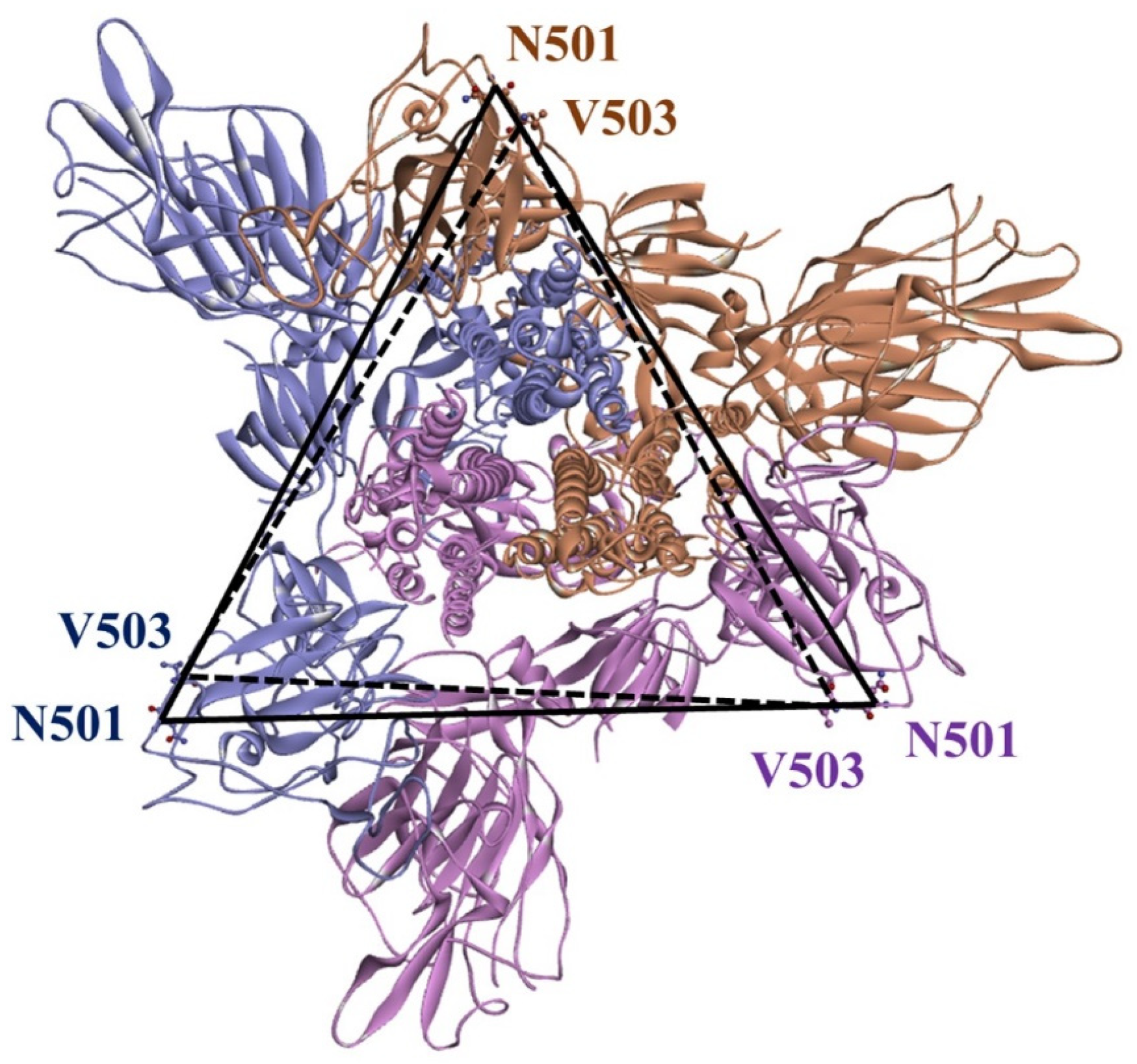
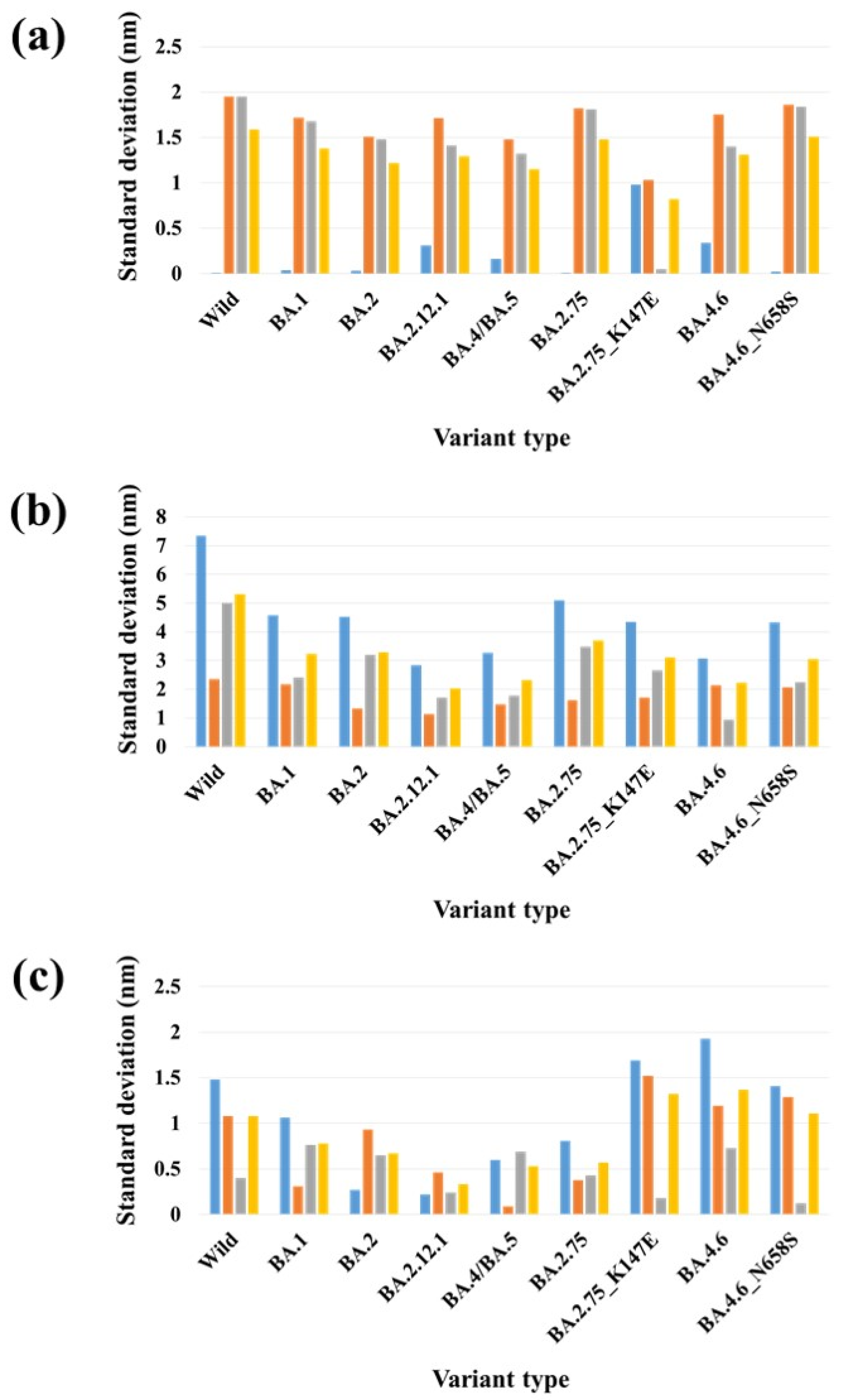
2.3. Stability Analysis of RBDs in Full-Length Trimeric S Protein Chains
3. Discussion
4. Materials and Methods
4.1. Ethical Considerations
4.2. Genome/Protein Sequence Investigation of Korean Patients with COVID-19
4.3. SARS-CoV-2 S Protein Structure
4.4. MD Simulation
4.5. Binding Free Energy Calculation
4.6. Distance Calculation
4.7. Interaction Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, V.; Singh, J.; Hasnain, S.E.; Sundar, D. Possible link between higher transmissibility of alpha, kappa and delta variants of SARS-CoV-2 and increased structural stability of its spike protein and hACE2 affinity. Int. J. Mol. Sci. 2021, 22, 9131. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Zhou, Z.; Dai, R.; Zhang, J.; Zhao, S.; Wu, X.; Lan, W.; Ren, Y.; Cui, L.; Lan, Q. V367F mutation in SARS-CoV-2 spike RBD emerging during the early transmission phase enhances viral infectivity through increased human ACE2 receptor binding affinity. J. Virol. 2021, 95, e00617-21. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi Gomari, M.; Rostami, N.; Omidi-Ardali, H.; Arab, S.S. Insight into molecular characteristics of SARS-CoV-2 spike protein following D614G point mutation, a molecular dynamics study. J. Biomol. Struct. Dyn. 2022, 40, 5634–5642. [Google Scholar] [CrossRef] [PubMed]
- Idowu, K.A.; Onyenaka, C.; Olaleye, O.A. A computational evaluation of structural stability of omicron and delta mutations of SARS-CoV-2 spike proteins and human ACE-2 interactions. Inform. Med. Unlocked 2022, 33, 101074. [Google Scholar] [CrossRef]
- Bazargan, M.; Elahi, R.; Esmaeilzadeh, A. OMICRON: Virology, immunopathogenesis, and laboratory diagnosis. J. Gene Med. 2022, 24, e3435. [Google Scholar] [CrossRef]
- Ghosh, N.; Nandi, S.; Saha, I. A review on evolution of emerging SARS-CoV-2 variants based on spike glycoprotein. Int. Immunopharmacol. 2022, 105, 108565. [Google Scholar] [CrossRef]
- Hossain, A.; Akter, S.; Rashid, A.A.; Khair, S.; Alam, A.R.U. Unique mutations in SARS-CoV-2 Omicron subvariants’ non-spike proteins: Potential impacts on viral pathogenesis and host immune evasion. Microb. Pathog. 2022, 170, 105699. [Google Scholar] [CrossRef]
- Ke, H.; Chang, M.R.; Marasco, W.A. Immune evasion of SARS-CoV-2 omicron subvariants. Vaccines 2022, 10, 1545. [Google Scholar] [CrossRef]
- Kumar, S.; Karuppanan, K.; Subramaniam, G. Omicron (BA. 1) and sub-variants (BA. 1.1, BA. 2, and BA. 3) of SARS-CoV-2 spike infectivity and pathogenicity: A comparative sequence and structural-based computational assessment. J. Med. Virol. 2022, 94, 4780–4791. [Google Scholar] [CrossRef]
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct. Target. Ther. 2022, 7, 138. [Google Scholar] [CrossRef]
- Cao, Y.; Song, W.; Wang, L.; Liu, P.; Yue, C.; Jian, F.; Yu, Y.; Yisimayi, A.; Wang, P.; Wang, Y. Characterization of the enhanced infectivity and antibody evasion of Omicron BA. 2.75. Cell Host Microbe 2022, 30, 1527–1539.e1525. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.; Dijokaite-Guraliuc, A.; Liu, C.; Zhou, D.; Ginn, H.M.; Das, R.; Supasa, P.; Selvaraj, M.; Nutalai, R.; Tuekprakhon, A. A delicate balance between antibody evasion and ACE2 affinity for Omicron BA. 2.75. Cell Rep. 2023, 42, 111903. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X. BA. 2.12. 1, BA. 4 and BA. 5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y. Antibody evasion by SARS-CoV-2 Omicron subvariants BA. 2.12. 1, BA. 4 and BA. 5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M. Antibody escape of SARS-CoV-2 Omicron BA. 4 and BA. 5 from vaccine and BA. 1 serum. Cell 2022, 185, 2422–2433.e13. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradník, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.; Ginn, H.M.; Mentzer, A.J.; Tuekprakhon, A. SARS-CoV-2 Omicron-B. 1.1. 529 leads to widespread escape from neutralizing antibody responses. Cell 2022, 185, 467–484.e15. [Google Scholar] [CrossRef]
- Nutalai, R.; Zhou, D.; Tuekprakhon, A.; Ginn, H.M.; Supasa, P.; Liu, C.; Huo, J.; Mentzer, A.J.; Duyvesteyn, H.M.; Dijokaite-Guraliuc, A. Potent cross-reactive antibodies following Omicron breakthrough in vaccinees. Cell 2022, 185, 2116–2131.e18. [Google Scholar] [CrossRef]
- Kim, S.; Liu, Y.; Ziarnik, M.; Seo, S.; Cao, Y.; Zhang, X.F.; Im, W. Binding of human ACE2 and RBD of omicron enhanced by unique interaction patterns among SARS-CoV-2 variants of concern. J. Comput. Chem. 2023, 44, 594–601. [Google Scholar] [CrossRef]
- Abas, A.H.; Marfuah, S.; Idroes, R.; Kusumawaty, D.; Fatimawali; Park, M.N.; Siyadatpanah, A.; Alhumaydhi, F.A.; Mahmud, S.; Tallei, T.E. Can the SARS-CoV-2 omicron variant confer natural immunity against COVID-19? Molecules 2022, 27, 2221. [Google Scholar] [CrossRef]
- Tallei, T.E.; Alhumaid, S.; AlMusa, Z.; Fatimawali; Kusumawaty, D.; Alynbiawi, A.; Alshukairi, A.N.; Rabaan, A.A. Update on the omicron sub-variants BA. 4 and BA. 5. Rev. Med. Virol. 2023, 33, e2391. [Google Scholar] [CrossRef]
- Rahmani, A.H.; Anwar, S.; Raut, R.; Almatroudi, A.; Babiker, A.Y.; Khan, A.A.; Alsahli, M.A.; Almatroodi, S.A. Therapeutic potential of myrrh, a natural resin, in health management through modulation of oxidative stress, inflammation, and advanced glycation end products formation using in vitro and in silico analysis. Appl. Sci. 2022, 12, 9175. [Google Scholar] [CrossRef]
- Dhama, K.; Nainu, F.; Frediansyah, A.; Yatoo, M.I.; Mohapatra, R.K.; Chakraborty, S.; Zhou, H.; Islam, M.R.; Mamada, S.S.; Kusuma, H.I. Global emerging Omicron variant of SARS-CoV-2: Impacts, challenges and strategies. J. Infect. Public Health 2023, 16, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Li, X.; Zhang, L.; Wan, S.; Zhang, L.; Zhou, F. SARS-CoV-2 Omicron variant: Recent progress and future perspectives. Signal Transduct. Target. Ther. 2022, 7, 141. [Google Scholar] [CrossRef]
- Silva, S.J.R.d.; Kohl, A.; Pena, L.; Pardee, K. Recent insights into SARS-CoV-2 omicron variant. Rev. Med. Virol. 2023, 33, e2373. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Bhattacharya, M.; Nag, S.; Dhama, K.; Chakraborty, C. A detailed overview of SARS-CoV-2 omicron: Its sub-variants, mutations and pathophysiology, clinical characteristics, immunological landscape, immune escape, and therapies. Viruses 2023, 15, 167. [Google Scholar] [CrossRef]
- Dhawan, M.; Saied, A.A.; Mitra, S.; Alhumaydhi, F.A.; Emran, T.B.; Wilairatana, P. Omicron variant (B. 1.1. 529) and its sublineages: What do we know so far amid the emergence of recombinant variants of SARS-CoV-2? Biomed. Pharmacother. 2022, 154, 113522. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 omicron variants BA. 1 to BA. 5: Implications for immune escape and transmission. Rev. Med. Virol. 2022, 32, e2381. [Google Scholar] [CrossRef]
- Xu, A.; Hong, B.; Lou, F.; Wang, S.; Li, W.; Shafqat, A.; An, X.; Zhao, Y.; Song, L.; Tong, Y. Sub-lineages of the SARS-CoV-2 Omicron variants: Characteristics and prevention. MedComm 2022, 3, e172. [Google Scholar] [CrossRef]
- Hachmann, N.P.; Miller, J.; Collier, A.-r.Y.; Barouch, D.H. Neutralization escape by SARS-CoV-2 Omicron subvariant BA. 4.6. N. Engl. J. Med. 2022, 387, 1904–1906. [Google Scholar] [CrossRef]
- Hachmann, N.P.; Miller, J.; Collier, A.-r.Y.; Ventura, J.D.; Yu, J.; Rowe, M.; Bondzie, E.A.; Powers, O.; Surve, N.; Hall, K. Neutralization escape by SARS-CoV-2 Omicron subvariants BA. 2.12. 1, BA. 4, and BA. 5. N. Engl. J. Med. 2022, 387, 86–88. [Google Scholar] [CrossRef]
- Kimura, I.; Yamasoba, D.; Tamura, T.; Nao, N.; Suzuki, T.; Oda, Y.; Mitoma, S.; Ito, J.; Nasser, H.; Zahradnik, J. Virological characteristics of the SARS-CoV-2 Omicron BA. 2 subvariants, including BA. 4 and BA. 5. Cell 2022, 185, 3992–4007.e3916. [Google Scholar] [CrossRef] [PubMed]
- Pastorio, C.; Zech, F.; Noettger, S.; Jung, C.; Jacob, T.; Sanderson, T.; Sparrer, K.M.; Kirchhoff, F. Determinants of Spike infectivity, processing, and neutralization in SARS-CoV-2 Omicron subvariants BA. 1 and BA. 2. Cell Host Microbe 2022, 30, 1255–1268.e1255. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-W.; Lim, B.-L.; Young, B.E.; Yeoh, A.Y.-Y.; Yung, C.-F.; Yap, W.-C.; Althaus, T.; Chia, W.-N.; Zhu, F.; Lye, D.C. Comparative neutralisation profile of SARS-CoV-2 omicron subvariants BA. 2.75 and BA. 5. Lancet Microbe 2022, 3, e898. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Nie, W.; Sun, Y.; Ye, Q. The epidemiological features of the SARS-CoV-2 Omicron subvariant BA. 5 and its evasion of the neutralizing activity of vaccination and prior infection. Vaccines 2022, 10, 1699. [Google Scholar] [CrossRef] [PubMed]
- Yamasoba, D.; Kosugi, Y.; Kimura, I.; Fujita, S.; Uriu, K.; Ito, J.; Sato, K. Neutralisation sensitivity of SARS-CoV-2 omicron subvariants to therapeutic monoclonal antibodies. Lancet Infect. Dis. 2022, 22, 942–943. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Das, N.C.; Patra, R.; Mukherjee, S. In silico analyses on the comparative sensing of SARS-CoV-2 mRNA by the intracellular TLRs of humans. J. Med. Virol. 2021, 93, 2476–2486. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, G.; Mukherjee, S. Chemotherapy vs. Immunotherapy in combating nCOVID19: An update. Hum. Immunol. 2021, 82, 649–658. [Google Scholar] [CrossRef]
- Choi, K.-E.; Kim, J.-M.; Rhee, J.; Park, A.K.; Kim, E.-J.; Kang, N.S. Molecular dynamics studies on the structural characteristics for the stability prediction of SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 8714. [Google Scholar] [CrossRef]
- Choi, K.-E.; Kim, J.-M.; Rhee, J.E.; Park, A.K.; Kim, E.-J.; Yoo, C.K.; Kang, N.S. Molecular dynamics studies on the structural stability prediction of SARS-CoV-2 variants including multiple mutants. Int. J. Mol. Sci. 2022, 23, 4956. [Google Scholar] [CrossRef]
- Singh, J.; Anantharaj, A.; Panwar, A.; Rani, C.; Bhardwaj, M.; Kumar, P.; Chattopadhyay, P.; Devi, P.; Maurya, R.; Mishra, P. BA. 1, BA. 2 and BA. 2.75 variants show comparable replication kinetics, reduced impact on epithelial barrier and elicit cross-neutralizing antibodies. PLoS Pathog. 2023, 19, e1011196. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.M.; Chakrabarti, J. Effect on the conformations of the spike protein of SARS-CoV-2 due to mutation. Biotechnol. Appl. Biochem. 2023, 70, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, E. Mutations in the SARS-CoV-2 spike protein may cause functional changes in the protein quaternary structure. Turk. J. Biochem. 2021, 46, 137–144. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, C.; Cao, X.; Gu, C.; Liu, H.; Jiang, M.; Wang, X.; Yuan, Q.; Wu, K.; Liu, J. Structural and biochemical mechanism for increased infectivity and immune evasion of Omicron BA. 2 variant compared to BA. 1 and their possible mouse origins. Cell Res. 2022, 32, 609–620. [Google Scholar] [CrossRef]
- Lee, M.; Major, M.; Hong, H. Distinct conformations of SARS-CoV-2 Omicron spike protein and its interaction with ACE2 and antibody. Int. J. Mol. Sci. 2023, 24, 3774. [Google Scholar] [CrossRef]
- Das, N.C.; Chakraborty, P.; Bayry, J.; Mukherjee, S. Comparative binding ability of human monoclonal antibodies against Omicron variants of SARS-CoV-2: An in silico investigation. Antibodies 2023, 12, 17. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Van Doremalen, N.; Bushmaker, T.; Morris, D.H.; Holbrook, M.G.; Gamble, A.; Williamson, B.N.; Tamin, A.; Harcourt, J.L.; Thornburg, N.J.; Gerber, S.I. Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1. N. Engl. J. Med. 2020, 382, 1564–1567. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Thai, N.Q.; Nguyen, P.H.; Li, M.S. SARS-CoV-2 omicron variant binds to human cells more strongly than the wild type: Evidence from molecular dynamics simulation. J. Phys. Chem. B 2022, 126, 4669–4678. [Google Scholar] [CrossRef]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.-D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Celik, I.; Abdellattif, M.H.; Tallei, T.E. An insight based on computational analysis of the interaction between the receptor-binding domain of the omicron variants and human angiotensin-converting enzyme 2. Biology 2022, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Kim, J.-M.; Chung, Y.-S.; Jo, H.J.; Lee, N.-J.; Kim, M.S.; Woo, S.H.; Park, S.; Kim, J.W.; Kim, H.M.; Han, M.-G. Identification of coronavirus isolated from a patient in Korea with COVID-19. Osong Public Health Res. Perspect. 2020, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—Distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM force field in GROMACS: Analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Maiorov, V.N.; Crippen, G.M. Size-independent comparison of protein three-dimensional structures. Proteins 1995, 22, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson− Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Luzar, A.; Chandler, D. Hydrogen-bond kinetics in liquid water. Nature 1996, 379, 55–57. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant Type | Total Binding Energy | Average Total Binding Energy | ||
|---|---|---|---|---|
| One-Open- Complex Form | Two-Open- Complex Form | Three-Open- Complex Form | All Complex Forms | |
| Wild | −11,773.3 | −20,757.0 | −17,384.2 | −16,638.2 |
| BA.1 | −16,980.7 | −37,287.8 | −45,273.1 | −33,180.5 |
| BA.2 | −15,130.2 | −37,202.6 | −45,986.4 | −32,773.1 |
| BA.2.12.1 | −16,047.9 | −34,681.3 | −47,719.7 | −32,816.3 |
| BA.4/BA.5 | −16,364.2 | −36,144.4 | −47,427.2 | −33,311.9 |
| BA.2.75 | −18,635.3 | −42,068.4 | −58,801.1 | −39,834.9 |
| BA.2.75_K147E | −14,639.9 | −31,729.8 | −47,497.0 | −31,288.9 |
| BA.4.6 | −14,997.9 | −31,413.1 | −45,491.2 | −30,634.1 |
| BA.4.6_N658S | −14,704.4 | −29,389.6 | −39,351.5 | −27,815.2 |
| PDB | Variant Type | A-B | A-C | B-C | SD (A-B) & (A-C) | SD (A-B) & (B-C) | SD (A-C) & (B-C) | SD (A-B-C) |
|---|---|---|---|---|---|---|---|---|
| One-open- complex form | Wild | 3.00 | 3.01 | 0.25 | 0.01 | 1.95 | 1.95 | 1.59 |
| BA.1 | 3.33 | 3.28 | 0.91 | 0.04 | 1.72 | 1.68 | 1.38 | |
| BA.2 | 2.42 | 2.37 | 0.28 | 0.03 | 1.51 | 1.48 | 1.22 | |
| BA.2.12.1 | 2.61 | 2.18 | 0.19 | 0.31 | 1.71 | 1.41 | 1.29 | |
| BA.4/BA.5 | 2.35 | 2.12 | 0.26 | 0.16 | 1.48 | 1.32 | 1.15 | |
| BA.2.75 | 3.00 | 2.99 | 0.43 | 0.01 | 1.82 | 1.81 | 1.48 | |
| BA.2.75_K147E | 2.47 | 1.08 | 1.01 | 0.98 | 1.03 | 0.05 | 0.82 | |
| BA.4.6 | 2.77 | 2.29 | 0.30 | 0.34 | 1.75 | 1.40 | 1.31 | |
| BA.4.6_N658S | 2.92 | 2.89 | 0.29 | 0.02 | 1.86 | 1.84 | 1.51 | |
| Two-open- complex form | Wild | 13.96 | 3.57 | 10.63 | 7.35 | 2.36 | 5.00 | 5.31 |
| BA.1 | 9.81 | 3.34 | 6.73 | 4.57 | 2.18 | 2.40 | 3.24 | |
| BA.2 | 9.67 | 3.28 | 7.81 | 4.52 | 1.32 | 3.20 | 3.29 | |
| BA.2.12.1 | 7.42 | 3.40 | 5.81 | 2.84 | 1.14 | 1.70 | 2.02 | |
| BA.4/BA.5 | 7.77 | 3.16 | 5.67 | 3.26 | 1.48 | 1.77 | 2.31 | |
| BA.2.75 | 9.78 | 2.57 | 7.50 | 5.10 | 1.62 | 3.48 | 3.69 | |
| BA.2.75_K147E | 9.31 | 3.15 | 6.90 | 4.35 | 1.70 | 2.65 | 3.10 | |
| BA.4.6 | 7.88 | 3.53 | 4.86 | 3.07 | 2.14 | 0.94 | 2.23 | |
| BA.4.6_N658S | 8.98 | 2.87 | 6.05 | 4.32 | 2.07 | 2.25 | 3.06 | |
| Three-open- complex form | Wild | 8.23 | 10.32 | 9.75 | 1.48 | 1.08 | 0.40 | 1.08 |
| BA.1 | 10.32 | 8.81 | 9.89 | 1.06 | 0.31 | 0.76 | 0.78 | |
| BA.2 | 8.87 | 8.48 | 7.56 | 0.27 | 0.93 | 0.65 | 0.67 | |
| BA.2.12.1 | 9.19 | 8.88 | 8.53 | 0.22 | 0.46 | 0.24 | 0.33 | |
| BA.4/BA.5 | 8.53 | 9.39 | 8.40 | 0.60 | 0.09 | 0.69 | 0.53 | |
| BA.2.75 | 8.89 | 7.75 | 8.36 | 0.81 | 0.38 | 0.43 | 0.57 | |
| BA.2.75_K147E | 11.10 | 8.70 | 8.95 | 1.69 | 1.52 | 0.18 | 1.32 | |
| BA.4.6 | 11.45 | 8.73 | 9.77 | 1.93 | 1.19 | 0.73 | 1.37 | |
| BA.4.6_N658S | 7.81 | 9.81 | 9.64 | 1.41 | 1.29 | 0.12 | 1.11 |
| PDB | Variant Type | A-B | A-C | B-C | SD (A-B) & (A-C) | SD (A-B) & (B-C) | SD (A-C) & (B-C) | SD (A-B-C) |
|---|---|---|---|---|---|---|---|---|
| One-open- complex form | Wild | 3.33 | 2.91 | 0.43 | 0.30 | 2.05 | 1.75 | 1.57 |
| BA.1 | 3.57 | 2.85 | 1.16 | 0.51 | 1.71 | 1.19 | 1.24 | |
| BA.2 | 2.99 | 2.06 | 0.56 | 0.66 | 1.72 | 1.06 | 1.23 | |
| BA.2.12.1 | 3.01 | 1.42 | 0.98 | 1.13 | 1.44 | 0.31 | 1.07 | |
| BA.4/BA.5 | 2.56 | 1.75 | 0.62 | 0.57 | 1.37 | 0.80 | 0.98 | |
| BA.2.75 | 2.98 | 2.08 | 0.81 | 0.64 | 1.53 | 0.89 | 1.09 | |
| BA.2.75_K147E | 3.36 | 1.47 | 1.67 | 1.33 | 1.19 | 0.14 | 1.04 | |
| BA.4.6 | 2.95 | 2.33 | 0.70 | 0.44 | 1.59 | 1.15 | 1.16 | |
| BA.4.6_N658S | 2.81 | 2.12 | 0.48 | 0.49 | 1.65 | 1.16 | 1.20 | |
| Two-open- complex form | Wild | 13.93 | 3.10 | 11.08 | 7.66 | 2.02 | 5.64 | 4.66 |
| BA.1 | 8.99 | 3.02 | 6.57 | 4.22 | 1.71 | 2.50 | 3.00 | |
| BA.2 | 10.71 | 2.96 | 8.98 | 5.48 | 1.23 | 4.26 | 4.07 | |
| BA.2.12.1 | 8.08 | 2.96 | 6.50 | 3.62 | 1.11 | 2.51 | 2.62 | |
| BA.4/BA.5 | 8.62 | 2.87 | 6.65 | 4.07 | 1.39 | 2.68 | 2.92 | |
| BA.2.75 | 10.85 | 2.50 | 8.83 | 5.90 | 1.43 | 4.47 | 4.35 | |
| BA.2.75_K147E | 10.07 | 2.55 | 8.23 | 5.31 | 1.30 | 4.02 | 3.92 | |
| BA.4.6 | 8.64 | 2.81 | 6.27 | 4.12 | 1.68 | 2.45 | 2.93 | |
| BA.4.6_N658S | 9.11 | 2.89 | 6.59 | 4.40 | 1.78 | 2.62 | 3.13 | |
| Three-open- complex form | Wild | 8.04 | 10.29 | 9.56 | 1.59 | 1.07 | 0.52 | 1.15 |
| BA.1 | 9.71 | 8.98 | 9.69 | 0.52 | 0.02 | 0.50 | 0.42 | |
| BA.2 | 9.72 | 9.14 | 8.72 | 0.41 | 0.71 | 0.30 | 0.50 | |
| BA.2.12.1 | 10.44 | 10.01 | 9.25 | 0.30 | 0.84 | 0.54 | 0.60 | |
| BA.4/BA.5 | 9.86 | 9.97 | 9.31 | 0.07 | 0.39 | 0.47 | 0.36 | |
| BA.2.75 | 9.42 | 8.74 | 9.53 | 0.48 | 0.08 | 0.56 | 0.43 | |
| BA.2.75_K147E | 11.82 | 9.69 | 10.17 | 1.51 | 1.17 | 0.34 | 1.12 | |
| BA.4.6 | 12.60 | 9.66 | 11.01 | 2.08 | 1.12 | 0.95 | 1.47 | |
| BA.4.6_N658S | 7.99 | 10.73 | 9.21 | 1.94 | 0.87 | 1.07 | 1.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balupuri, A.; Kim, J.-M.; Choi, K.-E.; No, J.S.; Kim, I.-H.; Rhee, J.E.; Kim, E.-J.; Kang, N.S. Comparative Computational Analysis of Spike Protein Structural Stability in SARS-CoV-2 Omicron Subvariants. Int. J. Mol. Sci. 2023, 24, 16069. https://doi.org/10.3390/ijms242216069
Balupuri A, Kim J-M, Choi K-E, No JS, Kim I-H, Rhee JE, Kim E-J, Kang NS. Comparative Computational Analysis of Spike Protein Structural Stability in SARS-CoV-2 Omicron Subvariants. International Journal of Molecular Sciences. 2023; 24(22):16069. https://doi.org/10.3390/ijms242216069
Chicago/Turabian StyleBalupuri, Anand, Jeong-Min Kim, Kwang-Eun Choi, Jin Sun No, Il-Hwan Kim, Jee Eun Rhee, Eun-Jin Kim, and Nam Sook Kang. 2023. "Comparative Computational Analysis of Spike Protein Structural Stability in SARS-CoV-2 Omicron Subvariants" International Journal of Molecular Sciences 24, no. 22: 16069. https://doi.org/10.3390/ijms242216069
APA StyleBalupuri, A., Kim, J.-M., Choi, K.-E., No, J. S., Kim, I.-H., Rhee, J. E., Kim, E.-J., & Kang, N. S. (2023). Comparative Computational Analysis of Spike Protein Structural Stability in SARS-CoV-2 Omicron Subvariants. International Journal of Molecular Sciences, 24(22), 16069. https://doi.org/10.3390/ijms242216069