
CRISPR-Cas9-Based Gene Knockout of Immune Checkpoints in Expanded NK Cells


 ,
,
Abstract
:1. Introduction
2. Results
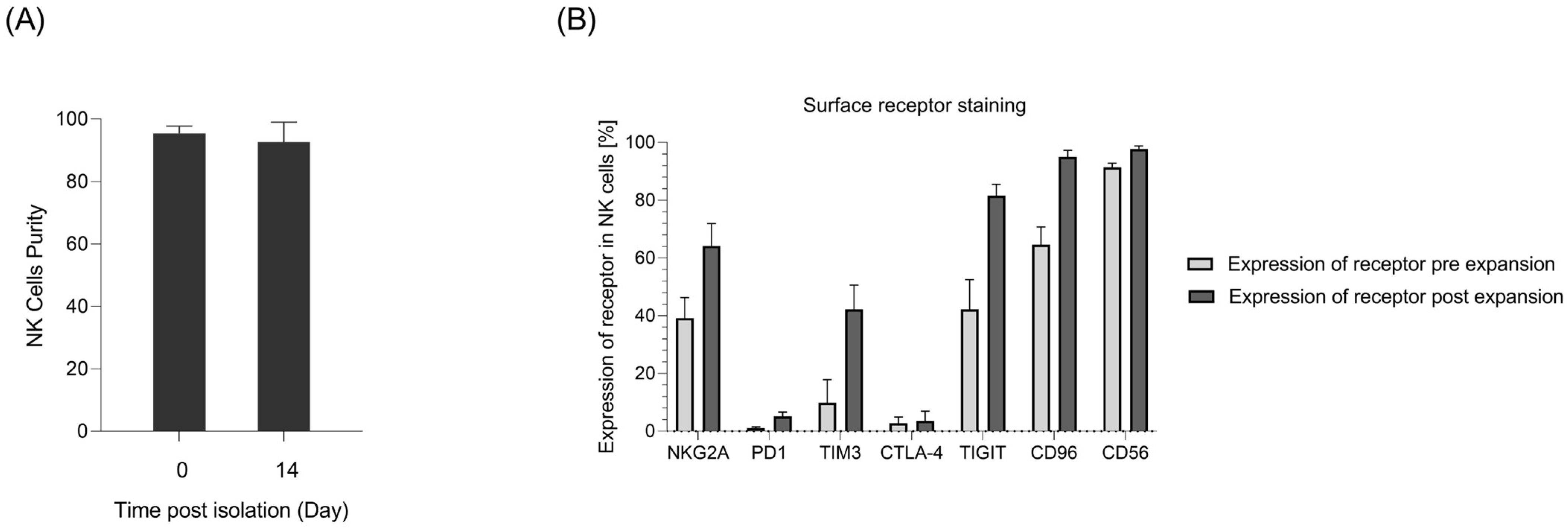
2.1. Primary NK Cell Isolation, Expansion, and Receptor Profile Characterization
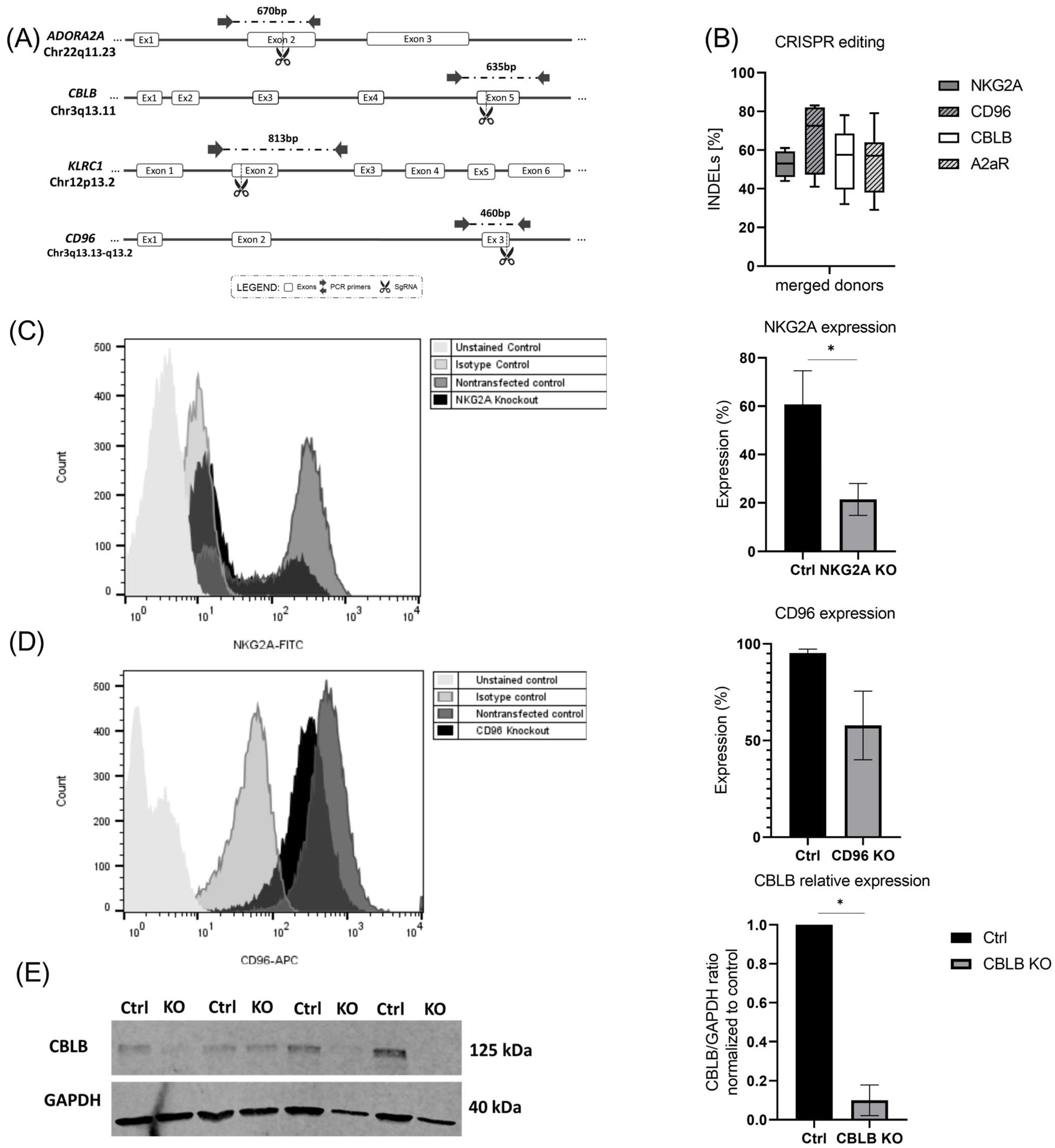
2.2. Targeting Inhibitory Signals Involved in NK Cell Function Using CRISPR-Cas9 System
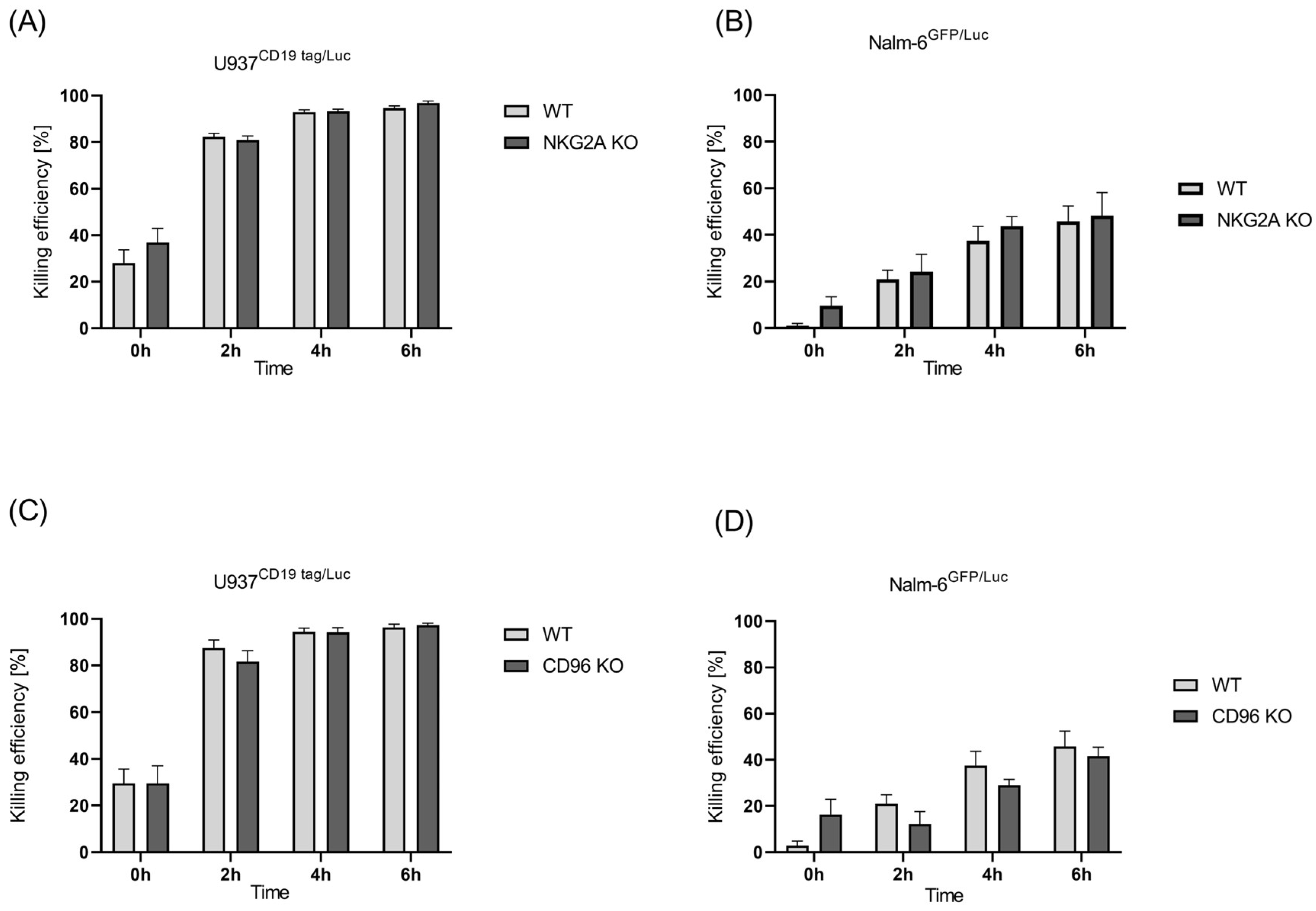
2.3. CRISPR-Mediated Blocking of CD96 and NKG2A, Two Inhibitory Receptors in NK Cells
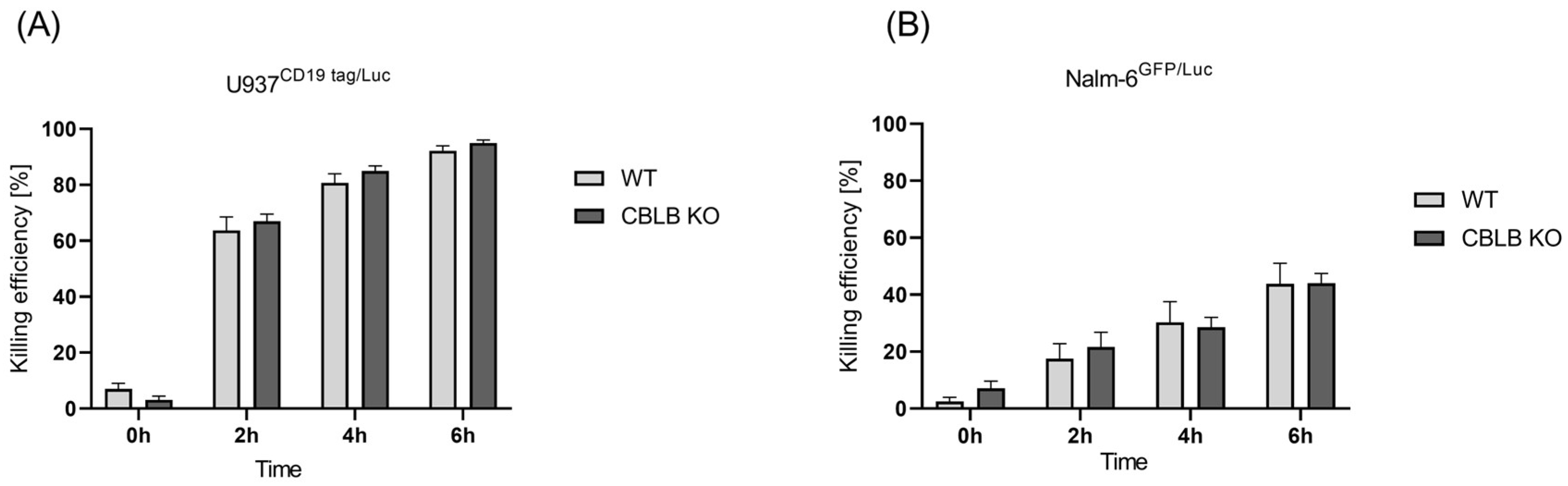
2.4. CRISPR-Mediated Blocking of CBLB as Intracellular Regulator of NK Cells
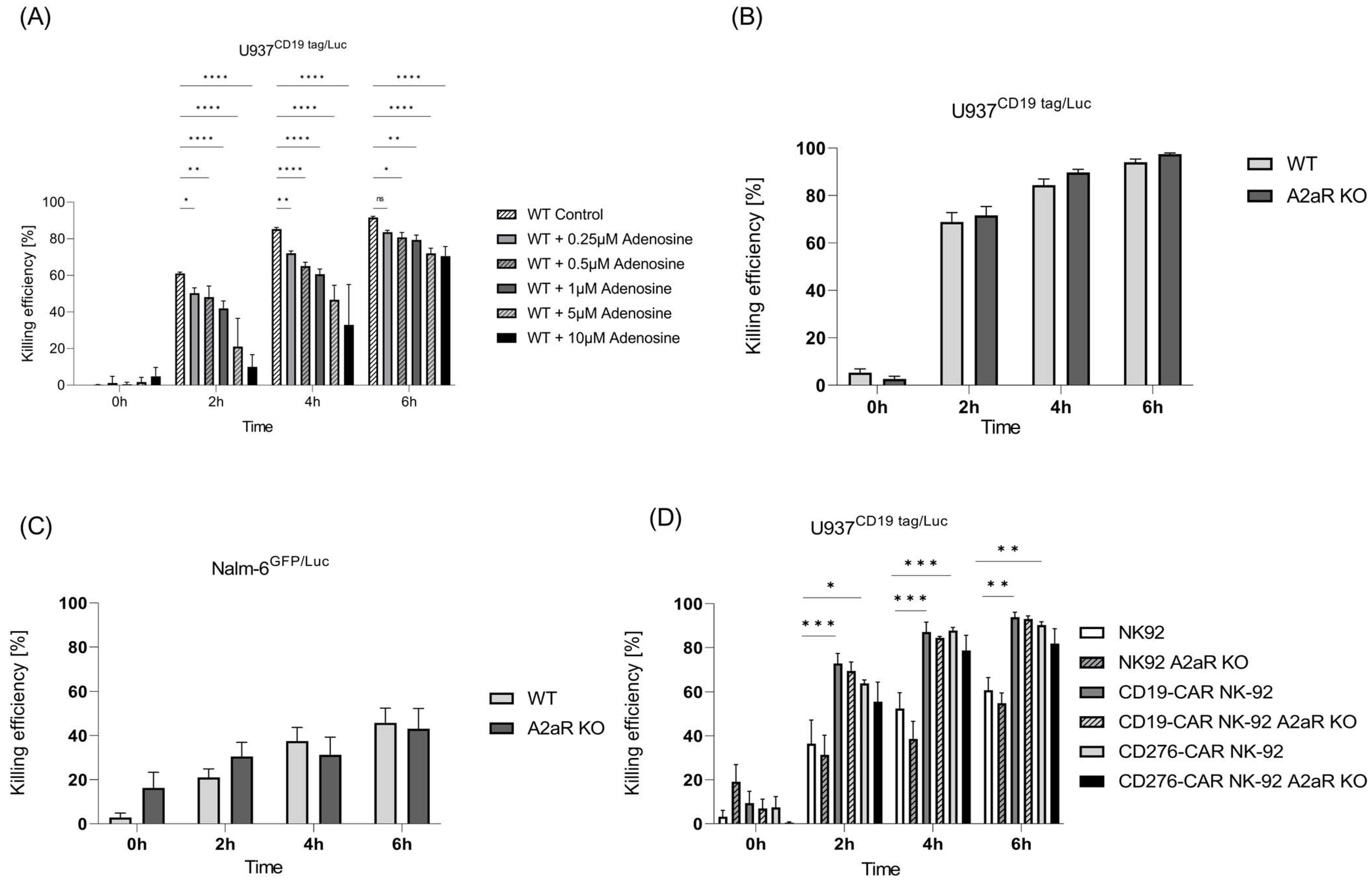
2.5. The Effect of Blocking Metabolic Purinergic Signaling on Antileukemic Function of NK Cells
3. Discussion
4. Materials and Methods
4.1. Cell Line Cultures
4.2. Primary Human NK Cell Isolation and Culture
4.3. Flow Cytometry
4.4. CRISPR-Cas9 Transfection
4.5. Evaluation of CRISPR-Cas9-Induced Knockout
4.6. Western Blot
4.7. Luciferase-Based Cytotoxicity Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Du, N.; Guo, F.; Wang, Y.; Cui, J. NK Cell Therapy: A Rising Star in Cancer Treatment. Cancers 2021, 13, 4129. [Google Scholar] [CrossRef] [PubMed]
- Lamers-Kok, N.; Panella, D.; Georgoudaki, A.M.; Liu, H.; Özkazanc, D.; Kučerová, L.; Duru, A.D.; Spanholtz, J.; Raimo, M. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J. Hematol. Oncol. 2022, 15, 164. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The Natural Cytotoxicity Receptors in Health and Disease. Front. Immunol. 2019, 10, 909. [Google Scholar] [CrossRef]
- Maddineni, S.; Silberstein, J.L.; Sunwoo, J.B. Emerging NK cell therapies for cancer and the promise of next generation engineering of iPSC-derived NK cells. J. ImmunoTherapy Cancer 2022, 10, e004693. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Kang, S.; Gao, X.; Zhang, L.; Yang, E.; Li, Y.; Yu, L. The Advances and Challenges of NK Cell-Based Cancer Immunotherapy. Curr. Oncol. 2021, 28, 1077–1093. [Google Scholar] [CrossRef]
- Cózar, B.; Greppi, M.; Carpentier, S.; Narni-Mancinelli, E.; Chiossone, L.; Vivier, E. Tumor-Infiltrating Natural Killer Cells. Cancer Discov. 2021, 11, 34–44. [Google Scholar] [CrossRef]
- Seliger, B.; Koehl, U. Underlying mechanisms of evasion from NK cells as rationale for improvement of NK cell-based immunotherapies. Front. Immunol. 2022, 13, 910595. [Google Scholar] [CrossRef]
- Allison, M.; Mathews, J.; Gilliland, T.; Mathew, S.O. Natural Killer Cell-Mediated Immunotherapy for Leukemia. Cancers 2022, 14, 843. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Jafari, D.; Mousavi, M.J.; Keshavarz Shahbaz, S.; Jafarzadeh, L.; Tahmasebi, S.; Spoor, J.; Esmaeilzadeh, A. E3 ubiquitin ligase Casitas B lineage lymphoma-b and its potential therapeutic implications for immunotherapy. Clin. Exp. Immunol. 2021, 204, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.S.; Atfield, A.; Venuprasad, K.; Krawczyk, C.; Sarao, R.; Elly, C.; Yang, C.; Arya, S.; Bachmaier, K.; Su, L.; et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity 2004, 21, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Reddi, A.L.; Ghosh, A.; Dimri, M.; Band, H. The Cbl family and other ubiquitin ligases: Destructive forces in control of antigen receptor signaling. Immunity 2004, 21, 7–17. [Google Scholar] [CrossRef]
- Lu, T.; Chen, L.; Mansour, A.G.; Yu, M.J.; Brooks, N.; Teng, K.Y.; Li, Z.; Zhang, J.; Barr, T.; Yu, J.; et al. Cbl-b Is Upregulated and Plays a Negative Role in Activated Human NK Cells. J. Immunol. 2021, 206, 677–685. [Google Scholar] [CrossRef]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef]
- Chirino, L.M.; Kumar, S.; Okumura, M.; Sterner, D.E.; Mattern, M.; Butt, T.R.; Kambayashi, T. TAM receptors attenuate murine NK-cell responses via E3 ubiquitin ligase Cbl-b. Eur. J. Immunol. 2020, 50, 48–55. [Google Scholar] [CrossRef]
- Liu, F.; Huang, J.; He, F.; Ma, X.; Fan, F.; Meng, M.; Zhuo, Y.; Zhang, L. CD96, a new immune checkpoint, correlates with immune profile and clinical outcome of glioma. Sci. Rep. 2020, 10, 10768. [Google Scholar] [CrossRef]
- Zhang, W.; Shao, Z.; Fu, R.; Wang, H.; Li, L.; Liu, H. Expressions of CD96 and CD123 in Bone Marrow Cells of Patients with Myelodysplastic Syndromes. Clin. Lab. 2015, 61, 1429–1434. [Google Scholar] [CrossRef]
- Blake, S.J.; Dougall, W.C.; Miles, J.J.; Teng, M.W.; Smyth, M.J. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 5183–5188. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731.e1713–1743.e1713. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Seow, S.V.; Wong, D.; Robinson, M.; Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Invest. 2019, 129, 2094–2106. [Google Scholar] [CrossRef] [PubMed]
- Ureña-Bailén, G.; Dobrowolski, J.M.; Hou, Y.; Dirlam, A.; Roig-Merino, A.; Schleicher, S.; Atar, D.; Seitz, C.; Feucht, J.; Antony, J.S.; et al. Preclinical Evaluation of CRISPR-Edited CAR-NK-92 Cells for Off-the-Shelf Treatment of AML and B-ALL. Int. J. Mol. Sci. 2022, 23, 12828. [Google Scholar] [CrossRef]
- Seth, R.; Singh, A. Leukemias in Children. Indian J. Pediatr. 2015, 82, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef]
- Arachchige, A.S.P.M. Human NK cells: From development to effector functions. Innate Immun. 2021, 27, 212–229. [Google Scholar] [CrossRef]
- Lapteva, N.; Szmania, S.M.; van Rhee, F.; Rooney, C.M. Clinical grade purification and expansion of natural killer cells. Crit. Rev. Oncog. 2014, 19, 121–132. [Google Scholar] [CrossRef]
- Grote, S.; Ureña-Bailén, G.; Chan, K.C.; Baden, C.; Mezger, M.; Handgretinger, R.; Schleicher, S. In Vitro Evaluation of CD276-CAR NK-92 Functionality, Migration and Invasion Potential in the Presence of Immune Inhibitory Factors of the Tumor Microenvironment. Cells 2021, 10, 1020. [Google Scholar] [CrossRef]
- Guo, X.; Mahlakõiv, T.; Ye, Q.; Somanchi, S.; He, S.; Rana, H.; DiFiglia, A.; Gleason, J.; van der Touw, W.; Hariri, R.; et al. CBLB ablation with CRISPR/Cas9 enhances cytotoxicity of human placental stem cell-derived NK cells for cancer immunotherapy. J. Immunother. Cancer 2021, 9, e001975. [Google Scholar] [CrossRef]
- Sundström, C.; Nilsson, K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 1976, 17, 565–577. [Google Scholar] [CrossRef]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H.J. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef] [PubMed]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Childs, R.W.; Berg, M. Bringing natural killer cells to the clinic: Ex vivo manipulation. Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; van Hoef, V.; Zhang, X.; Wennerberg, E.; Lorent, J.; Witt, K.; Masvidal, L.; Liang, S.; Murray, S.; Larsson, O.; et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood 2016, 128, 1475–1489. [Google Scholar] [CrossRef]
- Sportoletti, P.; De Falco, F.; Del Papa, B.; Baldoni, S.; Guarente, V.; Marra, A.; Dorillo, E.; Rompietti, C.; Adamo, F.M.; Ruggeri, L.; et al. NK Cells in Chronic Lymphocytic Leukemia and Their Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 6665. [Google Scholar] [CrossRef]
- Khaldoyanidi, S.; Nagorsen, D.; Stein, A.; Ossenkoppele, G.; Subklewe, M. Immune Biology of Acute Myeloid Leukemia: Implications for Immunotherapy. J. Clin. Oncol. 2021, 39, 419–432. [Google Scholar] [CrossRef]
- Bexte, T.; Alzubi, J.; Reindl, L.M.; Wendel, P.; Schubert, R.; Salzmann-Manrique, E.; von Metzler, I.; Cathomen, T.; Ullrich, E. CRISPR-Cas9-based gene editing of the immune checkpoint NKG2A enhances NK cell mediated cytotoxicity against multiple myeloma. Oncoimmunology 2022, 11, 2081415. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.C.; Mittal, D.; Aguilera, A.R.; Miles, J.J.; Lutzky, V.P.; de Andrade, L.F.; et al. Suppression of Metastases Using a New Lymphocyte Checkpoint Target for Cancer Immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Faroudi, M.; Utzny, C.; Salio, M.; Cerundolo, V.; Guiraud, M.; Müller, S.; Valitutti, S. Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: Manifestation of a dual activation threshold. Proc. Natl. Acad. Sci. USA 2003, 100, 14145–14150. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Arruga, F.; Serra, S.; Vitale, N.; Guerra, G.; Papait, A.; Baffour Gyau, B.; Tito, F.; Efremov, D.; Vaisitti, T.; Deaglio, S. Targeting of the A2A adenosine receptor counteracts immunosuppression in vivo in a mouse model of chronic lymphocytic leukemia. Haematologica 2021, 106, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, J.S.; Laursen, L.G.; Schuster, M.B.; Pundhir, S.; Schoof, E.; Ge, Y.; d’Altri, T.; Vitting-Seerup, K.; Rapin, N.; Gentil, C.; et al. Mutant CEBPA directly drives the expression of the targetable tumor-promoting factor CD73 in AML. Sci. Adv. 2019, 5, eaaw4304. [Google Scholar] [CrossRef]
- Brauneck, F.; Seubert, E.; Wellbrock, J.; Wiesch, J.S.Z.; Duan, Y.; Magnus, T.; Bokemeyer, C.; Koch-Nolte, F.; Menzel, S.; Fiedler, W. Combined Blockade of TIGIT and CD39 or A2AR Enhances NK-92 Cell-Mediated Cytotoxicity in AML. Int. J. Mol. Sci. 2021, 22, 12919. [Google Scholar] [CrossRef]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; Lai, J.; Chen, A.X.Y.; Meyran, D.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Mølck, C.; et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat. Commun. 2021, 12, 3236. [Google Scholar] [CrossRef]
- Arnold, D.P.; Xu, Y.; Takatori, S.C. Antibody binding reports spatial heterogeneities in cell membrane organization. Nat. Commun. 2023, 14, 2884. [Google Scholar] [CrossRef]
- Jiang, N.; Chen, W.; Jothikumar, P.; Patel, J.M.; Shashidharamurthy, R.; Selvaraj, P.; Zhu, C. Effects of anchor structure and glycosylation of Fcγ receptor III on ligand binding affinity. Mol. Biol. Cell 2016, 27, 3449–3458. [Google Scholar] [CrossRef]
- Van Audenaerde, J.R.M.; De Waele, J.; Marcq, E.; Van Loenhout, J.; Lion, E.; Van den Bergh, J.M.J.; Jesenofsky, R.; Masamune, A.; Roeyen, G.; Pauwels, P.; et al. Interleukin-15 stimulates natural killer cell-mediated killing of both human pancreatic cancer and stellate cells. Oncotarget 2017, 8, 56968–56979. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Kumar, R.; Kumar Singh, A.; Tsakem, E.L.; Kathania, M.; Riese, M.J.; Theiss, A.L.; Davila, M.L.; Venuprasad, K. Deletion of Cbl-b inhibits CD8(+) T-cell exhaustion and promotes CAR T-cell function. J. Immunother. Cancer 2021, 9, e001688. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Ureña-Bailén, G.; Mohammadian Gol, T.; Gratz, P.G.; Gratz, H.P.; Roig-Merino, A.; Antony, J.S.; Lamsfus-Calle, A.; Daniel-Moreno, A.; Handgretinger, R.; et al. Challenges in Gene Therapy for Somatic Reverted Mosaicism in X-Linked Combined Immunodeficiency by CRISPR/Cas9 and Prime Editing. Genes 2022, 13, 2348. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.R.; Choi, U.; Gao, J.-L.; Thompson, R.D.; Rodman, L.E.; Malech, H.L.; Kang, E.M. A Novel Method for Screening Adenosine Receptor Specific Agonists for Use in Adenosine Drug Development. Sci. Rep. 2017, 7, 44816. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammadian Gol, T.; Kim, M.; Sinn, R.; Ureña-Bailén, G.; Stegmeyer, S.; Gratz, P.G.; Zahedipour, F.; Roig-Merino, A.; Antony, J.S.; Mezger, M. CRISPR-Cas9-Based Gene Knockout of Immune Checkpoints in Expanded NK Cells. Int. J. Mol. Sci. 2023, 24, 16065. https://doi.org/10.3390/ijms242216065
Mohammadian Gol T, Kim M, Sinn R, Ureña-Bailén G, Stegmeyer S, Gratz PG, Zahedipour F, Roig-Merino A, Antony JS, Mezger M. CRISPR-Cas9-Based Gene Knockout of Immune Checkpoints in Expanded NK Cells. International Journal of Molecular Sciences. 2023; 24(22):16065. https://doi.org/10.3390/ijms242216065
Chicago/Turabian StyleMohammadian Gol, Tahereh, Miso Kim, Ralph Sinn, Guillermo Ureña-Bailén, Sarah Stegmeyer, Paul Gerhard Gratz, Fatemeh Zahedipour, Alicia Roig-Merino, Justin S. Antony, and Markus Mezger. 2023. "CRISPR-Cas9-Based Gene Knockout of Immune Checkpoints in Expanded NK Cells" International Journal of Molecular Sciences 24, no. 22: 16065. https://doi.org/10.3390/ijms242216065
APA StyleMohammadian Gol, T., Kim, M., Sinn, R., Ureña-Bailén, G., Stegmeyer, S., Gratz, P. G., Zahedipour, F., Roig-Merino, A., Antony, J. S., & Mezger, M. (2023). CRISPR-Cas9-Based Gene Knockout of Immune Checkpoints in Expanded NK Cells. International Journal of Molecular Sciences, 24(22), 16065. https://doi.org/10.3390/ijms242216065