
Estrobolome and Hepatocellular Adenomas—Connecting the Dots of the Gut Microbial β-Glucuronidase Pathway as a Metabolic Link

 , , and
, , and 
Abstract
1. Introduction
2. Methodology
3. Hepatocellular Adenomas

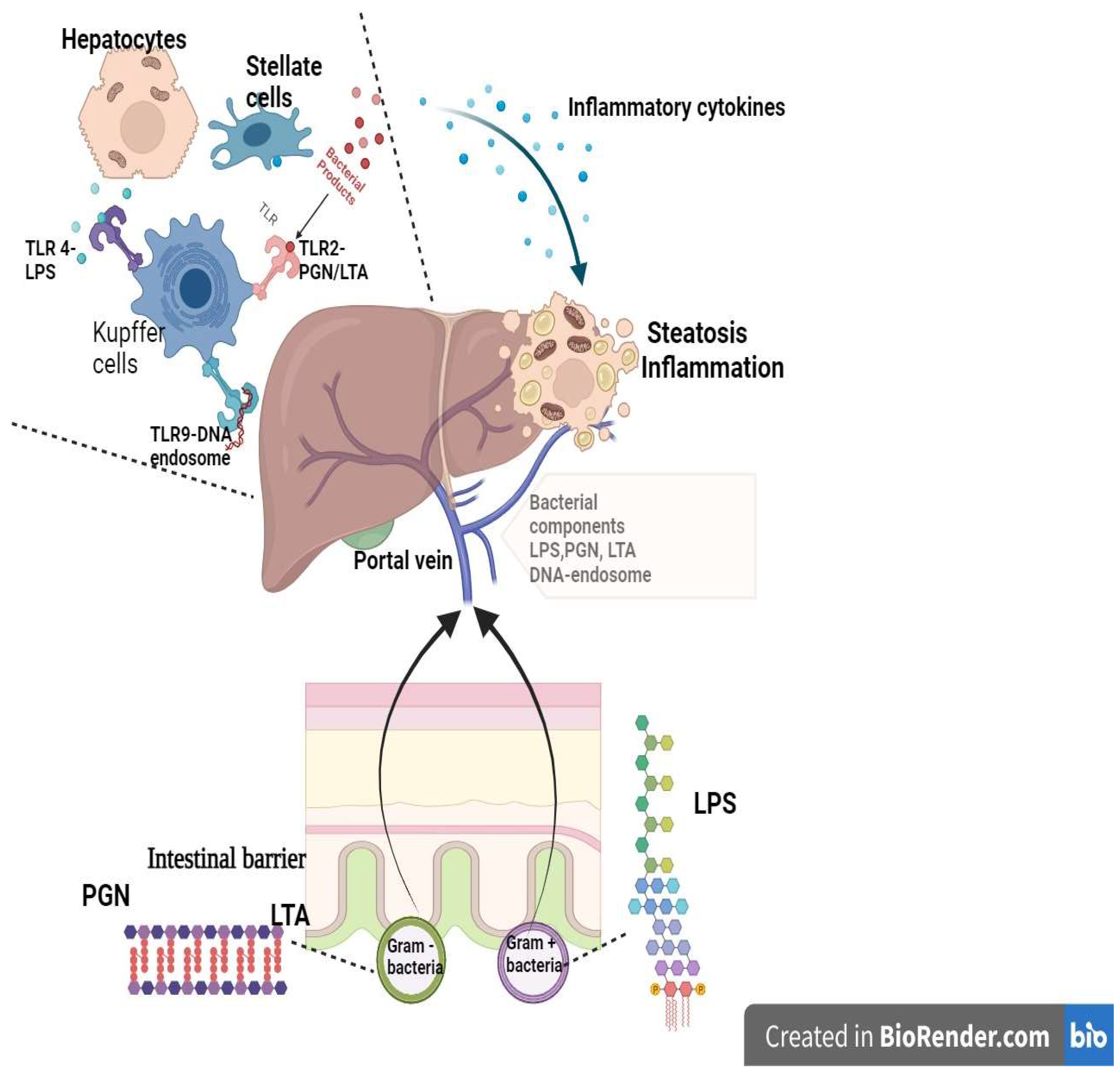{kind=link}
| HCA Classification | Incidence | Risk Factors | Clinical Characteristics | Complications |
|---|---|---|---|---|
| HNF1α-HCA (H-HCA) | 30–35% | Somatic or germline mutations of HNF1α; Over-exposure to exogenous estrogen [14] | Female; Familial adenomatosis; Fatty liver disease MODY 3 diabetes; | HCA Ø < 5 cm lower chance of malignancy; Lowest malignancy potential vs. other HCA subtypes; Rarely bleeding; |
| Inflammatory-HCA (I-HCA) | 35–45% the majority cases of HCA | Deletions of IL6ST gene; Mutations of IL-6/JAK/STAT2 family; Over-exposure to exogenous estrogen [14]; Metabolic syndrome and, or Obesity [14], Alcoholism | Obesity; Inflammatory phenotype: appearance of high serum amyloid (SAA) and C-reactive protein (CRP); | Rarely or not associated with malignancy potential; |
| Sonic hedgehog HCA | 4% | Over-exposure to exogenous estrogen [14]; Metabolic syndrome and, or Obesity [14] | Obesity | Greater possibility of bleeding |
| Unclassified HCA | 5–10% | - | - | - |
| β1-catenin (cadherin-associated protein) HCA [35] | ||||
| β-catenin HCA exon 3 [33] | 10–15% | Mutation in CTNNB1 gene exon [3]; High blood androgen exposure [14]; Liver vascular disease | Often in males [10]; Younger individuals; Frequently isolated tumors; | Highest chance of Hepatocellular carcinoma (HCC) [10]; Potential of bleeding [37]; |
| β-catenin HCA exon 7–8 [33] | 5–10% | Over-exposure to exogenous estrogen [14] | Often one tumor; Younger individuals; Inflammatory phenotype; | Low risk of HCC; Greater possibility of bleeding |
| Rare HCA subtypes [33] | ||||
| Pigmented HCA | very rare | Various genetic mutations, with pigment deposition of lipofuscin | High-level chance of carcinogenesis | |
| Myxoid HCA | Rare variant of H-HCA with extra mutations/separate subtype of HCA | Loss of L-FABP gene expression and/or HNF-1 α mutation; Recurrent mutations in PKA regulation of pathway (GNAS, CDKN1B and RNF123) or mutations of PKA pathway; | Older age | High-level chance of carcinogenesis |
| Atypical HCA/Borderline HCA/HUMP | - | - | Individuals > 50 years | Need more studies |
| I-HCA in cirrhotic liver | Need more studies | Fatty liver disease; Liver vascular disease; Metabolic syndrome; Alcoholic cirrhosis | - | Need more studies |
4. Estrogen Receptors and Evolution to Malignancy
5. Gastrointestinal Microbiome, Estrobolome and Bacterial Species Possessing β-Glucuronidase Enzymes
6. Estrogen-Microbiota Gut–Liver Axis
6.1. Microbiota in the Gut–Liver Axis
6.2. Estrogen-Microbiota-Gut Axis
7. The Estrobolome and Hepatocellular Adenomas
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, S.; Homaei, A.; Raju, A.B.; Meher, B.R. Estrogen: The Necessary Evil for Human Health, and Ways to Tame It. Biomed. Pharmacother. 2018, 102, 403–411. [Google Scholar] [CrossRef]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw029. [Google Scholar] [CrossRef]
- Plottel, C.S.; Blaser, M.J. Microbiome and Malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef]
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut Microbial β-Glucuronidases Reactivate Estrogens as Components of the Estrobolome That Reactivate Estrogens. J. Biol. Chem. 2019, 294, 18586–18599. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Wu, J.; Chen, J. The Role of Gut Microbial β-Glucuronidase in Estrogen Reactivation and Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 631552. [Google Scholar] [CrossRef]
- Jariwala, P.B.; Pellock, S.J.; Goldfarb, D.; Cloer, E.W.; Artola, M.; Simpson, J.B.; Bhatt, A.P.; Walton, W.G.; Roberts, L.R.; Major, M.B.; et al. Discovering the Microbial Enzymes Driving Drug Toxicity with Activity-Based Protein Profiling. ACS Chem. Biol. 2020, 15, 217–225. [Google Scholar] [CrossRef]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen–Gut Microbiome Axis: Physiological and Clinical Implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef]
- Mani, S.; Boelsterli, U.A.; Redinbo, M.R. Understanding and Modulating Mammalian-Microbial Communication for Improved Human Health. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 559–580. [Google Scholar] [CrossRef] [PubMed]
- Stoot, J.H.M.B.; Coelen, R.J.S.; De Jong, M.C.; Dejong, C.H.C. Malignant Transformation of Hepatocellular Adenomas into Hepatocellular Carcinomas: A Systematic Review Including More than 1600 Adenoma Cases. HPB 2010, 12, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Védie, A.-L.; Sutter, O.; Ziol, M.; Nault, J.-C. Molecular Classification of Hepatocellular Adenomas: Impact on Clinical Practice. Hepat. Oncol. 2018, 5, HEP04. [Google Scholar] [CrossRef]
- Renzulli, M.; Clemente, A.; Tovoli, F.; Cappabianca, S.; Bolondi, L.; Golfieri, R. Hepatocellular Adenoma: An Unsolved Diagnostic Enigma. World J. Gastroenterol. 2019, 25, 2442–2449. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X. Hepatocellular Adenoma: Where Are We Now? World J. Gastroenterol. 2022, 28, 1384–1393. [Google Scholar] [CrossRef] [PubMed]
- Giannitrapani, L.; Soresi, M.; La Spada, E.; Cervello, M.; D’Alessandro, N.; Montalto, G. Sex Hormones and Risk of Liver Tumor. Ann. N. Y. Acad. Sci. 2006, 1089, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Jargalsaikhan, O.; Ichimura-Shimizu, M.; Cai, Q.; Ogawa, H.; Miyakami, Y.; Atsumi, K.; Tomita, M.; Sutoh, M.; Toyohara, S.; et al. Spontaneous Occurrence of Various Types of Hepatocellular Adenoma in the Livers of Metabolic Syndrome-Associated Steatohepatitis Model TSOD Mice. Int. J. Mol. Sci. 2022, 23, 11923. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Pollet, R.M.; D’Agostino, E.H.; Walton, W.G.; Xu, Y.; Little, M.S.; Biernat, K.A.; Pellock, S.J.; Patterson, L.M.; Creekmore, B.C.; Isenberg, H.N.; et al. An Atlas of β-Glucuronidases in the Human Intestinal Microbiome. Structure 2017, 25, 967–977.e5. [Google Scholar] [CrossRef] [PubMed]
- Haro, C.; Rangel-Zúñiga, O.A.; Alcalá-Díaz, J.F.; Gómez-Delgado, F.; Pérez-Martínez, P.; Delgado-Lista, J.; Quintana-Navarro, G.M.; Landa, B.B.; Navas-Cortés, J.A.; Tena-Sempere, M.; et al. Intestinal Microbiota Is Influenced by Gender and Body Mass Index. PLoS ONE 2016, 11, e0154090. [Google Scholar] [CrossRef]
- Org, E.; Mehrabian, M.; Parks, B.W.; Shipkova, P.; Liu, X.; Drake, T.A.; Lusis, A.J. Sex Differences and Hormonal Effects on Gut Microbiota Composition in Mice. Gut Microbes 2016, 7, 313–322. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Lay, C.; Rigottier-Gois, L.; Holmstrøm, K.; Rajilic, M.; Vaughan, E.E.; de Vos, W.M.; Collins, M.D.; Thiel, R.; Namsolleck, P.; Blaut, M.; et al. Colonic Microbiota Signatures across Five Northern European Countries. Appl. Environ. Microbiol. 2005, 71, 4153–4155. [Google Scholar] [CrossRef]
- Mueller, S.; Saunier, K.; Hanisch, C.; Norin, E.; Alm, L.; Midtvedt, T.; Cresci, A.; Silvi, S.; Orpianesi, C.; Verdenelli, M.C.; et al. Differences in Fecal Microbiota in Different European Study Populations in Relation to Age, Gender, and Country: A Cross-Sectional Study. Appl. Environ. Microbiol. 2006, 72, 1027–1033. [Google Scholar] [CrossRef]
- Sinha, T.; Vich Vila, A.; Garmaeva, S.; Jankipersadsing, S.A.; Imhann, F.; Collij, V.; Bonder, M.J.; Jiang, X.; Gurry, T.; Alm, E.J.; et al. Analysis of 1135 Gut Metagenomes Identifies Sex-Specific Resistome Profiles. Gut Microbes 2019, 10, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, B.; Zhang, M.; Rantalainen, M.; Wang, S.; Zhou, H.; Zhang, Y.; Shen, J.; Pang, X.; Zhang, M.; et al. Symbiotic Gut Microbes Modulate Human Metabolic Phenotypes. Proc. Natl. Acad. Sci. USA 2008, 105, 2117–2122. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, M.; Xue, J.; Huang, J.; Zhuang, R.; Zhou, X.; Zhang, H.; Fu, Q.; Hao, Y. Body Mass Index Differences in the Gut Microbiota Are Gender Specific. Front. Microbiol. 2018, 9, 1250. [Google Scholar] [CrossRef] [PubMed]
- Dominianni, C.; Sinha, R.; Goedert, J.J.; Pei, Z.; Yang, L.; Hayes, R.B.; Ahn, J. Sex, Body Mass Index, and Dietary Fiber Intake Influence the Human Gut Microbiome. PLoS ONE 2015, 10, e0124599. [Google Scholar] [CrossRef]
- Singh, P.; Manning, S.D. Impact of Age and Sex on the Composition and Abundance of the Intestinal Microbiota in Individuals with and without Enteric Infections. Ann. Epidemiol. 2016, 26, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Borgo, F.; Garbossa, S.; Riva, A.; Severgnini, M.; Luigiano, C.; Benetti, A.; Pontiroli, A.E.; Morace, G.; Borghi, E. Body Mass Index and Sex Affect Diverse Microbial Niches within the Gut. Front. Microbiol. 2018, 9, 213. [Google Scholar] [CrossRef]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Dohi, O.; Yoshida, N.; Kamada, K.; et al. Differences in Gut Microbiota Associated with Age, Sex, and Stool Consistency in Healthy Japanese Subjects. J. Gastroenterol. 2019, 54, 53–63. [Google Scholar] [CrossRef]
- Kovacs, A.; Ben-Jacob, N.; Tayem, H.; Halperin, E.; Iraqi, F.A.; Gophna, U. Genotype Is a Stronger Determinant than Sex of the Mouse Gut Microbiota. Microb. Ecol. 2011, 61, 423–428. [Google Scholar] [CrossRef]
- Human Microbiome Project Consortium. Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Duncan, S.H.; Lobley, G.E.; Holtrop, G.; Ince, J.; Johnstone, A.M.; Louis, P.; Flint, H.J. Human Colonic Microbiota Associated with Diet, Obesity and Weight Loss. Int. J. Obes. 2008, 32, 1720–1724. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial Ecology: Human Gut Microbes Associated with Obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Bioulac-Sage, P.; Sempoux, C.; Balabaud, C. Hepatocellular Adenoma: Classification, Variants and Clinical Relevance. Semin. Diagn. Pathol. 2017, 34, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, Y.N. Hepatocellular Adenomas: Recent Updates. J. Pathol. Transl. Med. 2021, 55, 171–180. [Google Scholar] [CrossRef]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouzé, E.; Imbeaud, S.; Pilati, C.; Nault, J.-C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-Phenotype Correlation of CTNNB1 Mutations Reveals Different ß-Catenin Activity Associated with Liver Tumor Progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL). EASL Clinical Practice Guidelines on the Management of Benign Liver Tumours. J. Hepatol. 2016, 65, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.-C.; Couchy, G.; Balabaud, C.; Morcrette, G.; Caruso, S.; Blanc, J.-F.; Bacq, Y.; Calderaro, J.; Paradis, V.; Ramos, J.; et al. Molecular Classification of Hepatocellular Adenoma Associates with Risk Factors, Bleeding, and Malignant Transformation. Gastroenterology 2017, 152, 880–894.e6. [Google Scholar] [CrossRef]
- Shafizadeh, N.; Genrich, G.; Ferrell, L.; Kakar, S. Hepatocellular Adenomas in a Large Community Population, 2000 to 2010: Reclassification per Current World Health Organization Classification and Results of Long-Term Follow-Up. Hum. Pathol. 2014, 45, 976–983. [Google Scholar] [CrossRef]
- Haring, M.P.D.; Gouw, A.S.H.; de Haas, R.J.; Cuperus, F.J.C.; de Jong, K.P.; de Meijer, V.E. The Effect of Oral Contraceptive Pill Cessation on Hepatocellular Adenoma Diameter: A Retrospective Cohort Study. Liver Int. 2019, 39, 905–913. [Google Scholar] [CrossRef]
- Klompenhouwer, A.J.; van Rosmalen, B.V.; Haring, M.P.D.; Thomeer, M.G.J.; Doukas, M.; Verheij, J.; de Meijer, V.E.; van Gulik, T.M.; Takkenberg, R.B.; Kazemier, G.; et al. A Multicentre Retrospective Analysis on Growth of Residual Hepatocellular Adenoma after Resection. Liver Int. 2020, 40, 2272–2278. [Google Scholar] [CrossRef]
- Krause, K.; Tanabe, K.K. A Shifting Paradigm in Diagnosis and Management of Hepatic Adenoma. Ann. Surg. Oncol. 2020, 27, 3330–3338. [Google Scholar] [CrossRef] [PubMed]
- Torbenson, M. Hepatic Adenomas. Surg. Pathol. Clin. 2018, 11, 351–366. [Google Scholar] [CrossRef]
- Villemain, L.; Prigent, S.; Abou-Lovergne, A.; Pelletier, L.; Chiral, M.; Pontoglio, M.; Foufelle, F.; Caruso, S.; Pineau, R.; Rebouissou, S.; et al. Sigma 1 Receptor Is Overexpressed in Hepatocellular Adenoma: Involvement of ERα and HNF1α. Cancers 2020, 12, 2213. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T. The Sigma-1 Receptor in Cellular Stress Signaling. Front. Neurosci. 2019, 13, 733. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Cao, J.; Shi, H. Effects of Estrogen and Estrogen Receptors on Transcriptomes of HepG2 Cells: A Preliminary Study Using RNA Sequencing. Int. J. Endocrinol. 2018, 2018, 5789127. [Google Scholar] [CrossRef] [PubMed]
- Uebi, T.; Umeda, M.; Imai, T. Estrogen Induces Estrogen Receptor Alpha Expression and Hepatocyte Proliferation in the Livers of Male Mice. Genes Cells 2015, 20, 217–223. [Google Scholar] [CrossRef]
- Guillaume, M.; Riant, E.; Fabre, A.; Raymond-Letron, I.; Buscato, M.; Davezac, M.; Tramunt, B.; Montagner, A.; Smati, S.; Zahreddine, R.; et al. Selective Liver Estrogen Receptor α Modulation Prevents Steatosis, Diabetes, and Obesity Through the Anorectic Growth Differentiation Factor 15 Hepatokine in Mice. Hepatol. Commun. 2019, 3, 908–924. [Google Scholar] [CrossRef]
- Lau, M.Y.; Han, H.; Hu, J.; Ji, C. Association of Cyclin D and Estrogen Receptor A36 with Hepatocellular Adenomas of Female Mice under Chronic Endoplasmic Reticulum Stress: Hepatocellular Adenomas of Female Mice. J. Gastroenterol. Hepatol. 2013, 28, 576–583. [Google Scholar] [CrossRef]
- Pelletier, L.; Rebouissou, S.; Paris, A.; Rathahao-Paris, E.; Perdu, E.; Bioulac-Sage, P.; Imbeaud, S.; Zucman-Rossi, J. Loss of Hepatocyte Nuclear Factor 1α Function in Human Hepatocellular Adenomas Leads to Aberrant Activation of Signaling Pathways Involved in Tumorigenesis. Hepatology 2010, 51, 557–566. [Google Scholar] [CrossRef]
- Longcope, C.; Gorbach, S.; Goldin, B.; Woods, M.; Dwyer, J.; Warram, J. The Metabolism of Estradiol; Oral Compared to Intravenous Administration. J. Steroid Biochem. 1985, 23, 1065–1070. [Google Scholar] [CrossRef]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal Microbial Determinants of Fecal and Systemic Estrogens and Estrogen Metabolites: A Cross-Sectional Study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef]
- Lephart, E.D.; Naftolin, F. Estrogen Action and Gut Microbiome Metabolism in Dermal Health. Dermatol. Ther. 2022, 12, 1535–1550. [Google Scholar] [CrossRef]
- Rose, D.P. Diet, Hormones, and Cancer. Annu. Rev. Public Health 1993, 14, 1–17. [Google Scholar] [CrossRef][Green Version]
- Zhong, Z.; Sun, B.; Shu, N.; Xie, Q.; Tang, X.; Ling, Z.; Wang, F.; Zhao, K.; Xu, P.; Zhang, M.; et al. Ciprofloxacin Blocked Enterohepatic Circulation of Diclofenac and Alleviated NSAID-Induced Enteropathy in Rats Partly by Inhibiting Intestinal β-Glucuronidase Activity. Acta Pharmacol. Sin. 2016, 37, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Jardou, M.; Brossier, C.; Guiyedi, K.; Faucher, Q.; Lawson, R. Pharmacological Hypothesis: A Recombinant Probiotic for Taming Bacterial Β-glucuronidase in Drug-induced Enteropathy. Pharmacol. Res. Perspect. 2022, 10, e00998. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.P.; Pellock, S.J.; Biernat, K.A.; Walton, W.G.; Wallace, B.D.; Creekmore, B.C.; Letertre, M.M.; Swann, J.R.; Wilson, I.D.; Roques, J.R.; et al. Targeted Inhibition of Gut Bacterial β-Glucuronidase Activity Enhances Anticancer Drug Efficacy. Proc. Natl. Acad. Sci. USA 2020, 117, 7374–7381. [Google Scholar] [CrossRef]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The Gut-Liver Axis and the Intersection with the Microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.-H.; Jin, Z.; Yang, X.-X.; Lou, J.; Shan, W.-X.; Hu, Y.-X.; Du, Q.; Liao, Q.-S.; Xie, R.; Xu, J.-Y. Role of Gut Microbiota via the Gut-Liver-Brain Axis in Digestive Diseases. World J. Gastroenterol. 2020, 26, 6141–6162. [Google Scholar] [CrossRef]
- Wang, R.; Tang, R.; Li, B.; Ma, X.; Schnabl, B.; Tilg, H. Gut Microbiome, Liver Immunology, and Liver Diseases. Cell Mol. Immunol. 2021, 18, 4–17. [Google Scholar] [CrossRef]
- Ji, Y.; Yin, Y.; Li, Z.; Zhang, W. Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2019, 11, 1712. [Google Scholar] [CrossRef]
- Alferink, L.J.M.; Radjabzadeh, D.; Erler, N.S.; Vojinovic, D.; Medina-Gomez, C.; Uitterlinden, A.G.; De Knegt, R.J.; Amin, N.; Ikram, M.A.; Janssen, H.L.A.; et al. Microbiomics, Metabolomics, Predicted Metagenomics, and Hepatic Steatosis in a Population-Based Study of 1355 Adults. Hepatology 2021, 73, 968–982. [Google Scholar] [CrossRef] [PubMed]
- Samavat, H.; Kurzer, M.S. Estrogen Metabolism and Breast Cancer. Cancer Lett. 2015, 356 Part A, 231–243. [Google Scholar] [CrossRef]
- An, S.-L.; Wang, L.-M.; Rong, W.-Q.; Wu, F.; Sun, W.; Yu, W.-B.; Feng, L.; Liu, F.-Q.; Tian, F.; Wu, J.-X. Hepatocellular Adenoma with Malignant Transformation in Male Patients with Non-Cirrhotic Livers. Chin. J. Cancer 2015, 34, 17. [Google Scholar] [CrossRef] [PubMed]
- Van Rosmalen, B.V.; Furumaya, A.; Klompenhouwer, A.J.; Tushuizen, M.E.; Braat, A.E.; Reinten, R.J.; Ligthart, M.A.P.; Haring, M.P.D.; De Meijer, V.E.; Van Voorthuizen, T.; et al. Hepatocellular Adenoma in Men: A Nationwide Assessment of Pathology and Correlation with Clinical Course. Liver Int. 2021, 41, 2474–2484. [Google Scholar] [CrossRef] [PubMed]
- Patricia Sîrbu Boeți, M.; Tivadar, B.; Lupescu, I.G.; Herlea, V.; Boroș, M.; Tomescu, D.; Brașoveanu, V. Challenging Issues in Hepatic Adenoma. In Liver Disease and Surgery; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bucurica, S.; Lupanciuc, M.; Ionita-Radu, F.; Stefan, I.; Munteanu, A.E.; Anghel, D.; Jinga, M.; Gaman, E.L. Estrobolome and Hepatocellular Adenomas—Connecting the Dots of the Gut Microbial β-Glucuronidase Pathway as a Metabolic Link. Int. J. Mol. Sci. 2023, 24, 16034. https://doi.org/10.3390/ijms242216034
Bucurica S, Lupanciuc M, Ionita-Radu F, Stefan I, Munteanu AE, Anghel D, Jinga M, Gaman EL. Estrobolome and Hepatocellular Adenomas—Connecting the Dots of the Gut Microbial β-Glucuronidase Pathway as a Metabolic Link. International Journal of Molecular Sciences. 2023; 24(22):16034. https://doi.org/10.3390/ijms242216034
Chicago/Turabian StyleBucurica, Sandica, Mihaela Lupanciuc, Florentina Ionita-Radu, Ion Stefan, Alice Elena Munteanu, Daniela Anghel, Mariana Jinga, and Elena Laura Gaman. 2023. "Estrobolome and Hepatocellular Adenomas—Connecting the Dots of the Gut Microbial β-Glucuronidase Pathway as a Metabolic Link" International Journal of Molecular Sciences 24, no. 22: 16034. https://doi.org/10.3390/ijms242216034
APA StyleBucurica, S., Lupanciuc, M., Ionita-Radu, F., Stefan, I., Munteanu, A. E., Anghel, D., Jinga, M., & Gaman, E. L. (2023). Estrobolome and Hepatocellular Adenomas—Connecting the Dots of the Gut Microbial β-Glucuronidase Pathway as a Metabolic Link. International Journal of Molecular Sciences, 24(22), 16034. https://doi.org/10.3390/ijms242216034