1.1. Background
Time-lapse microscopy is essential for studying cell proliferation, migration, and other dynamic cellular processes. It helps to understand fundamental biological mechanisms, such as tissue formation and repair, wound healing, and tumor therapy [
1]. Detecting and tracking cellular behavior is crucial when studying the biological characteristics of many cells, especially during the early embryonic stages [
2]. However, it is difficult to extract and locate the iPS progenitor cells, namely, iPS-forming cells, during the early reprogramming stage; this is because no known biomarkers are available to map iPS progenitor cells [
3]. However, after six days of induction, iPS cells can be tested with fluorescent probes, and the proportion of iPS progenitor cells in the early stage of reprogramming is less than 5%. This indicates that the number of iPS cells available during the experimental determination stage is deficient.
Different imaging modalities, including magnetic resonance imaging, mammography, breast sonography, and magnetic resonance tomography, have found extensive applications in medical diagnostics, particularly in histopathology studies [
4]. Mammograms, which are valued for their cost-effective high sensitivity, are the preferred method for early detection; they excel in detecting masses and microcalcifications, while maintaining reliability through reduced radiation exposure, thereby contributing significantly to histopathology studies [
5]. Recently, a novel framework utilizing neural network concepts and reduced feature vectors, combined with ensemble learning, was introduced, achieving superior accuracy in classifying mitotic and non-mitotic cells in breast cancer histology images, thereby contributing to enhanced cancer diagnoses [
6]. Moreover, Labrada and Barkana introduced a breast cancer diagnosis methodology using histopathology images, achieving robust classification via three feature sets and four machine learning (ML) algorithms [
7]. Chowanda investigated optimal deep learning (DL) parameters for breast cancer classifications; they introduced a modified architecture and evaluated ML algorithms using mammogram images [
8].
Recently, several studies on iPS cell identification during the early stages of reprogramming focused on the analysis of tracking and detection results, as well as the characteristics of cell migration trajectories, so that researchers can understand the characteristics of iPS cells [
9,
10]. However, modern imaging techniques have created large volumes of data that cannot be analyzed manually. Additionally, it is challenging for specialized personnel to detect low-resolution objects. Traditional cell detection and tracking methods need to design feature extraction operators to capture the unique pattern of each cell type, which usually requires professional knowledge and complex adjustment processes [
11]. As presented in natural image tracking benchmarks, traditional methods have dominated cell detection and tracking due to the need for more high-quality annotations [
12,
13].
Cell detection methods can be classified into three major categories. The first category consists of thresholding methods that separate the cell’s foreground and background in different pixel value ranges [
14]. The second category consists of feature extraction methods based on extraction operators, such as the scale-invariant feature transform, followed by a self-labeling algorithm, and two clustering steps to detect unstained cells [
15]. The third category consists of edge detection methods that use watersheds to detect cell edges based on gradient changes and corresponding connected regions [
16]. A review of cell detection methods for label-free contrast microscopy is provided in Ref. [
17].
The two most frequently used approaches for cell tracking are tracking via model evolution and tracking via detection; each approach tackles some challenges more efficiently than the other [
18]. Tracking via model evolution method combines segmentation and tracking; this is achieved by describing cell contours over time using an evolving mathematical representation [
19]. Despite the ability to produce more accurate contours and individual cell tracks in some applications, these approaches require the initial seeding of cell contours. They could be more computationally efficient for multicell tracking. As examples, parametric models, including an active contour-based “snakes” model in two dimensions [
20], and a dynamic mesh or a deformable model in three dimensions [
21], may be used. Implicit methods such as the advanced level-set-based multicell segmentation and tracking algorithm and the Chan–Vese Model, may also be used; these methods naturally manage splits, and they merge the new appearances of the cells [
22,
23].
In tracking by detection, cells are first detected using image segmentation and then tracked to establish correspondence between cells across all frames [
19]. Segmentation procedures can be performed via gradient features, intensity features, wavelet decomposition, and region-based or edge-based features [
24]. Comparing cell tracking algorithms revealed that most tracking approaches use nearest neighbors, graph-based linking, or multiple hypotheses [
13]. The Viterbi algorithm was proposed to link cell outlines generated by a segmentation method into tracks [
25]. In Ref. [
26], a joint segmentation-tracking algorithm was presented in which the model parameters are learned using Bayesian risk minimization. A comprehensive review of computational methods utilized in cell tracking is provided in Ref. [
27].
Artificial intelligence (AI)-/DL-based detection and tracking approaches have made significant progress in this area and have demonstrated better performance when compared to traditional methods [
28]. Despite the success of DL methods in tracking multiple objects on natural images, researchers have developed only a few DL approaches for tracking individual cells. Payer et al. [
29] presented a method that simultaneously segments and tracks cells using a pixel-wise metric embedding learning strategy combined with a recurrent hourglass network. In Ref. [
30], the authors combined a CNN-based observation algorithm with a particle-filter-based method to track non-rigid and non-significant cells.
CNN-based models have demonstrated the ability to perform better in many computer vision tasks without manual annotations [
31]. They can work well with prior knowledge on specific tasks to achieve almost the same performance as models trained with manually labeled data [
32]. Nishimura et al. proposed weakly supervised cell tracking, which uses the detection results (i.e., the coordinates of cell positions) to train the CNN model without association information, in which nuclear staining can be used to quickly determine cell positions [
33]. Similarly, a semi-automated cell tracker was developed using a CNN-based ML algorithm to detect cell nuclei [
34]. The detection results were linked at different time points using a minimum-cost flow solver to form cell trajectories [
35].
Moreover, a DL technique called cross-modality inference or transformation accurately predicted fluorescent labels from transmitted-light microscopy images of unlabeled biological samples using the training data composed of a pair of images from different imaging modalities [
36]. The authors in Ref. [
37] revealed that the training dataset formed by a couple of brightfield and fluorescence image modalities of the same cells could be used to train the robust network. This network can learn to predict fluorescent labels from electron microscope images and alleviate the need to acquire the corresponding fluorescence images [
37]. In addition, this capability is especially suitable for long-term live-cell imaging, where low phototoxicity acquisitions offer significant benefits. The same method performed better using a small dataset of around 30 to 40 images [
28] and could differentiate cell type and subcellular structures [
38].
However, state-of-the-art traditional and DL cell tracking methods still rely on the supervision of manual annotations, which is a time-consuming task requiring qualified professional experimenters’ participation [
39]. In particular, cell detection and tracking require further development for low signal-to-noise ratio and three-dimensional data [
13]. Moreover, the cells may adhere to each other due to persistent cell migration, making it challenging to determine cell boundaries. Similarly, cell division and differentiation processes may lead to continuous changes in appearance, making it difficult to label the cells precisely. Since iPS cells are cultured in a nutrient solution, there are complex texture features in the background of the cell image and the phototoxicity of fluorescence, which can lead to a significant decline in imaging performance. Furthermore, brightfield images are sometimes taken at high magnification, which causes some impurities and bubbles in the images and consequently causes difficulties in cell labeling because experimenters are required to correct repeated errors.
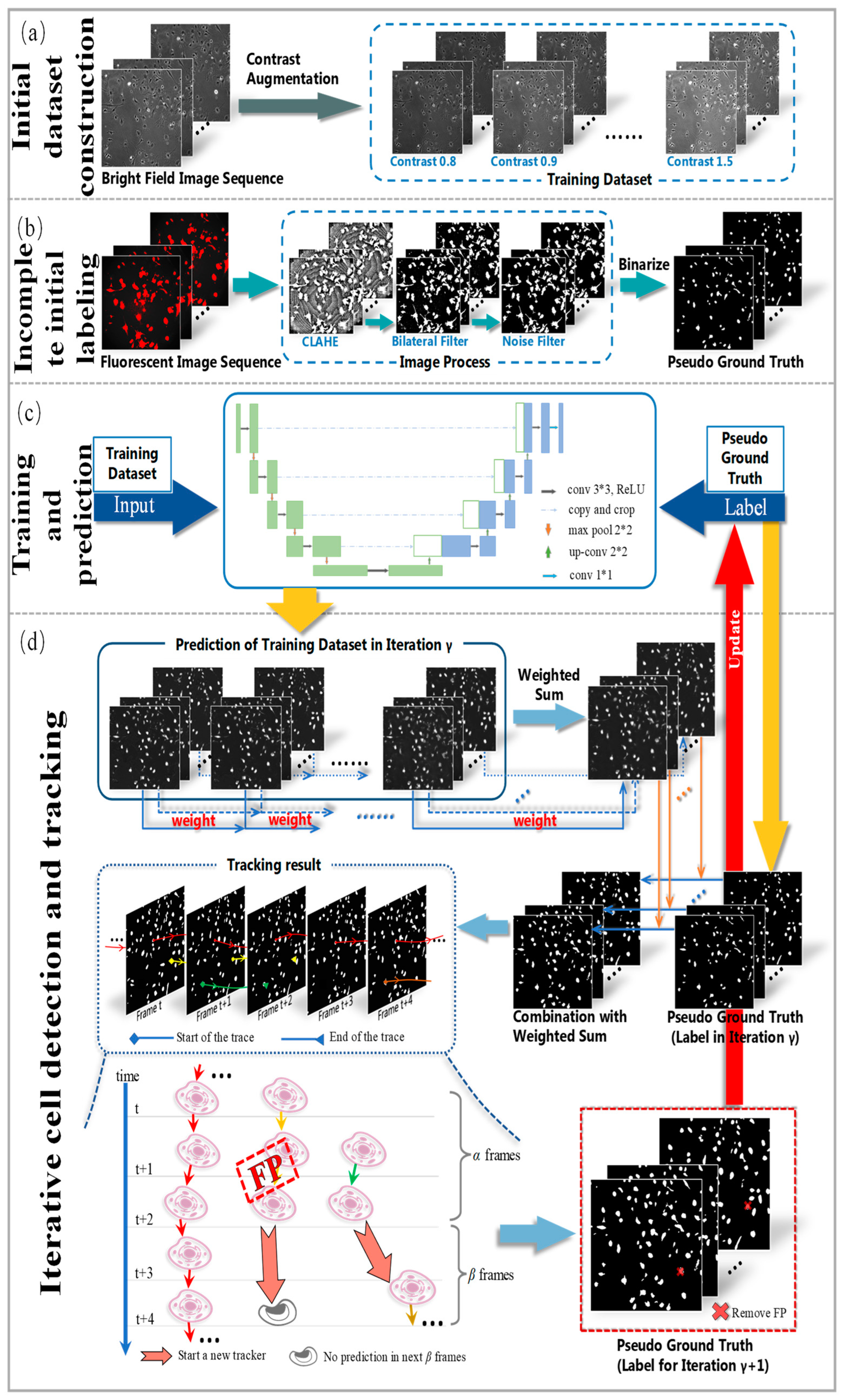
To address these issues, we propose a novel weakly supervised learning method utilizing incomplete initial annotations for automated cell detection and tracking on the iPS cell dataset including brightfield and fluorescence images in the early reprogramming stage. Specifically, we aim to investigate whether this approach can successfully handle the complexities posed by dynamic changes in cell morphology, shape, and thickness during processes such as cell division and differentiation, ultimately achieving performance comparable to or exceeding traditional fully supervised methods. By integrating morphological shape and thickness analysis into the training process, the proposed method demonstrated robust performance, even with incomplete initial annotations, and outperformed traditional fully supervised methods that rely on extensive manual annotations. Inspired by automated curriculum learning [
40], the training process starts with incomplete initial annotations generated from paired red fluorescent images taken under the point light. This is similar to beginning training with simple samples in the early stages of curriculum learning. Although all cells cannot be labeled at this stage, we train the model iteratively, where the analysis of cell tracking results is used to update optimized labels in the subsequent training process. Similar to the continuous addition of complex samples in curriculum learning, the updated results are used to continue the training process in our method. Therefore, we updated and improved the incomplete initial labels using the tracking-by-detection algorithm to develop a robust cell detector. This study contributes to advancing the automated microscopy image analysis field, with potential implications for a wide range of biomedical research applications. The code and data for our work, along with more technical details, can be found by visiting the following link:
https://github.com/jovialniyo93/cell-detection-and-tracking (accessed on 21 June 2023).
The contributions and novelties of this paper can be summarized as follows:
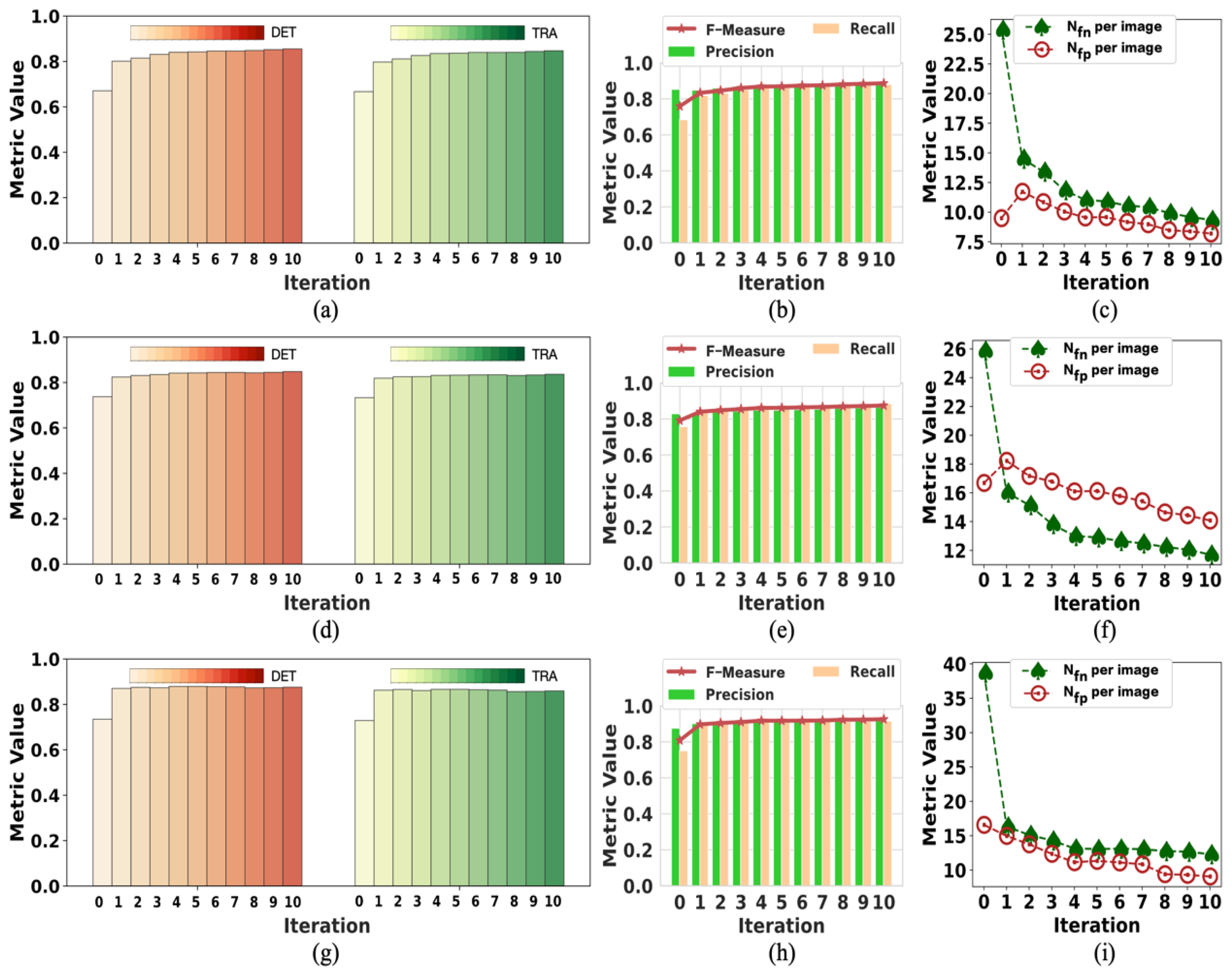
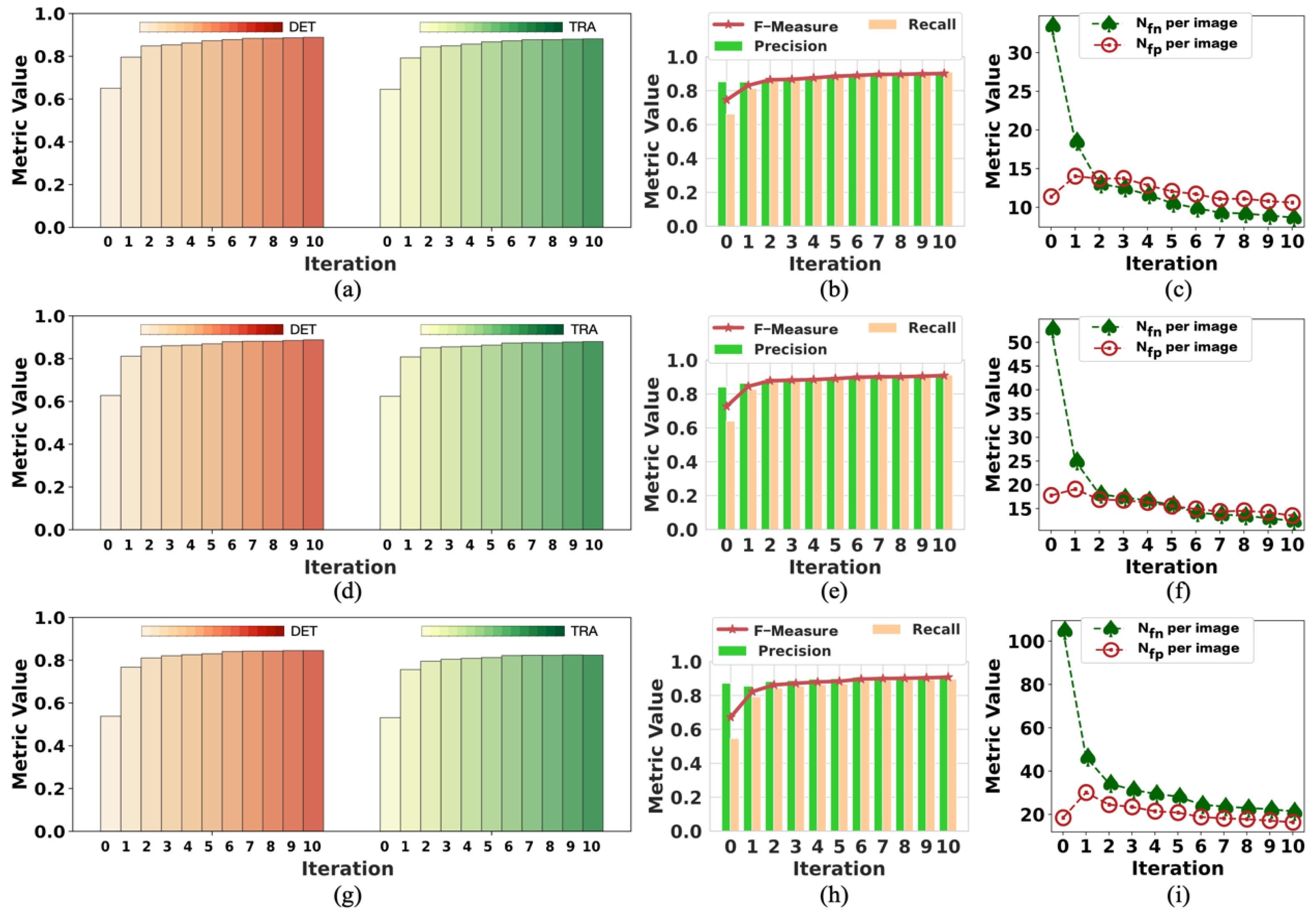
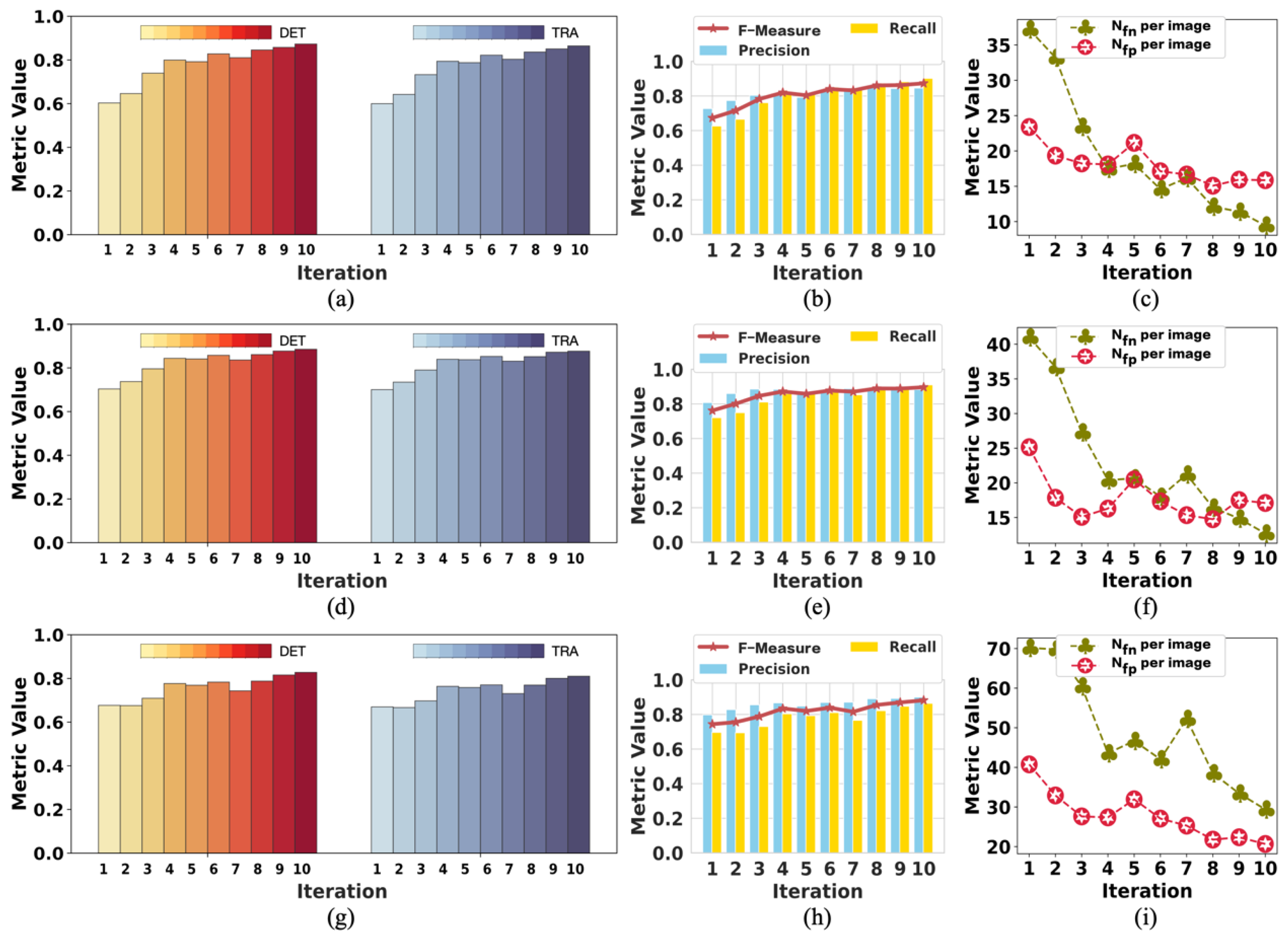
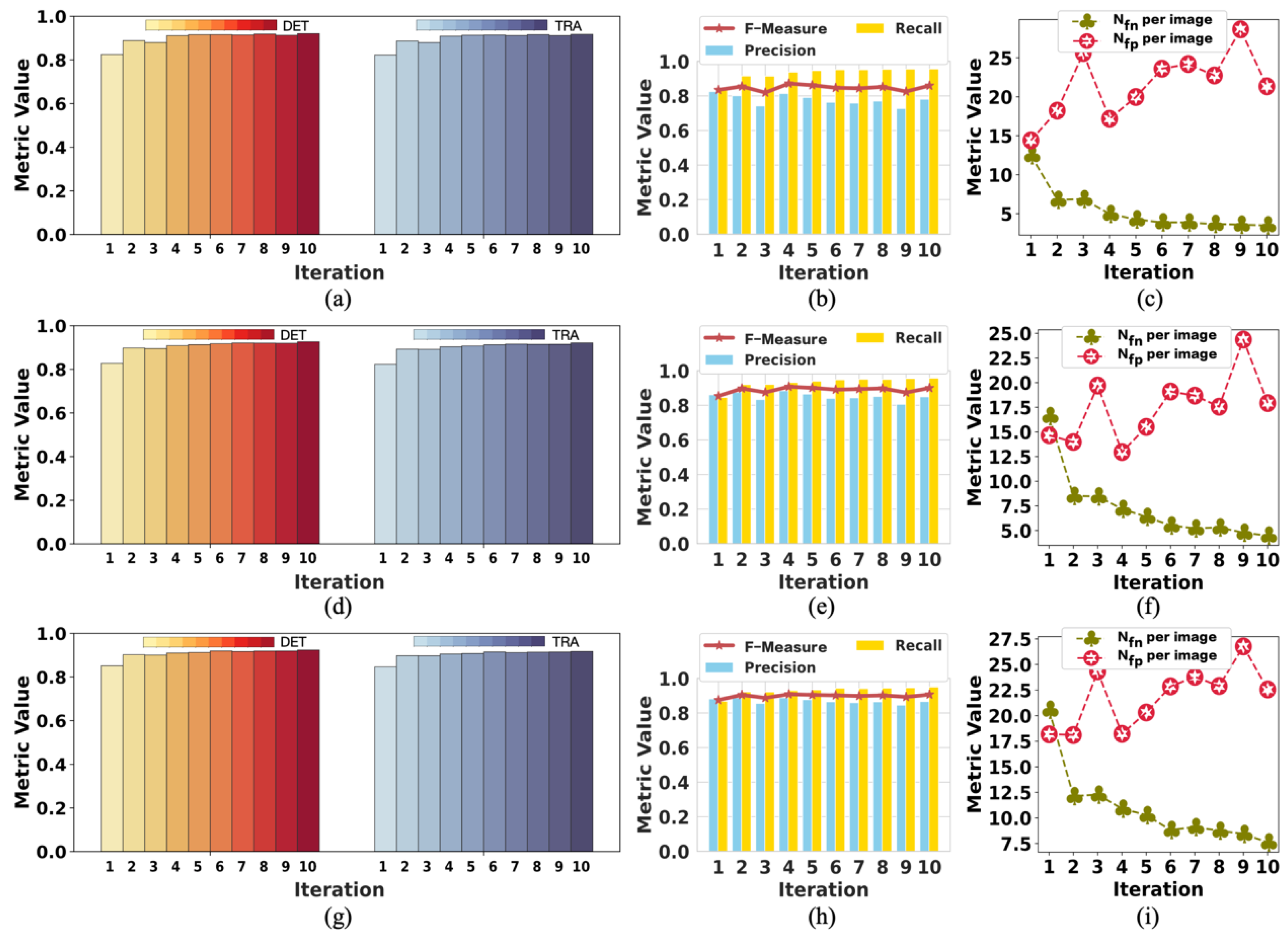
To the best of our knowledge, this pioneering study successfully utilizes incomplete initial annotations to develop a robust and universal weakly supervised learning method for automated cell detection and tracking on a brand-new iPS cell reprogramming dataset. The experimental results demonstrate that this study’s developed method achieves a justifiable performance (0.862 and 0.924 DET on two fields of iPS) comparable to the state-of-the-art supervised learning approaches.
We propose a procedure to obtain incomplete initial labels by removing the possible point light interference from paired red fluorescent images. On other microscopy image datasets, incomplete initial annotations can be obtained using different unsupervised cell detectors such as the Gaussian filter, Cellbow magnification, and automatic thresholding methods provided in ImageJ (version 2.1.0, open source software available at
https://github.com/imagej/imagej (accessed on 21 June 2023)), and bilateral filter with prior knowledge of cell shape.
We demonstrate that our method achieves competitive detection and tracking performance on the public dataset FluoN2DH-GOWT1 from CTC, which contains two datasets with reference annotations. To simulate the incomplete initial annotations, we randomly removed 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, and 90% of the cell markers to train the model. The prediction performance on all metrics using partial point annotation is comparable to the model trained using the conventional supervised learning method.
Since there are few relevant evaluation indicators in cell detection and tracking, we referred to CTC to select the appropriate evaluation metrics to assess the proposed method’s prediction performance.
1.2. Literature Review
The precise detection and tracking of cells in microscopy images play a crucial role in various biological tasks [
39]. Multiple techniques have been introduced for cell detection and tracking. Recently, the prevailing methods follow a two-step approach: Firstly, they detect cells within each frame, and subsequently, they establish connections between the detected cells across consecutive frames [
19]. Various methods have been suggested for detecting cell regions within individual frames, including graph cuts and deep learning [
41,
42]. The process of associating cells is tackled through linear programming optimization [
43]. Alternatively, graph-based optimization techniques have been introduced, wherein an entire sequence of images is represented as a graph, with cells as nodes and association hypotheses as edges, thereby solving for comprehensive solutions [
44]. However, these methods rely on the proximity of cell positions for association scores, rendering them ineffective at low frame rates when cells exhibit significant movement.
Recent advancements involve data-driven approaches that employ deep neural networks to estimate cell motion or optical flow. He et al. [
30] evaluated motion attributes such as cell motion direction, enlargement, and shape alteration. Nonetheless, this method falters when dealing with densely populated cell scenarios, as it is designed for single-object tracking. Various CNN-based methods for estimating optical flow, like FlowNet [
45], have been introduced. Yet, challenges persist in establishing accurate ground-truth data for flow in cell images, and even when estimated, the application of flow in tracking multiple cells within dense environments remains unclear.
The application of AI and DL techniques has shown remarkable progress across various medical domains. Ryu et al. presented SegR-Net, a DL model that combines multiscale feature fusion, deep feature magnification, and precise interference for retinal vessel segmentation [
46]. Their framework outperforms existing models in accuracy and sensitivity, and it holds promise for enhancing retinal disease diagnosis and clinical decision-making. Introducing an efficient DL technique, Mangj et al. [
47] utilized a multiscale DCNN to detect brain tumors, with a remarkable 97% accuracy on MRI scans of meningioma and glioma cases, demonstrating superiority over prior ML and DL models for tumor classification.
In Ref. [
48], the authors introduced RAAGR2-Net, an encoder–decoder-based brain tumor segmentation network leveraging a residual spatial pyramid pooling module and attention gate module to enhance accuracy on multimodal MRI images, surpassing existing methods on the BraTS benchmark. Attallah and Zaghlool [
49] introduced a pioneering AI-based pipeline that merges textural analysis and DL to enhance the precision of categorizing pediatric medulloblastoma subtypes from histopathological images. This innovative approach holds significant promise for advancing individualized therapeutic strategies and risk assessment in pediatric brain tumors, underscoring the transformative potential of AI in medical diagnosis and treatment [
49].
Recently, U-Net and its variants have become well known, particularly for medical imaging, revolutionizing the way we diagnose and treat. Their innovative architecture empowers precise analysis, aiding doctors’ expertise. With exceptional detection and segmentation capabilities, they unravel intricate details, enhancing our understanding of conditions. A novel modified U-Net architecture was introduced for the accurate and automatic segmentation of dermoscopic skin lesions, incorporating feature map dimension modifications and increased kernels for precise nodule extraction [
50].
Similarly, a modified U-Net was introduced to precisely segment diabetic retinopathy lesions, utilizing residual networks and sub-pixel convolution [
51]. Rehman et al. [
52] proposed BrainSeg-Net, an encoder–decoder model with a feature enhancer block, enhancing spatial detail retention for accurate MR brain tumor segmentation. A UAV-based weed density evaluation method utilizing a modified U-Net is presented, facilitating precise field management [
53]. Furthermore, a modified U-Net architecture known as BU-Net was introduced, enhancing accurate brain tumor segmentation through residual extended skip and wide context modifications, along with a customized loss function, thereby improving feature diversity and contextual information and enhancing segmentation performance [
54].
In this study, we introduce an innovative, weakly supervised learning method designed for detecting and tracking cells, leveraging incomplete initial annotations. This approach adds to the current spectrum of AI models and directly confronts the labor-intensive demands inherent in conventional, fully supervised training. Building upon these advancements, our work extends the research frontier by presenting a novel strategy that effectively harnesses the potential of incomplete initial labels for training purposes. This strategic enhancement significantly alleviates the burden of extensive manual annotations, thus enabling a more streamlined and efficient approach to developing accurate cell detection and tracking models.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}