Abstract
Alzheimer’s disease (AD) represents a major diagnostic challenge, as early detection is crucial for effective intervention. This review examines the diagnostic challenges facing current AD evaluations and explores the emerging field of retinal alterations as early indicators. Recognizing the potential of the retina as a noninvasive window to the brain, we emphasize the importance of identifying retinal biomarkers in the early stages of AD. However, the examination of AD is not without its challenges, as the similarities shared with other retinal diseases introduce complexity in the search for AD-specific markers. In this review, we address the relevance of using the retina for the early diagnosis of AD and the complex challenges associated with the search for AD-specific retinal biomarkers. We provide a comprehensive overview of the current landscape and highlight avenues for progress in AD diagnosis by retinal examination.
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative brain disorder and the most common cause of dementia [1,2]. Recently, AD has been recognized as a global public health priority by the World Health Organization [3]. It is characterized by the presence of extracellular amyloid-β (Aβ) plaques and intraneuronal neurofibrillary tangles (NFTs) consisting of hyperphosphorylated tau protein (pTau) [4,5].
The amyloid cascade hypothesis postulates that altered production and clearance of Aβ is the major driving factor for AD pathogenesis in the brain, as abnormal accumulation of Aβ disrupts synaptic function, leading to neuronal loss and the characteristic neurodegeneration [6,7]. It is also known that the blood-brain barrier is compromised in AD, allowing the extravasation of fibrinogen and inflammatory mediators that activate microglial cells [8,9]. Microglia act as resident phagocytes and mediate synapse pruning by engulfing synapses [10], and once activated, microglia tend to form clusters around Aβ plaques [11], which increases microglial proliferation [12]. In addition, microglial exosomes have been shown to propagate pTau [12,13], while the downregulation of microglial homeostatic genes involved in regulation has been reported to correlate with neuronal loss [14].
Activated microglia secrete inflammatory factors [10,15], including reactive oxygen species (ROS) [16]. Under normal physiological function, the reactive species generated can be controlled by antioxidant defenses [17]. However, in the early stages of AD, oxidative stress is present, and glia and neurons are highly sensitive to this [17], as brain cells respond to oxidative stress, they enter a cycle of increased ROS generation, causing more oxidative stress and mitochondrial damage, ultimately leading to cell death [18,19].
Screening for AD involves several techniques, including cognitive tests, neuroimaging, cerebrospinal fluid (CSF) analysis, blood tests, and genetic testing [20,21,22]. Cognitive tests assess memory and cognitive function, while neuroimaging techniques such as magnetic resonance imaging (MRI) and positron emission tomography (PET) can detect structural and protein changes in the brain [23]. CSF analysis and blood tests provide information on biomarkers associated with AD [21,24], and genetic testing can identify mutations in familial forms of the disease [25]. However, a definitive diagnosis can only be made at autopsy [26].
There are several difficulties with diagnosing AD. The most important is that early diagnosis remains challenging, as clinical symptoms often appear late in the disease [27]. In addition, the overlap of symptoms with other neurodegenerative diseases makes differentiation difficult [20], and biomarkers may lack specificity and be present in individuals without AD [28]. Accurate and widespread diagnosis is further complicated by access to specialized testing, ethical issues, and economic concerns. Despite these difficulties, research is underway to improve the accuracy and accessibility of AD detection. It also aims to find new ways to study the disease and detect it in its early stages.
Retinal alterations are one of the earliest signs of AD [29]. Several post-mortem studies have shown Aβ plaques and NFTs in the retinas of AD patients [29,30,31]. In parallel, the retinal glial cell types, such as microglia and macroglia, become activated and change their morphology and immune phenotype [32]. In particular, macroglia, namely Müller glia, and astrocytes, are important for retinal homeostasis [33]. Müller glia are the most abundant glial cell type in the retina [34] and require large amounts of energy to maintain their functions, such as ensuring the survival of retinal ganglion cells (RGCs) [19,35,36]. Increased levels of oxidative stress have been shown to decrease glutamate transport and to reduce both the glycolytic activity and the mitochondrial respiratory function in Müller glia [37]. As changes in glial cells and cellular energetics are implicated in both AD and retinal dysfunction, a growing body of research suggests that energetic instability in the retina may be a predictor of AD.
Refs. [38,39] Certain clinical signs have been observed in AD patients, including increased pupil size [40] and a less pronounced pupillary light reflex [41], and the presence of Aβ40 in the aqueous humour comparable to levels found in cerebrospinal fluid [42]. However, in post-mortem eyes of patients with AD, no deposits of Aβ have been reported in the lens [43]. Interestingly, increased cupping of the optic nerve has also been reported in AD [38], whereas a mouse study using Aβ injection found no axonal damage to the optic nerve [44]. In addition to anatomical manifestations, visual and motor impairments have also been reported in AD patients. These impairments include abnormal hypometric saccades, reduced visual acuity, impaired contrast sensitivity, stereopsis, lack of hand-eye coordination, impaired fixation of the eye, and difficulty in identifying objects [38,39,45] These changes provide predictive and progression information on neurodegenerative changes and represent potential ocular biomarkers of AD (Figure 1).
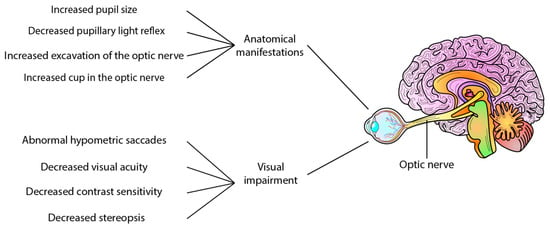
Figure 1.
Visual changes in Alzheimer’s disease and erase AD. Several clinical and functional alterations have been reported in Alzheimer’s disease patients, such as decreased pupillary light reflex, increased pupil size, and increased excavation of the optic nerve. Other features include decreased contrast sensitivity, decreased visual acuity, stereopsis, abnormal hypometric saccade, and reduced visual sensitivity, among other alterations.
The diagnosis of AD is challenging because of the subjectivity of the methods used and the presence of other conditions that present with similar cognitive impairment, such as vascular dementia [46]. This results in limited sensitivity and specificity, leading to an inaccurate diagnosis in 10–15% of AD cases [47]. In recent years, considerable progress has been made in techniques for detecting AD-related disorders, but many of these are invasive and expensive, so a new approach to detecting AD, such as retinal screening, should be considered. At present, there are non-invasive and low-cost techniques for the detection of retinal and ocular changes that are associated with AD [48]. However, the similarity of the pathology to other diseases compels us to continue the study of changes that can be considered as specific retinal and ocular biomarkers in AD. This would also allow us to study AD from a new perspective, as the ocular and retinal changes already observed could be starting in the brain, allowing us to make a timely diagnosis and perhaps stop the progression of AD, giving patients a better quality of life.
In this review, we highlight the compelling parallels between the retinal and cerebral manifestations of AD given their close association. We provide a detailed account of the retinal structure and cell alterations observed in the context of AD. Furthermore, we offer insights into the intricate interplay between retinal cells and the resulting energetic impairment, oxidative stress, and mitochondrial dysfunction. At the same time, we outline the major challenges in retinal AD research caused by shared pathophysiological mechanisms with certain retinal diseases.
Lastly, we discuss the promising diagnostic techniques already available for the detection of retinal and ocular alterations in AD, which offer the dual advantage of early diagnosis and potentially dynamic monitoring of disease progression. This offers an alternative perspective for studying AD through retinal and ocular changes. However, it should be noted that some of these innovative approaches are still in the research phase with encouraging preliminary results.
2. The Retina: A Window to the Brain
The retina is a part of the central nervous system (CNS), originating from an out-pouching of the diencephalon [49]. The retina contains a high density of neurons that form a sensory extension of the brain [50]. Visual input is converted into neuronal signals in the photoreceptor cells and transmitted to the bipolar cells and then to the RGCs, the output neurons of the retina. RGC axons remain unmyelinated in the retina and converge on the optic nerve, where they become myelinated and travel to the visual centers in the brain. This lack of myelination in the intraretinal portions of RGC axons, which is essential for light to travel unimpeded to the photoreceptors, combined with a semi-hypoxic environment, renders the retina susceptible to metabolic stress [19].
The retina is dependent on a well-regulated process for the maintenance of a proper blood supply for the support of cellular functions [51]. Studies have shown changes in the retinal vasculature, the mid-peripheral retina [52], and the foveal avascular zone (FAZ) in AD, including changes in vessel density, thickness, and integrity [53,54]. Angiogenesis can be studied in a non-invasive way and since it is associated with AD [55], it is considered a possible diagnostic biomarker, but it requires further standardization.
2.1. Outer Retinal Changes in AD Patients
The outer retina, the outermost layer of cells in the retina, consists of photoreceptor cells and the retinal pigment epithelium (RPE) [56,57], which has received increasing attention in AD. However, compared to the inner retina, its role in AD is less well understood. The photoreceptor cells, which are essential for the capture of light and the initiation of the visual process, have shown signs of progressive degeneration in AD mice [58], potentially contributing to the visual impairment associated with the disease.
Optical coherence tomography (OCT) studies have correlated volumetric changes in the outer retina, ellipsoid zone to the retinal pigmented epithelium, with changes in brain volume in patients with AD, reflecting photoreceptor outer segment shortening [59]. Photoreceptor dysfunction accompanied by Aβ deposition is a common observation in animal models of AD [60,61]. In addition, a clinical study including 39 patients with mild AD and 21 patients with moderate AD showed impaired color perception in addition to photoreceptor degeneration compared to controls by OCT [62].
Visual impairment, including problems with contrast sensitivity, color discrimination, and depth perception, is a recognized symptom of AD [38,45]. Although the exact mechanisms remain unclear, these visual problems suggest a link between AD and the outer retina. Further research is needed to establish a definitive link and to better understand the role of the outer retina in AD.
2.2. Inner Retinal Alterations
The inner retina, which extends from the inner limiting membrane to the outer plexiform layer, consists of RGCs, the nerve fiber layer, and bipolar cells [63]. The RGCs are responsible for the transmission of visual information from the retina to the brain and show signs of degeneration in people with AD [38]. The thinning of the nerve fiber layer (NFL) and ganglion cell layer (GCL) in people with AD has been demonstrated using advanced imaging techniques such as OCT [38,64,65], suggesting structural changes in the inner retina.
In addition, post-mortem examinations of the retinas of patients with AD show the presence of Aβ in the NFL and GCL [31,66]. The presence of Aβ deposits, Aβ40 and Aβ42 alloforms, vascularization, and inflammation have been reported in the retinas of AD patients and mice [30,66,67,68].
Furthermore, pTau accumulation has been found in the inner plexiform layer and in the GCL where it has been linked to RGCs loss and inflammation [29,38,69]. Several post-mortem studies have reported an increase in pTau in the peripheral retina of people with AD [70]. This information highlights the link between AD-related changes in the brain and manifestations in the retina.
2.3. Microglia Abnormalities
Microglia are the resident immune cells of the CNS, including the brain and retina, where they are involved in the maintenance of homeostasis [71,72]. Microglia play a vital role in brain plasticity and development [73] through the removal of synapses from neuronal cell bodies [74,75]. Furthermore, microglia are important contributors to proper synaptic processing in health but are also involved in pathological conditions by disrupting neuronal connections [76]. Although astrocytes and microglia do not yet appear to play a direct role in synapse degeneration, neurons appear to be more active in this process [77].
Under normal circumstances, both the Triggering Receptor Expressed in Myeloid Cells 2 (TREM2) and Apolipoprotein E (ApoE) are involved in microglial cells in the phagocytosis of apoptotic neurons in the brain [78]. However, mutations in TREM2 are associated with an increased risk of late-onset AD as Aβ is not cleared properly, resulting in increased accumulation [10,79]. Increased levels of pro-inflammatory mediators such as interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-alpha (TNF-α) have been found in the cerebrospinal fluid, contributing to neuroinflammation in AD [12,80]. Aβ activation of microglia requires the participation of the receptor complex CD36, α6β1 integrin, and CD47, which are involved in the internalization of Aβ [81], and receptors such as CD14 and Toll-like receptors 2 and 4 (TLR-2 and TLR-4) [82,83,84].
Under physiological conditions, microglia are found in the inner retina. As in the brain, they can be activated to remove debris and pathogens in response to injury or infection [71]. And in AD, activated microglia can infiltrate the outer retina [72].
2.4. Macroglia: The Importance of Müller Glia in AD
In addition to their homeostatic and metabolic support functions, Müller glia is also involved in the regulation of synaptic activity in the inner retina [15,85]. Müller glia are also sensitive to microglial changes [86] and can be activated in neurodegenerative diseases such as AD. Müller glia prevents the accumulation of glutamate in the synaptic clefts via excitatory amino acid transporters 1 and 2 (EAAT1 and EAAT2, also known as GLT-1), thereby preventing excitotoxicity [87]. Müller glia requires substantial bioenergetic support, provided primarily by a large number of mitochondria, to perform glutamate uptake and maintain retinal homeostasis [88].
The retina is one of the highest lactate-containing organs in the human body relative to size, and lactate has been shown to act as an energy substrate for retinal cells [34,89,90,91]. Müller glia and RGCs are the main cells responsible for lactate production in the retina [89]. Studies have shown increased cell survival in both cultures of isolated primary RGCs and Müller glia from neonatal mice (C57BL/6J) retinas when exposed to L-lactate. In addition to acting as an energy substrate, lactate also activates the G-coupled protein receptor 81 (GPR81) [34]. Lactate has been shown to have regulatory functions in the retina, such as increasing glutamate uptake, improving mitochondrial function and increasing glucose consumption, protecting against neuroinflammation, and providing neuroprotection in neurodegenerative conditions [89].
The bidirectional interaction between microglia and Müller glia is thought to act as a regulator in neuron-microglia interaction. Müller glia can detect neurotransmitter signals from neuronal and synaptic activity [86,87,92]. Adenosine triphosphate (ATP) is also known as a chemotactic agent [93], and Müller glia, via ATP secretion, can mediate activity dependent regulation of microglial dynamics [87,89], as well as regulate retinal blood flow [94]. The release of ATP from Müller glia through pannexin channels drives a resting state in microglia [95] (Figure 2).
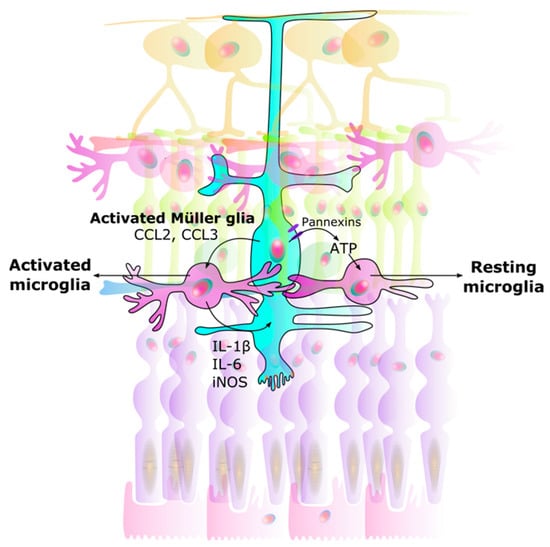
Figure 2.
Interaction between Müller glia and microglia. Signals from Müller glia induce changes in microglia, causing them to either rest or become activated: (1) Activated microglia: Once activated, Müller glia activate microglia by CCL2 and CCL3 secretion. (2) Neuroinflammation: Microglia in turn produce IL-1β, IL-6, and iNOS in a positive feedback loop. This results in unregulated over-activation leading to neurodegeneration. (3) Resting microglia: ATP secretion through Müller glial pannexin channels allows microglia to rest. CCL Chemokine (C-C motif) ligand. IL Interleukin. iNOS Inducible nitric oxide synthase. ATP Adenosine triphosphate.
3. Intricate Connection Means the Retina Mirrors the Brain
Findings from in vivo and in vitro studies in humans and animal models show the similarities in the manifestations of AD in the brain and retina. Post-mortem studies of the retina in humans and some animal models of AD have shown activation of microglia and Müller glia, as well as disturbances in energetic, metabolic, and mitochondrial processes. This suggests that Aβ accumulation triggers neurodegeneration in the retina similar to that observed in the brain [35,96] (Table 1).

Table 1.
Alzheimer’s-related findings in both brain and retina of human and animal models.
Aβ accumulation in the brain has been found decades before the onset of symptoms [136,137], and several reports indicate that retinal Aβ follows a similar time course to that in the brain [67,138]. Retinal abnormalities have also been associated with AD progression and cognitive decline. These include loss of RGCs [120], reduced thickness of the retinal NFL (RNFL) and RGC layer and reduced retinal blood flow [102,121,139], as well as decreased thickness in the macular area as a result of inner layer degeneration [62]. Taken together, this suggests that the eye can be considered a window to study AD.
3.1. Energetic Impairment
The brain has a remarkably high energy requirement, with glucose being the main energy substrate [104]. However, lactate, ketone bodies, glycogen, and amino acids can also be used as bioenergetic substrates under certain circumstances [118]. Pathogenic factors such as abnormalities in glucose transport, impaired glucose metabolism, and mitochondrial dysfunction in the brain occur in AD and have been characterized in animal models [108,118,140] (Figure 3).
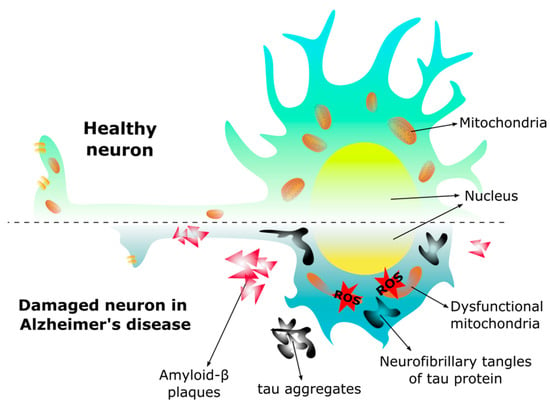
Figure 3.
Morphology in the healthy neuron and the neuron of Alzheimer’s disease. In Alzheimer’s disease the neuron changes morphology, the dendrites are shorter, the axon is reduced decreasing the synapse, and the mitochondria become dysfunctional due to the presence of amyloid-β (Aβ) plaques and neurofibrillary tangles, leading to an increase of reactive oxygen species (ROS), and subsequent cell death.
Like the brain, the retina is one of the most energy-demanding tissues in the body. To meet its vast energy demands [141], the retina is rich in mitochondria, particularly in the RPE, photoreceptors, and Müller glia, as well as in the RGC axons at the optic nerve head [102,142]. To achieve this, the structure and shape of the mitochondria are maintained by a balance of mitochondrial fission and fusion. Fission is the mitochondrial fragmentation that occurs when dynamin-related protein 1 (Drp1) and mitochondrial fission 1 protein (Fis1) are recruited to the outer mitochondrial membrane, resulting in smaller and more functional mitochondria [143]. Fusion allows mitochondria to support each other, compensating for defects in damaged mitochondria by physically fusing the outer and inner membranes of two different mitochondria [144]. This process also protects mitochondria from harmful DNA mutations and allows them to change shape for specific functions. The lack of mitochondrial fusion can therefore increase ROS production and decrease ATP synthesis [145]. Reports have suggested that alterations in these mitochondrial dynamics may be involved in neurodegenerative diseases. In AD, impaired fission-fusion dynamics can lead to mitochondrial dysfunction in the brain [146,147,148], with reduced levels of Drp1 and increased levels of Fis1 reported [149].
Deposition of Aβ has been shown to affect mitochondrial function. For instance, even mild increases in Aβ have been found to have detrimental effects on mitochondria [150]. In addition, several studies have indicated that mitochondrial morphology and function can be affected by inhibition of protein import [151], by the interaction of Aβ with the cyclophilin D-dependent mitochondrial permeability transition pore (mPTP) [152,153], and by its binding to mitochondrial Aβ-binding alcohol dehydrogenase (ABAD) [150,154]. Recent studies have also demonstrated that metabolic alterations in the retina of an AD mouse model, including decreased glutamine synthesis and glucose hypometabolism, provide insight into potential neuroprotective pathways [19].
3.2. Oxidative Stress and Mitochondrial Dysfunction
Oxidative stress and impaired Ca2+ homeostasis contribute to the pathogenesis and progression of AD [119,155]. Upregulation of oxidative phosphorylation (OXPHOS) activity can lead to an increase in ROS that directly affects neurons [6] and glia [112] in the AD brain.
Neurons are fundamentally dependent on mitochondrial OXPHOS to meet their energy needs. When neurons are damaged, they respond by upregulating OXPHOS, better known as the inverse Warburg effect [6]. It shifts from a healthy state to pathological aging with subsequent amyloid plaque accumulation, DNA damage, increased ROS production, and neuronal loss due to their high susceptibility to free radicals [113]. The inverse Warburg effect suggests that an imbalance between the fusion and fission of intact and damaged mitochondria may play an important role in the pathogenesis of AD [6,156]. In AD brain neurons, altered Ca2+ influx in dysfunctional mitochondria can lead to altered regulation of neurotransmitters, neurogenesis, neuronal plasticity, and lipid synthesis [157]. It has also been suggested that Aβ enters the mitochondria and interacts with mitochondrial proteins that produce ROS and free radicals [145].
Research has shown that at least 42 metabolic proteins, including glyceraldehyde-3-phosphate dehydrogenase (GAPDH), are downregulated in the AD brain. GAPDH is involved in glucose metabolism and has been shown to interact with amyloid precursor protein (APP). The non-glycolytic activity of GAPDH can be modulated by ROS and GAPDH activity is reduced in the AD brain, which may contribute to the loss of neuronal function and subsequent neurodegeneration [158,159].
It has been suggested that metabolic pathways become dysregulated with age, leading to mitochondrial dysfunction and reduced ATP production [160]. Mitochondrial changes in AD have also been shown to correlate with reduced energy metabolism and oxidative stress [150]. In this context, it is proposed that damaged neurons in an AD brain attempt to protect themselves and compensate for energy deprivation by switching to glycolysis with lactate and pyruvate to promote rapid ATP production. Residual lactate and pyruvate enter the mitochondria and the tricarboxylic acid (TCA) cycle for OXPHOS [161]. Competition for lactate also occurs between neurons, and neurons with upregulated OXPHOS outperform the rest. They have the advantage of using lactate from astrocytic glycolysis as an energy substrate, thus sparing glucose for healthy neurons [160,162]. In addition, a defect in cytochrome c oxidase has been implicated in the bioenergetic deficit of AD [127] as a result of mitochondrial DNA (mtDNA) mutations [163]. In aged animals, reduced glutathione levels have been associated with mitochondrial damage [87,105].
Similar to the brain, the chronic increase of ROS in the AD retina contributes to oxidative stress, mitochondrial dysfunction, and cell death [126,128] (Figure 4). This excessive ROS production is thought to be related to abnormal stimulation of NMDAR by Aβ oligomers [134] and mitochondrial dysfunction. Aβ aggregates bind redox-active metals such as iron and copper, sources of ROS, causing mitochondrial damage and subsequent neurotoxicity [122]. Aβ deposits and NFTs activate retinal astrocytes and microglia [164] with the production of inflammatory cytokines such as IL-1β, IL-6, and TNF-α [165], which together with ROS may cause neurotoxicity. RGCs appear to be particularly susceptible to this neurotoxic effect and thinning of the retinal nerve fiber layer is an increasingly recognized phenomenon in AD [122].
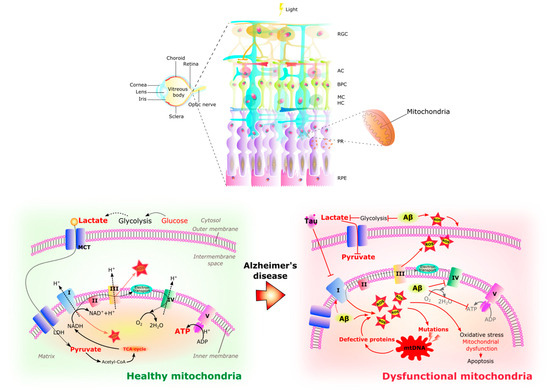
Figure 4.
Healthy and dysfunctional mitochondria in the retina. The RPE/photoreceptor complex resides in a highly oxidative environment. A large number of mitochondria are located in the outer end of Müller glia, in the inner segments of photoreceptors, and in the basal part of RPE cells. The retina is a highly energy demanding organ. In healthy retinal cells, glycolysis occurs in the cytoplasm, providing pyruvate and lactate to the mitochondria where they can be substrates for acetyl-CoA synthesis, fueling the TCA cycle and electron transport chain (complex I–V) activity with final energy production in the form of ATP. In the Alzheimer’s disease retina, glycolysis is affected by the amyloid-β (Aβ) peptide causing the increase in reactive oxidative species (ROS), while hyperphosphorylated tau (pTau) inhibits the electron transport chain, provoking the increase of ROS inside the matrix of the mitochondria. In a harmful cycle, the increase in ROS causes mutations in mitochondrial DNA that will produce defective proteins which in turn decreases ATP and increases ROS with the subsequent oxidative stress, mitochondrial damage, and apoptosis which contribute to the retinal pathology of AD. RGC Retinal ganglion cells. AC Amacrine cells. BC Bipolar cells. MC Müller cells. HC Horizontal cells. PR Photoreceptors. RPE Retinal pigmented epithelium. I NADH reductase. II Succinate dehydrogenase. III Cytochrome complex. IV Cytochrome C oxidase. V ATP synthase. TCA cycle Tricarboxylic acid cycle. MCT Monocarboxylate transporter. ROS Reactive oxygen species. mtDNA Human mitochondrial DNA. ATP Adenosine triphosphate. ADP Adenosine diphosphate.
Studies of the retina in mouse models of AD suggest that Aβ peptides are responsible for increased Ca2+ in mitochondria, leading to mitochondrial fragmentation, disruption of the fission-fusion balance, and mitochondrial dysfunction [102].
It is still unknown whether mitochondria play a primary or secondary role in the pathogenesis of AD, as it is debated as to whether this is a consequence of amyloid deposition, or whether it is directly involved in the early stages of AD. Therefore, an alternative hypothesis is that amyloid/tau pathology and mitochondrial dysfunction are correlated [166].
4. Similarities between Retinal Manifestations in Retinas from AD Patients and Retinal Manifestations in Other Eye Diseases
Studies of retinal diseases are therefore crucial in recognizing the potential of the retina as a window to the brain, providing new insights into neurodegenerative processes.
Common mechanisms and biomarkers of neuronal dysfunction can likely be identified by studying degeneration patterns, proteins, metabolic and energy pathways, and inflammatory responses in different retinal diseases. This approach will not only aid in the early diagnosis of AD but may also potentially improve the understanding of disease progression and the effects of therapeutic interventions (Table 2).
Table 2.
Comparison of Alzheimer’s disease with Diabetic Retinopathy, Glaucoma, and Age-Related Macular Degeneration.
4.1. Diabetic Retinopathy and AD
Diabetic retinopathy (DR) is a leading cause of vision loss and is expected to affect more than 190 million people by 2030 [186]. The increasing prevalence of DR is linked to the rise in cases of diabetes associated with the increasing prevalence of obesity and sedentary lifestyles [187]. Altered metabolic parameters and modifiable risk factors predisposing to type 2 diabetes mellitus (T2DM) have been associated with the risk of AD [128,188,189,190,191,192,193]. Similarly, both diabetes and diabetic retinopathy are associated with an increased risk of developing AD [194,195,196] and cognitive impairment [197,198,199,200].
Disruption of insulin signaling and abnormal activation of components of the insulin signaling pathway in the brain, features typical of diabetes, have been reported in AD [128,201,202,203]. The brain has a high density of insulin receptors [204,205], and some studies show that Aβ oligomers are involved in removing insulin receptors (IRs), thereby reducing the IR protein tyrosine kinase activity through TNF-α/c-Jun NH2-terminal kinase (JNK) activation [128,206]. In addition, the retina is also an insulin-sensitive tissue, and it has also been suggested that Müller cells may secrete insulin [207] and that a deficiency in receptor signaling may contribute to retinal cell death [208].
Intranasal insulin has been shown to improve memory in healthy adults, verbal memory in patients with memory impairment, and cognitive performance in patients with early AD and mild cognitive impairment [209,210,211]. Insulin has been shown to block the downregulation of IR, IR substrate-1pSer (IRS-1pSer), and oxidative stress, and to protect neurons against synapse loss induced by Aβ oligomers [128,212]. GLP-1R agonists activate insulin signaling pathways through G-protein-dependent signaling. This influences the regulation of glucose metabolism and impairs neurological and cognitive function [213,214]. In GLP-1R agonist-treated mice, reduction of Aβ plaques and microglial activation have been observed, thereby conferring neuroprotection [213].
4.2. Glaucoma and AD
Glaucoma is a progressive optic neuropathy, characterized by RGC loss and progressive visual field loss. It is one of the leading causes of blindness worldwide [33,215,216]. As is the case in AD, mitochondrial dysfunction, altered OXPHOS, and increased ROS have been reported to play a role in the pathogenesis of glaucoma [217,218]. In addition, similarities between AD and glaucoma have long been recognized, as the retinal neurodegeneration in AD appears to predominantly affect RGCs [139].
There is evidence that primary open-angle glaucoma (POAG) and AD may share genetic risk factors. For example, the optineurin (OPTN) gene encodes for the optineurin protein [219], which is expressed in the brain, retina, heart, placenta, kidney, and other tissues [220]. Overexpression of OPTN is strongly associated with TNF-α induced death of rat RGC-5 cells [221], suggesting an upregulation in AD [222]. Optineurin neurotoxicity may be a common risk factor in normal tension glaucoma (NTG) and AD [220].
Despite these pathogenic similarities, the association between AD and primary POAG remains controversial [219,223,224]. Some studies have found no association between POAG and AD [225,226], including a meta-analysis in 2021 that found no association between glaucoma and AD (risk ratio (RR): 1.03, 95% confidence interval (CI): 0.93–1.05; I2 = 83%, p= 0.55) [227]. On the other hand, some studies suggest an association between the two diseases [228,229], including a systematic review and meta-analysis in 2019 that showed an increased risk of AD in glaucoma patients (RR = 1.52; 95% CI: 1.41–1.63; I2 = 97%, p < 0.001) and an Asian study that found a significant association (RR = 2.03; 95% CI: 1.02–4.07) [230]. In addition, studies in Taiwan, Korea, and Canada have all shown that patients with POAG have a higher risk of developing AD [231,232,233], suggesting that POAG may be a predictor of AD in certain demographic groups. The contradictory evidence of associations between AD and glaucoma may be due to the diagnostic criteria used in each study, including diagnostic misclassification. In addition, the fact that some studies may have included only patients with severe dementia, the small size of the cohort in some studies, the inclusion of case-control studies, which may lead to a positive patient selection bias, and the different mean ages of the study participants may also explain the inconsistent conclusions. Future longitudinal studies are needed to better understand the associations between AD and glaucoma.
4.3. Age-Related Macular Degeneration and AD
Age-related macular degeneration (AMD) is another major cause of blindness in people over the age of 50 years [234]. AMD is a chronic degenerative disease of the macula that affects central vision, and it is characterized by the accumulation of drusen between the RPE and Bruch’s membrane [235].
Extracellular deposits of Aβ [236], iron accumulation [237], chronic inflammation, and oxidative stress [122] are common features in the retina of patients with AMD. This suggests a strong correlation and probably overlap in pathology between AD and AMD [15,122,238,239]. Li-Yen Wen et al. have shown a 1.23-fold increased risk in AD patients with AMD [240].
Based on the overlapping features of the diseases, iron chelating agents have been proposed as a potential strategy for the treatment of both AD and AMD [122,241,242], together with therapeutic elimination of Aβ accumulation with anti-Aβ antibodies or reduction of Aβ production with β-secretase inhibitors based on the overlapping mechanisms [138]. Trials of these therapeutic approaches are currently ongoing.
Overall, as mentioned above, some studies have shown an association between AMD and the risk of AD. However, a UK study of 2088 individuals aged 69 to 97 found a lack of a significant association between AD and early AMD, although AD and AMD have similar pathologies, ApoE status, and vascular risk factors. However, they also mention that although the study was large, only 145 of the 707 participants with dementia were considered to have AMD. Also, some of the people with AMD and AD may have died before the study was conducted. In addition, only one eye was examined in the retinal photography study. Finally, the study was a cross-sectional study which meant that it was impossible to know when the cognitive decline occurred in AMD [243].
5. Non-Invasive Retinal Imaging to Detect Features of AD
5.1. Optical Coherence Tomography (OCT)
The detection of Aβ and tau accumulation in the brain currently relies on the use of PET imaging or CSF analysis, both of which are expensive and invasive [244]. Assuming that AD can be predicted via the retina, it is relevant to explore methods for mapping retinal biomarkers [59]. In this context, OCT is a non-invasive technique that uses low-coherence interferometry to obtain cross-sectional three-dimensional images of the retina, allowing for the study of retinal thinning, macular thinning, and vascular changes [244].
The thickness of the macula as examined by OCT is a valuable indicator of AD. Studies have shown reduced macular volume and thickness in asymptomatic individuals with a high genetic risk of developing AD [245] and reduced central macular thickness in patients with AD compared to healthy controls [246].
In patients with AD, macular and peripapillary RNFL thinning has been reported in numerous studies using OCT [123,247], suggesting that OCT may be used to detect biomarkers for AD [122,239,248,249,250,251]. At present, these changes lack specificity for disease detection, as there is considerable overlap between OCT findings in AD and several other neurodegenerative diseases, including glaucoma. It should be noted, however, that some factors may affect the OCT measurements, such as elevated intraocular pressure, variable axial length, refractive error, systemic comorbidities, or other eye diseases due to similarities in the pathology [252].
Nevertheless, the use of OCT is a useful tool in the diagnosis and monitoring of retinal diseases and may therefore be a valuable resource in the diagnosis of neurodegenerative diseases, such as AD [123,253,254,255,256] (Figure 5).
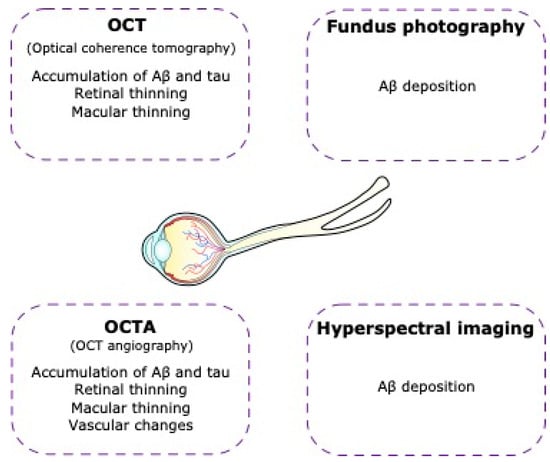
Figure 5.
Non-invasive imaging of the retina in Alzheimer’s disease. In Alzheimer’s disease patients there are visual alterations that can lead us to Alzheimer’s disease diagnosis using technological tools such as optical coherence tomography (OCT), optical coherence tomography angiography (OCTA), fundus photography, and hyperspectral imaging.
5.2. Optical Coherence Tomography Angiography (OCT-A)
Vascular changes in the brain, including cerebral amyloid angiopathy and blood-brain barrier dysfunction, are known to be associated with AD [257]. Similarly, AD has also been associated with vascular changes in the retina [252]. OCT-A provides a detailed non-invasive examination of the retinal vasculature without the use of contrast agents [258]. Studies have shown retinal microvascular network changes, with an enlargement of the FAZ, microvascular density lesions in the deep retinal capillary plexuses, and lower vascular density in whole macular, foveal, and parafoveal zones in people with AD compared to controls [259,260].
Thus, OCT-A may help us detect these changes in the retinal microvasculature before the clinical symptoms of AD appear, which may aid in predicting and evaluating the progression of the disease.
Errors in data acquisition or image quality, as well as ocular anatomy and diseases such as hypertension or diabetes, can affect OCT-A readings [261,262]. As with OCT, other eye diseases can also affect the readings [262]. Furthermore, it must be considered that OCT and OCT-A values may be altered by race, age, or gender [263].
5.3. Hyperspectral Imaging
The presence of Aβ deposits in the inner retinal layers of patients with AD has been reported in several studies [264,265]. Hyperspectral imaging is an imaging technique that acquires a series of images across many contiguous wavelengths of light to combine spectral and spatial information into a single data cube [266]. This emerging imaging technique has shown promise in distinguishing AD mice from wild-type mice [264,267,268]. Furthermore, recent studies have shown that hyperspectral imaging may be able to discriminate patients with AD pathology from healthy controls [264,265]. Larger replication studies are needed before it is clear whether this imaging technique has clinical value in AD.
5.4. Fundus Photography
Fundus photography is a non-invasive imaging technique that produces two-dimensional color images of the back of the eye, including the retina. This provides detailed images with high sensitivity and specificity. These include vessel caliber, tortuosity, and the global geometric branching network [269]. The microvascular changes in the retinas of patients with AD include narrower retinal venules and more tortuous retinal vessels, which may reflect changes in vascular structure and function that are associated with the brain pathology of AD and dementia [270,271,272].
Although not a standalone diagnostic tool, fundus photography contributes to our understanding of retinal changes in AD and may complement other diagnostic and follow-up approaches in AD.
6. Other Approaches to Detect Biomarkers of AD in the Eye
6.1. Aβ and pTau in the Vitreous Humor
Reduced levels of Aβ and pTau have been reported in the vitreous humor and correlated with poor cognitive function. Therefore, whilst it may be technically feasible to use the presence of these proteins as a biomarker for AD [273,274], the method’s applicability is limited as it requires surgery to collect intraocular fluid.
6.2. Fluorescent Signal of Ligand Bound to Aβ in the Lens
A combination of a fluorescent ligand with an affinity for Aβ and a laser scanning device have been proposed as a means to detect the accumulation of Aβ in the crystalline lens of the eye following application as an eyedrop [275,276], with significant correlations between eye and brain measurements [277]. Further studies of this imaging method are in progress for the detection of AD.
6.3. Corneal Confocal Microscopy (CCM)
Corneal confocal microscopy (CCM) is a non-invasive ophthalmic imaging technique that has shown promise in the detection of neuronal loss in neurodegenerative diseases. It can detect changes in corneal neurons and is useful in detecting neuropathies such as diabetic neuropathy [278] and can also identify corneal nerve changes in Parkinson’s disease [279] and multiple sclerosis [280]. The neurotransmitter acetylcholine (ACh) has been shown to play an important role in maintaining the corneal epithelium. Furthermore, ACh is deficient in AD, so changes in corneal structure could be used for the detection and monitoring of AD. Therefore, by analyzing the micromorphology of the corneal subbasal nerve plexus (SNP), CCM may provide insight into peripheral and central neurodegeneration in AD [281].
6.4. Ocular Manifestations
Anatomic changes in AD extend beyond the retinal layers to include changes in the size of the pupil and optic nerve. Pupillary abnormalities have been observed in individuals with AD, often manifesting as varying responses of the pupil to constrict and dilate in response to light stimulation [282]. This may be indicative of underlying neurological dysfunction, as the pupillary reflex is influenced by the sympathetic (adrenergic) and parasympathetic (cholinergic) autonomic nervous systems [283], memory function [284], and brain control of the pupillary muscles [283]. To detect these changes in the pupil, pupillometry provides a non-invasive method and a potential biomarker for AD [285].
Another important site of anatomical change in AD is the optic nerve, which connects the eye to the brain. Although not directly observable with routine retinal imaging, there is evidence of retinal ganglion cell degeneration and a widespread axonal degeneration in the optic nerve [286], nerve fiber damage, increased cup-to-disc ratio [287], and with the use of a retinograph, detection of papillary paleness in the optic disk in AD [288]. These changes may be the result of axonal and neuronal degeneration in the optic nerve head [39].
7. Conclusions
AD is a neurodegenerative disorder characterized by the abnormal accumulation of Aβ peptides and tau protein in the brain. Several studies have shown that AD patients have abnormal accumulations of both Aβ and tau in the RGC layer and the inner retina, suggesting a possible link between retinal pathology and AD. Accumulating research suggests that these pathological changes occur in the retina over a similar timeframe as in the brain, making the retina an attractive approach to AD diagnosis.
In AD, mitochondrial dysfunction disrupts energy metabolism, leading to impaired cellular function and an increased susceptibility to neurodegeneration. This impairment extends to retinal cells and may contribute to the retinal degeneration seen in AD. Another consequence of mitochondrial dysfunction is an increase in the production of ROS. The accumulation of ROS in the retina can lead to oxidative stress and further exacerbate retinal degeneration. Taken together, retinal Aβ and tau accumulation, along with reduced mitochondrial function, impaired energy metabolism, and increased ROS production, are likely to contribute to retinal degeneration in AD.
Retinal tests, such as OCT and hyperspectral imaging, offer several advantages in detecting AD compared to traditional clinical brain diagnostic methods. Retinal assays are non-invasive, low-cost, time-efficient, and well-tolerated by patients, making them accessible tools for early detection and disease monitoring. They can detect subtle retinal changes, even before the onset of cognitive symptoms in AD.
It is important to note that there are differences between retinal imaging techniques, such as OCT or OCT-A, which provide more detailed information on retinal structure, thickness, and microvascular changes compared to traditional fundus photography or hyperspectral imaging. However, the combination of some techniques, such as hyperspectral imaging and OCT, may lead to a better and faster diagnosis of AD [289]. Other ocular methods, such as CCM, provide insight into the corneal nerve morphology related to AD but do not offer insight into the retina. On the other hand, detection of Aβ by fluorescent signaling in the lens may not provide a complete picture of AD-related changes, and detection of Aβ and pTau in the vitreous humor is an invasive tool requiring surgery.
There are many factors to consider when comparing the cost of retinal techniques for the detection of AD. Methods such as fundus photography are in general more affordable, which makes them more accessible for routine screening. The integration of artificial intelligence (AI) algorithms into these techniques may increase their cost but bring significant benefits such as accuracy and precision. AI can analyze retinal data more comprehensively, potentially leading to the detection of subtle changes associated with early AD. As a result, early detection of AD, early intervention, and increased treatment efficacy would justify this investment.
While retinal and ocular tests are promising, at present they are not standalone diagnostic tools but rather complements to existing clinical techniques. Integration of retinal evaluation with MRI, PET, and other biomarker-based diagnostic approaches may improve the sensitivity and specificity of AD detection, providing a diagnostic strategy that incorporates both brain and retinal pathology.
An understanding of the mechanisms in Alzheimer’s retinas could provide valuable insights into the pathophysiology of AD and potentially facilitate the development of novel approaches for early diagnosis and therapeutic strategies for the disease. The U.S. Food and Drug Administration recently approved two anti-amyloid antibodies (Lecanemab and Aducanumab) as disease-modifying therapies for AD. With this treatment, it stands to reason that the implementation of retinal screening for early diagnosis could serve to initiate treatment at a stage when the damage has not yet caused cognitive impairment in the brain, thus providing neuroprotection.
In summary, there is compelling evidence to support retinal changes as biomarkers for the diagnosis and potential prediction of AD. However, there is a lack of consensus on how to implement such a strategy in clinical practice and further research is needed to improve the understanding of the relationship between retinal manifestations and AD.
Author Contributions
Writing—original draft preparation, M.Y.G.-B.; writing—review and editing, all authors; visualization, M.Y.G.-B.; supervision, M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Department of Drug Design and Pharmacology at the University of Copenhagen, the BRIDGE-Translational Excellence Program funded by the Novo Nordisk Foundation (Grant agreement no. NNF18SA0034956), the Foundation for Research in Neurology and Innovation Foundation Denmark grant 2078-00002B StemScreen (K. Freude), and the Savværksejer Jeppe Juhls og hustru Ovita Juhls Mindelegat. The present review is part of the Doctoral studies of M. Y. García-Bermúdez MSc, who is a student at the University of Copenhagen. She was supported by the ILF group and CONACYT doctoral scholarship CVU: 859376.
Conflicts of Interest
P. van Wijngaarden is co-founder of Enlighten Imaging, a medical technology company focused on retinal biomarkers of diseases including Alzheimer’s disease. K. Martin is a Director of Enlighten Imaging on behalf of the Centre for Eye Research Australia. The other authors declare no conflict of interest.
References
- Wilson, R.S.; Segawa, E.; Boyle, P.A.; Anagnos, S.E.; Hizel, L.P.; Bennett, D.A. The natural history of cognitive decline in Alzheimer’s disease. Psychol. Aging 2012, 27, 1008–1017. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Revi, M. Alzheimer’s Disease Therapeutic Approaches. In GeNeDis 2018; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2018; pp. 105–116. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef]
- Demetrius, L.A.; Driver, J. Alzheimer’s as a metabolic disease. Biogerontology 2013, 14, 641–649. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Mendiola, A.S.; Cardona, A.E. The IL-1beta phenomena in neuroinflammatory diseases. J. Neural Transm. 2018, 125, 781–795. [Google Scholar] [CrossRef]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Tejera, D.; Heneka, M.T. Microglia in Neurodegenerative Disorders. Methods Mol. Biol. 2019, 2034, 57–67. [Google Scholar] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef] [PubMed]
- Madeira, M.H.; Boia, R.; Santos, P.F.; Ambrosio, A.F.; Santiago, A.R. Contribution of microglia-mediated neuroinflammation to retinal degenerative diseases. Mediat. Inflamm. 2015, 2015, 673090. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Adam, R.H.I.; Broersen, K. Molecular Mechanisms and Genetics of Oxidative Stress in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 72, 981–1017. [Google Scholar] [CrossRef]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; Lopez-Cuenca, I.; Rojas, P.; Trivino, A.; Ramirez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef]
- Tams, A.L.M.; Sanz-Morello, B.; Westi, E.W.; Mouhammad, Z.A.; Andersen, J.V.; Freude, K.K.; Vohra, R.; Hannibal, J.; Aldana, B.I.; Kolko, M. Decreased Glucose Metabolism and Glutamine Synthesis in the Retina of a Transgenic Mouse Model of Alzheimer’s Disease. Cell. Mol. Neurobiol. 2021, 42, 291–303. [Google Scholar] [CrossRef]
- Compta, Y.; Revesz, T. Neuropathological and Biomarker Findings in Parkinson’s Disease and Alzheimer’s Disease: From Protein Aggregates to Synaptic Dysfunction. J. Park. Dis. 2021, 11, 107–121. [Google Scholar] [CrossRef]
- Hansson, O.; Edelmayer, R.M.; Boxer, A.L.; Carrillo, M.C.; Mielke, M.M.; Rabinovici, G.D.; Salloway, S.; Sperling, R.; Zetterberg, H.; Teunissen, C.E. The Alzheimer’s Association appropriate use recommendations for blood biomarkers in Alzheimer’s disease. Alzheimer’s Dement. 2022, 18, 2669–2686. [Google Scholar] [CrossRef]
- Tahami Monfared, A.A.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer’s Disease: Epidemiology and Clinical Progression. Neurol. Ther. 2022, 11, 553–569. [Google Scholar] [CrossRef]
- Zamrini, E.; De Santi, S.; Tolar, M. Imaging is superior to cognitive testing for early diagnosis of Alzheimer’s disease. Neurobiol. Aging 2004, 25, 685–691. [Google Scholar] [CrossRef]
- Lleo, A.; Nunez-Llaves, R.; Alcolea, D.; Chiva, C.; Balateu-Panos, D.; Colom-Cadena, M.; Gomez-Giro, G.; Munoz, L.; Querol-Vilaseca, M.; Pegueroles, J.; et al. Changes in Synaptic Proteins Precede Neurodegeneration Markers in Preclinical Alzheimer’s Disease Cerebrospinal Fluid. Mol. Cell Proteom. 2019, 18, 546–560. [Google Scholar] [CrossRef] [PubMed]
- Di Resta, C.; Ferrari, M. New molecular approaches to Alzheimer’s disease. Clin. Biochem. 2019, 72, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Trojanowski, J.Q.; Schmidt, M.L.; Shin, R.W.; Bramblett, G.T.; Rao, D.; Lee, V.M. Altered tau and neurofilament proteins in neuro-degenerative diseases: Diagnostic implications for Alzheimer’s disease and Lewy body dementias. Brain Pathol. 1993, 3, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S.; Sabbagh, M.N.; Rubino, I. Diagnosis of Early Alzheimer’s Disease: Clinical Practice in 2021. J. Prev. Alzheimers Dis. 2021, 8, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Hart, N.J.; Koronyo, Y.; Black, K.L.; Koronyo-Hamaoui, M. Ocular indicators of Alzheimer’s: Exploring disease in the retina. Acta Neuropathol. 2016, 132, 767–787. [Google Scholar] [CrossRef]
- Koronyo, Y.; Salumbides, B.C.; Black, K.L.; Koronyo-Hamaoui, M. Alzheimer’s disease in the retina: Imaging retinal abeta plaques for early diagnosis and therapy assessment. Neurodegener. Dis. 2012, 10, 285–293. [Google Scholar] [CrossRef]
- Koronyo, Y.; Biggs, D.; Barron, E.; Boyer, D.S.; Pearlman, J.A.; Au, W.J.; Kile, S.J.; Blanco, A.; Fuchs, D.T.; Ashfaq, A.; et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight 2017, 2, e93621. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, R.; Luo, X.; Wang, F.; Sun, X. The Interaction Between Microglia and Macroglia in Glaucoma. Front. Neurosci. 2021, 15, 610788. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bermudez, M.Y.; Freude, K.K.; Mouhammad, Z.A.; van Wijngaarden, P.; Martin, K.K.; Kolko, M. Glial Cells in Glaucoma: Friends, Foes, and Potential Therapeutic Targets. Front. Neurol. 2021, 12, 624983. [Google Scholar] [CrossRef] [PubMed]
- Vohra, R.; Aldana, B.I.; Waagepetersen, H.; Bergersen, L.H.; Kolko, M. Dual Properties of Lactate in Muller Cells: The Effect of GPR81 Activation. Investig. Ophthalmol. Vis. Sci. 2019, 60, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Winkler, B.S.; Arnold, M.J.; Brassell, M.A.; Puro, D.G. Energy metabolism in human retinal Muller cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3183–3190. [Google Scholar]
- Toft-Kehler, A.K.; Skytt, D.M.; Svare, A.; Lefevere, E.; Van Hove, I.; Moons, L.; Waagepetersen, H.S.; Kolko, M. Mitochondrial function in Muller cells–Does it matter? Mitochondrion 2017, 36, 43–51. [Google Scholar] [CrossRef]
- Toft-Kehler, A.K.; Gurubaran, I.S.; Desler, C.; Rasmussen, L.J.; Skytt, D.M.; Kolko, M. Oxidative Stress-Induced Dysfunction of Müller Cells During Starvation. Investig. Opthalmol. Vis. Sci. 2016, 57, 2721. [Google Scholar] [CrossRef]
- Javaid, F.Z.; Brenton, J.; Guo, L.; Cordeiro, M.F. Visual and Ocular Manifestations of Alzheimer’s Disease and Their Use as Biomarkers for Diagnosis and Progression. Front. Neurol. 2016, 7, 55. [Google Scholar] [CrossRef]
- Romaus-Sanjurjo, D.; Regueiro, U.; Lopez-Lopez, M.; Vazquez-Vazquez, L.; Ouro, A.; Lema, I.; Sobrino, T. Alzheimer’s Disease Seen through the Eye: Ocular Alterations and Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 2486. [Google Scholar] [CrossRef]
- Granholm, E.L.; Panizzon, M.S.; Elman, J.A.; Jak, A.J.; Hauger, R.L.; Bondi, M.W.; Lyons, M.J.; Franz, C.E.; Kremen, W.S. Pupillary Responses as a Biomarker of Early Risk for Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 56, 1419–1428. [Google Scholar] [CrossRef]
- Chougule, P.S.; Najjar, R.P.; Finkelstein, M.T.; Kandiah, N.; Milea, D. Light-Induced Pupillary Responses in Alzheimer’s Disease. Front. Neurol. 2019, 10, 360. [Google Scholar] [CrossRef]
- Goldstein, L.E.; Muffat, J.A.; Cherny, R.A.; Moir, R.D.; Ericsson, M.H.; Huang, X.; Mavros, C.; Coccia, J.A.; Faget, K.Y.; Fitch, K.A.; et al. Cytosolic beta-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer’s disease. Lancet 2003, 361, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-Y.; Troncoso, J.C.; Knox, D.; Stark, W.; Eberhart, C.G. Beta-Amyloid, Phospho-Tau and Alpha-Synuclein Deposits Similar to Those in the Brain Are Not Identified in the Eyes of Alzheimer’s and Parkinson’s Disease Patients. Brain Pathol. 2014, 24, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-W.; Nishioka, C.; Labib, W.; Liang, H.-F. Axonal Terminals Exposed to Amyloid-β May Not Lead to Pre-Synaptic Axonal Damage. J. Alzheimer’s Dis. 2015, 45, 1139–1148. [Google Scholar] [CrossRef]
- Chang, L.Y.L.; Lowe, J.; Ardiles, A.; Lim, J.; Grey, A.C.; Robertson, K.; Danesh-Meyer, H.; Palacios, A.G.; Acosta, M.L. Alzheimer’s disease in the human eye. Clinical tests that identify ocular and visual information processing deficit as biomarkers. Alzheimer’s Dement. 2014, 10, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, W.J.; Grossberg, G.T. Behavioral and psychological symptoms in Alzheimer’s dementia and vascular dementia. Handb. Clin. Neurol. 2019, 165, 5–32. [Google Scholar]
- Singh, A.K.; Verma, S. Use of ocular biomarkers as a potential tool for early diagnosis of Alzheimer’s disease. Indian. J. Ophthalmol. 2020, 68, 555–561. [Google Scholar] [CrossRef]
- Majeed, A.; Marwick, B.; Yu, H.; Fadavi, H.; Tavakoli, M. Ophthalmic Biomarkers for Alzheimer’s Disease: A Review. Front. Aging Neurosci. 2021, 13, 720167. [Google Scholar] [CrossRef]
- Kashani, A.H.; Asanad, S.; Chan, J.W.; Singer, M.B.; Zhang, J.; Sharifi, M.; Khansari, M.M.; Abdolahi, F.; Shi, Y.; Biffi, A.; et al. Past, present and future role of retinal imaging in neurodegenerative disease. Prog. Retin. Eye Res. 2021, 83, 100938. [Google Scholar] [CrossRef]
- Byerly, M.S.; Blackshaw, S. Vertebrate retina and hypothalamus development. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 380–389. [Google Scholar] [CrossRef]
- Dreyfuss, J.L.; Giordano, R.J.; Regatieri, C.V. Ocular Angiogenesis. J. Ophthalmol. 2015, 2015, 892043. [Google Scholar] [CrossRef]
- Pead, E.; Thompson, A.C.; Grewal, D.S.; McGrory, S.; Robbins, C.B.; Ma, J.P.; Johnson, K.G.; Liu, A.J.; Hamid, C.; Trucco, E.; et al. Retinal Vascular Changes in Alzheimer’s Dementia and Mild Cognitive Impairment: A Pilot Study Using Ultra-Widefield Imaging. Transl. Vis. Sci. Technol. 2023, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Rifai, O.M.; McGrory, S.; Robbins, C.B.; Grewal, D.S.; Liu, A.; Fekrat, S.; MacGillivray, T.J. The application of optical coherence tomography angiography in Alzheimer’s disease: A systematic review. Alzheimer’s Dement. 2021, 13, e12149. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Koronyo, Y.; Rentsendorj, A.; Fuchs, D.T.; Sheyn, J.; Black, K.L.; Mirzaei, N.; Koronyo-Hamaoui, M. Retinal Vasculopathy in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 731614. [Google Scholar] [CrossRef] [PubMed]
- Govindpani, K.; McNamara, L.G.; Smith, N.R.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It? J. Clin. Med. 2019, 8, 651. [Google Scholar] [CrossRef]
- Simo, R.; Villarroel, M.; Corraliza, L.; Hernandez, C.; Garcia-Ramirez, M. The retinal pigment epithelium: Something more than a constituent of the blood-retinal barrier--implications for the pathogenesis of diabetic retinopathy. J. Biomed. Biotechnol. 2010, 2010, 190724. [Google Scholar] [CrossRef]
- Gupta, M.P.; Herzlich, A.A.; Sauer, T.; Chan, C.C. Retinal Anatomy and Pathology. Dev. Ophthalmol. 2016, 55, 7–17. [Google Scholar] [PubMed]
- Zhang, J.; Gao, F.; Ma, Y.; Xue, T.; Shen, Y. Identification of early-onset photoreceptor degeneration in transgenic mouse models of Alzheimer’s disease. iScience 2021, 24, 103327. [Google Scholar] [CrossRef]
- Uchida, A.; Pillai, J.A.; Bermel, R.; Jones, S.E.; Fernandez, H.; Leverenz, J.B.; Srivastava, S.K.; Ehlers, J.P. Correlation between brain volume and retinal photoreceptor outer segment volume in normal aging and neurodegenerative diseases. PLoS ONE 2020, 15, e0237078. [Google Scholar] [CrossRef]
- Lim, J.K.H.; Li, Q.X.; He, Z.; Vingrys, A.J.; Chinnery, H.R.; Mullen, J.; Bui, B.V.; Nguyen, C.T.O. Retinal Functional and Structural Changes in the 5xFAD Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 862. [Google Scholar] [CrossRef]
- Perez, S.E.; Lumayag, S.; Kovacs, B.; Mufson, E.J.; Xu, S. Beta-amyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer’s disease. Investig. Ophthalmol. Vis. Sci. 2009, 50, 793–800. [Google Scholar] [CrossRef]
- Salobrar-García, E.; De Hoz, R.; Ramírez, A.I.; López-Cuenca, I.; Rojas, P.; Vazirani, R.; Amarante, C.; Yubero, R.; Gil, P.; Pinazo-Durán, M.D.; et al. Changes in visual function and retinal structure in the progression of Alzheimer’s disease. PLoS ONE 2019, 14, e0220535. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, N.; Shi, H.; Oviatt, M.; Doustar, J.; Rentsendorj, A.; Fuchs, D.T.; Sheyn, J.; Black, K.L.; Koronyo, Y.; Koronyo-Hamaoui, M. Alzheimer’s Retinopathy: Seeing Disease in the Eyes. Front. Neurosci. 2020, 14, 921. [Google Scholar] [CrossRef] [PubMed]
- La Morgia, C.; Ross-Cisneros, F.N.; Koronyo, Y.; Hannibal, J.; Gallassi, R.; Cantalupo, G.; Sambati, L.; Pan, B.X.; Tozer, K.R.; Barboni, P.; et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann. Neurol. 2016, 79, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Lopez-de-Eguileta, A.; Cervero, A.; Ruiz de Sabando, A.; Sanchez-Juan, P.; Casado, A. Ganglion Cell Layer Thinning in Alzheimer’s Disease. Medicina 2020, 56, 553. [Google Scholar] [CrossRef]
- Lee, S.; Jiang, K.; McIlmoyle, B.; To, E.; Xu, Q.A.; Hirsch-Reinshagen, V.; Mackenzie, I.R.; Hsiung, G.R.; Eadie, B.D.; Sarunic, M.V.; et al. Amyloid Beta Immunoreactivity in the Retinal Ganglion Cell Layer of the Alzheimer’s Eye. Front. Neurosci. 2020, 14, 758. [Google Scholar] [CrossRef] [PubMed]
- Doustar, J.; Rentsendorj, A.; Torbati, T.; Regis, G.C.; Fuchs, D.T.; Sheyn, J.; Mirzaei, N.; Graham, S.L.; Shah, P.K.; Mastali, M.; et al. Parallels between retinal and brain pathology and response to immunotherapy in old, late-stage Alzheimer’s disease mouse models. Aging Cell 2020, 19, e13246. [Google Scholar] [CrossRef]
- Grimaldi, A.; Brighi, C.; Peruzzi, G.; Ragozzino, D.; Bonanni, V.; Limatola, C.; Ruocco, G.; Di Angelantonio, S. Inflammation, neurodegeneration and protein aggregation in the retina as ocular biomarkers for Alzheimer’s disease in the 3xTg-AD mouse model. Cell Death Dis. 2018, 9, 685. [Google Scholar] [CrossRef]
- Koronyo-Hamaoui, M.; Koronyo, Y.; Ljubimov, A.V.; Miller, C.A.; Ko, M.K.; Black, K.L.; Schwartz, M.; Farkas, D.L. Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 2011, 54 (Suppl. S1), S204–S217. [Google Scholar] [CrossRef]
- den Haan, J.; Morrema, T.H.J.; Verbraak, F.D.; de Boer, J.F.; Scheltens, P.; Rozemuller, A.J.; Bergen, A.A.B.; Bouwman, F.H.; Hoozemans, J.J. Amyloid-beta and phosphorylated tau in post-mortem Alzheimer’s disease retinas. Acta Neuropathol. Commun. 2018, 6, 147. [Google Scholar] [CrossRef]
- Ginhoux, F.; Prinz, M. Origin of microglia: Current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 2015, 7, a020537. [Google Scholar] [CrossRef]
- Fan, W.; Huang, W.; Chen, J.; Li, N.; Mao, L.; Hou, S. Retinal microglia: Functions and diseases. Immunology 2022, 166, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.E.; Green, K.N. Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. Brain Behav. Immun. 2017, 61, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Blinzinger, K.; Kreutzberg, G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z. Zellforsch. Mikrosk. Anat. 1968, 85, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef]
- Di Liberto, G.; Pantelyushin, S.; Kreutzfeldt, M.; Page, N.; Musardo, S.; Coras, R.; Steinbach, K.; Vincenti, I.; Klimek, B.; Lingner, T.; et al. Neurons under T Cell Attack Coordinate Phagocyte-Mediated Synaptic Stripping. Cell 2018, 175, 458–471.e419. [Google Scholar] [CrossRef]
- Perry, V.H.; O’Connor, V. The role of microglia in synaptic stripping and synaptic degeneration: A revised perspective. ASN Neuro 2010, 2, e00047. [Google Scholar] [CrossRef]
- Atagi, Y.; Liu, C.C.; Painter, M.M.; Chen, X.F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef]
- Arcuri, C.; Mecca, C.; Bianchi, R.; Giambanco, I.; Donato, R. The Pathophysiological Role of Microglia in Dynamic Surveillance, Phagocytosis and Structural Remodeling of the Developing CNS. Front. Mol. Neurosci. 2017, 10, 191. [Google Scholar] [CrossRef]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef]
- Doens, D.; Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef]
- Fiala, M.; Veerhuis, R. Biomarkers of inflammation and amyloid-beta phagocytosis in patients at risk of Alzheimer disease. Exp. Gerontol. 2010, 45, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Graca, A.B.; Hippert, C.; Pearson, R.A. Müller Glia Reactivity and Development of Gliosis in Response to Pathological Conditions; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 303–308. [Google Scholar]
- Wang, M.; Wong, W.T. Microglia-Müller Cell Interactions in the Retina; Springer: New York, NY, USA, 2014; pp. 333–338. [Google Scholar]
- Reichenbach, A.; Bringmann, A. New functions of Muller cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef] [PubMed]
- Vohra, R.; Kolko, M. Neuroprotection of the inner retina: Muller cells and lactate. Neural Regen. Res. 2018, 13, 1741–1742. [Google Scholar] [PubMed]
- Vohra, R.; Kolko, M. Lactate: More Than Merely a Metabolic Waste Product in the Inner Retina. Mol. Neurobiol. 2020, 57, 2021–2037. [Google Scholar] [CrossRef] [PubMed]
- Vohra, R.; Aldana, B.I.; Bulli, G.; Skytt, D.M.; Waagepetersen, H.; Bergersen, L.H.; Kolko, M. Lactate-Mediated Protection of Retinal Ganglion Cells. J. Mol. Biol. 2019, 431, 1878–1888. [Google Scholar] [CrossRef]
- Vohra, R.; Aldana, B.I.; Skytt, D.M.; Freude, K.; Waagepetersen, H.; Bergersen, L.H.; Kolko, M. Essential Roles of Lactate in Muller Cell Survival and Function. Mol. Neurobiol. 2018, 55, 9108–9121. [Google Scholar] [CrossRef]
- Bringmann, A.; Grosche, A.; Pannicke, T.; Reichenbach, A. GABA and Glutamate Uptake and Metabolism in Retinal Glial (Muller) Cells. Front. Endocrinol. 2013, 4, 48. [Google Scholar] [CrossRef]
- Li, Y.; Du, X.-F.; Liu, C.-S.; Wen, Z.-L.; Du, J.-L. Reciprocal Regulation between Resting Microglial Dynamics and Neuronal Activity In Vivo. Dev. Cell 2012, 23, 1189–1202. [Google Scholar] [CrossRef]
- Ernst, C.; Skov Jensen, P.; Aalkjaer, C.; Bek, T. Differential Effects of Intra- and Extravascular ATP on the Diameter of Porcine Vessels at Different Branching Levels Ex Vivo. Investig. Ophthalmol. Vis. Sci. 2020, 61, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wong, W.T. Microglia-Muller cell interactions in the retina. Adv. Exp. Med. Biol. 2014, 801, 333–338. [Google Scholar] [PubMed]
- Salobrar-Garcia, E.; Rodrigues-Neves, A.C.; Ramirez, A.I.; de Hoz, R.; Fernandez-Albarral, J.A.; Lopez-Cuenca, I.; Ramirez, J.M.; Ambrosio, A.F.; Salazar, J.J. Microglial Activation in the Retina of a Triple-Transgenic Alzheimer’s Disease Mouse Model (3xTg-AD). Int. J. Mol. Sci. 2020, 21, 816. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.M.; Rodríguez, J.J.; Gutierrez-Lanza, R.; Yates, J.; Verkhratsky, A.; Lutty, G.A. Retinal macroglia changes in a triple transgenic mouse model of Alzheimer’s disease. Exp. Eye Res. 2014, 127, 252–260. [Google Scholar] [CrossRef]
- Chidlow, G.; Wood, J.P.; Manavis, J.; Finnie, J.; Casson, R.J. Investigations into Retinal Pathology in the Early Stages of a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 56, 655–675. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef]
- Chiasseu, M.; Alarcon-Martinez, L.; Belforte, N.; Quintero, H.; Dotigny, F.; Destroismaisons, L.; Vande Velde, C.; Panayi, F.; Louis, C.; Di Polo, A. Tau accumulation in the retina promotes early neuronal dysfunction and precedes brain pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 58. [Google Scholar] [CrossRef]
- He, Y.; Zhao, H.; Su, G. Ginsenoside Rg1 Decreases Neurofibrillary Tangles Accumulation in Retina by Regulating Activities of Neprilysin and PKA in Retinal Cells of AD Mice Model. J. Mol. Neurosci. 2014, 52, 101–106. [Google Scholar] [CrossRef]
- Chiu, K.; Chan, T.F.; Wu, A.; Leung, I.Y.; So, K.F.; Chang, R.C. Neurodegeneration of the retina in mouse models of Alzheimer’s disease: What can we learn from the retina? Age 2012, 34, 633–649. [Google Scholar] [CrossRef]
- Oliveira-Souza, F.G.; Deramus, M.L.; Van Groen, T.; Lambert, A.E.; Bolding, M.S.; Strang, C.E. Retinal changes in the Tg-SwDI mouse model of Alzheimer’s disease. Neuroscience 2017, 354, 43–53. [Google Scholar] [CrossRef]
- Country, M.W. Retinal metabolism: A comparative look at energetics in the retina. Brain Res. 2017, 1672, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Paasche, G.; Gartner, U.; Germer, A.; Grosche, J.; Reichenbach, A. Mitochondria of retinal Muller (glial) cells: The effects of aging and of application of free radical scavengers. Ophthalmic Res. 2000, 32, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Kolko, M.; Vosborg, F.; Henriksen, U.L.; Hasan-Olive, M.M.; Diget, E.H.; Vohra, R.; Gurubaran, I.R.S.; Gjedde, A.; Mariga, S.T.; Skytt, D.M.; et al. Lactate Transport and Receptor Actions in Retina: Potential Roles in Retinal Function and Disease. Neurochem. Res. 2016, 41, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Datta, D. APP Modulates Abeta-Induced Activation of Microglia in Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 238–240. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Westi, E.W.; Diaz-delCastillo, M.; Tanila, H.; Schousboe, A.; Aldana, B.I.; Waagepetersen, H.S. Deficient astrocyte metabolism impairs glutamine synthesis and neurotransmitter homeostasis in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2021, 148, 105198. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 30–40. [Google Scholar] [CrossRef]
- Hernández-Zimbrón, L.F.; Perez-Hernández, M.; Torres-Romero, A.; Gorostieta-Salas, E.; Gonzalez-Salinas, R.; Gulias-Cañizo, R.; Quiroz-Mercado, H.; Zenteno, E. Markers of Alzheimer’s Disease in Primary Visual Cortex in Normal Aging in Mice. BioMed Res. Int. 2017, 2017, 3706018. [Google Scholar] [CrossRef]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.-I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e496. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1977–1987. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.-J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Vina, J. Abeta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef]
- Blanks, J.C.; Hinton, D.R.; Sadun, A.A.; Miller, C.A. Retinal ganglion cell degeneration in Alzheimer’s disease. Brain Res. 1989, 501, 364–372. [Google Scholar] [CrossRef]
- Lopez-de-Eguileta, A.; Lage, C.; Lopez-Garcia, S.; Pozueta, A.; Garcia-Martinez, M.; Kazimierczak, M.; Bravo, M.; de Arcocha-Torres, M.; Banzo, I.; Jimenez-Bonilla, J.; et al. Ganglion cell layer thinning in prodromal Alzheimer’s disease defined by amyloid PET. Alzheimer’s Dement. 2019, 5, 570–578. [Google Scholar] [CrossRef]
- Ashok, A.; Singh, N.; Chaudhary, S.; Bellamkonda, V.; Kritikos, A.E.; Wise, A.S.; Rana, N.; McDonald, D.; Ayyagari, R. Retinal Degeneration and Alzheimer’s Disease: An Evolving Link. Int. J. Mol. Sci. 2020, 21, 7290. [Google Scholar] [CrossRef]
- Ascaso, F.J.; Cruz, N.; Modrego, P.J.; Lopez-Anton, R.; Santabarbara, J.; Pascual, L.F.; Lobo, A.; Cristobal, J.A. Retinal alterations in mild cognitive impairment and Alzheimer’s disease: An optical coherence tomography study. J. Neurol. 2014, 261, 1522–1530. [Google Scholar] [CrossRef]
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10, 1975. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Tammineni, P. Mitochondrial Aspects of Synaptic Dysfunction in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1087–1103. [Google Scholar] [CrossRef]
- Maurer, I.; Zierz, S.; Moller, H.J. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Eckert, G.P.; Renner, K.; Eckert, S.H.; Eckmann, J.; Hagl, S.; Abdel-Kader, R.M.; Kurz, C.; Leuner, K.; Muller, W.E. Mitochondrial Dysfunction—A Pharmacological Target in Alzheimer’s Disease. Mol. Neurobiol. 2012, 46, 136–150. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. Alzheimer’s disease: Diverse aspects of mitochondrial malfunctioning. Int. J. Clin. Exp. Pathol. 2010, 3, 570–581. [Google Scholar]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef]
- Ghoshal, N.; García-Sierra, F.; Fu, Y.; Beckett, L.A.; Mufson, E.J.; Kuret, J.; Berry, R.W.; Binder, L.I. Tau-66: Evidence for a novel tau conformation in Alzheimer’s disease. J. Neurochem. 2001, 77, 1372–1385. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Local neuroinflammation and the progression of Alzheimer’s disease. J. Neurovirol. 2002, 8, 529–538. [Google Scholar] [CrossRef]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Nakamoto, Y.; Yoneda, M. PET Imaging for Oxidative Stress in Neurodegenerative Disorders Associated with Mitochondrial Dysfunction. Antioxidants 2020, 9, 861. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Albert, M.S.; Knopman, D.S.; McKhann, G.M.; Sperling, R.A.; Carrillo, M.C.; Thies, B.; Phelps, C.H. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Rudkin, A.; Agzarian, M.; Patel, S.; Lake, S.; Chen, C. Retinal Vascular Abnormalities in Patients with Cerebral Amyloid Angiopathy. Cerebrovasc. Dis. 2009, 28, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Mao, X. Role of Retinal Amyloid-beta in Neurodegenerative Diseases: Overlapping Mechanisms and Emerging Clinical Applications. Int. J. Mol. Sci. 2021, 22, 2360. [Google Scholar] [CrossRef] [PubMed]
- Blanks, J.C.; Schmidt, S.Y.; Torigoe, Y.; Porrello, K.V.; Hinton, D.R.; Blanks, R.H. Retinal pathology in Alzheimer’s disease. II. Regional neuron loss and glial changes in GCL. Neurobiol. Aging 1996, 17, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Skotte, N.H.; Christensen, S.K.; Polli, F.S.; Shabani, M.; Markussen, K.H.; Haukedal, H.; Westi, E.W.; Diaz-delCastillo, M.; Sun, R.C.; et al. Hippocampal disruptions of synaptic and astrocyte metabolism are primary events of early amyloid pathology in the 5xFAD mouse model of Alzheimer’s disease. Cell Death Dis. 2021, 12, 954. [Google Scholar] [CrossRef]
- Viegas, F.O.; Neuhauss, S.C.F. A Metabolic Landscape for Maintaining Retina Integrity and Function. Front. Mol. Neurosci. 2021, 14, 656000. [Google Scholar] [CrossRef]
- Lefevere, E.; Toft-Kehler, A.K.; Vohra, R.; Kolko, M.; Moons, L.; Van Hove, I. Mitochondrial dysfunction underlying outer retinal diseases. Mitochondrion 2017, 36, 66–76. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Mitochondrial medicine for aging and neurodegenerative diseases. Neuromol. Med. 2008, 10, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Wang, X.; Perry, G.; Smith, M.A.; Zhu, X. Mitochondrial dynamics in Alzheimer’s disease: Opportunities for future treatment strategies. Drugs Aging 2010, 27, 181–192. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 109 (Suppl. S1), 153–159. [Google Scholar] [CrossRef] [PubMed]
- Hroudova, J.; Singh, N.; Fisar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Stockburger, C.; Gold, V.A.; Pallas, T.; Kolesova, N.; Miano, D.; Leuner, K.; Muller, W.E. A cell model for the initial phase of sporadic Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42, 395–411. [Google Scholar] [CrossRef]
- Tillement, L.; Lecanu, L.; Papadopoulos, V. Alzheimer’s disease: Effects of β-amyloid on mitochondria. Mitochondrion 2011, 11, 13–21. [Google Scholar] [CrossRef]
- Stockburger, C.; Eckert, S.; Eckert, G.P.; Friedland, K.; Muller, W.E. Mitochondrial Function, Dynamics, and Permeability Transition: A Complex Love Triangle as A Possible Target for the Treatment of Brain Aging and Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, S455–S467. [Google Scholar] [CrossRef]
- Guo, L.; Du, H.; Yan, S.; Wu, X.; McKhann, G.M.; Chen, J.X.; Yan, S.S. Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer’s neurons. PLoS ONE 2013, 8, e54914. [Google Scholar] [CrossRef]
- Lustbader, J.W. ABAD Directly Links A to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Hardas, S.S.; Lange, M.L. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: Many pathways to neurodegeneration. J. Alzheimer’s Dis. 2010, 20, 369–393. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.D. Glyceraldehyde-3-phosphate dehydrogenase as a target for small-molecule disease-modifying therapies in human neurodegenerative disorders. J. Psychiatry Neurosci. 2004, 29, 337–345. [Google Scholar]
- Demetrius, L.A.; Simon, D.K. An inverse-Warburg effect and the origin of Alzheimer’s disease. Biogerontology 2012, 13, 583–594. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Maguire, D.J.; Maguire, R.S.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Cybrids in Alzheimer’s disease: A cellular model of the disease? Neurology 1997, 49, 918–925. [Google Scholar] [CrossRef]
- Fernandez-Albarral, J.A.; Salobrar-Garcia, E.; Martinez-Paramo, R.; Ramirez, A.I.; de Hoz, R.; Ramirez, J.M.; Salazar, J.J. Retinal glial changes in Alzheimer’s disease—A review. J. Optom. 2019, 12, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Nilson, A.N.; English, K.C.; Gerson, J.E.; Barton Whittle, T.; Nicolas Crain, C.; Xue, J.; Sengupta, U.; Castillo-Carranza, D.L.; Zhang, W.; Gupta, P.; et al. Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neurodegenerative Diseases. J. Alzheimer’s Dis. 2017, 55, 1083–1099. [Google Scholar] [CrossRef] [PubMed]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.M. Retinal Neurodegeneration in Diabetes: An Emerging Concept in Diabetic Retinopathy. Curr. Diabetes Rep. 2021, 21, 65. [Google Scholar] [CrossRef]
- Barresi, C.; Chhablani, J.; Dolz-Marco, R.; Gallego-Pinazo, R.; Berni, A.; Bandello, F.; Borrelli, E. Retinal neurodegeneration in age-related macular degeneration. Eur. J. Ophthalmol. 2023, 11206721231186166. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, L.; Shen, Y. The retina: A window in which to view the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101590. [Google Scholar] [CrossRef]
- Nagai, N.; Ito, Y.; Tanino, T. Effect of high glucose levels on amyloid beta production in retinas of spontaneous diabetes mellitus Otsuka Long-Evans Tokushima fatty rats. Biol. Pharm. Bull. 2015, 38, 601–610. [Google Scholar] [CrossRef]
- Inyushin, M.; Zayas-Santiago, A.; Rojas, L.; Kucheryavykh, Y.; Kucheryavykh, L. Platelet-generated amyloid beta peptides in Alzheimer’s disease and glaucoma. Histol. Histopathol. 2019, 34, 843–856. [Google Scholar]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Hill, J.M.; Clement, C.; Sambamurti, K.; Dua, P.; Lukiw, W.J. Beta-Amyloid Precursor Protein (betaAPP) Processing in Alzheimer’s Disease (AD) and Age-Related Macular Degeneration (AMD). Mol. Neurobiol. 2015, 52, 533–544. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, W.; Zhao, Y.; Shu, X.; Wang, W.; Wang, D.; Yang, Y.; He, Z.; Wang, X.; Ying, Y. GSK3β-mediated tau hyperphosphorylation triggers diabetic retinal neurodegeneration by disrupting synaptic and mitochondrial functions. Mol. Neurodegener. 2018, 13, 62. [Google Scholar] [CrossRef]
- Chiasseu, M.; Cueva Vargas, J.L.; Destroismaisons, L.; Vande Velde, C.; Leclerc, N.; Di Polo, A. Tau Accumulation, Altered Phosphorylation, and Missorting Promote Neurodegeneration in Glaucoma. J. Neurosci. 2016, 36, 5785–5798. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lo, A.C.Y. Diabetic Retinopathy: Pathophysiology and Treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Li, Y.N.; Chen, A.Q.; Hong, C.D.; Zhang, C.L.; Wang, H.L.; Zhou, Y.F.; Li, P.C.; Wang, Y.; Mao, L.; et al. Inhibition of Sema4D/PlexinB1 signaling alleviates vascular dysfunction in diabetic retinopathy. EMBO Mol. Med. 2020, 12, e10154. [Google Scholar] [CrossRef]
- Urbonaviciute, D.; Buteikiene, D.; Januleviciene, I. A Review of Neovascular Glaucoma: Etiology, Pathogenesis, Diagnosis, and Treatment. Medicina 2022, 58, 1870. [Google Scholar] [CrossRef]
- Lip, P.L.; Blann, A.D.; Hope-Ross, M.; Gibson, J.M.; Lip, G.Y. Age-related macular degeneration is associated with increased vascular endothelial growth factor, hemorheology and endothelial dysfunction. Ophthalmology 2001, 108, 705–710. [Google Scholar] [CrossRef]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Mlynarczyk, M.; Falkowska, M.; Micun, Z.; Obuchowska, I.; Kochanowicz, J.; Socha, K.; Konopinska, J. Diet, Oxidative Stress, and Blood Serum Nutrients in Various Types of Glaucoma: A Systematic Review. Nutrients 2022, 14, 1421. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Rolev, K.D.; Shu, X.S.; Ying, Y. Targeted pharmacotherapy against neurodegeneration and neuroinflammation in early diabetic retinopathy. Neuropharmacology 2021, 187, 108498. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Li, Y.; Chen, Z.; Xiong, Y.; Zhou, T.; Tao, W.; Xu, F.; Yang, H.; Yla-Herttuala, S.; et al. MicroRNA-15b Targets VEGF and Inhibits Angiogenesis in Proliferative Diabetic Retinopathy. J. Clin. Endocrinol. Metab. 2020, 105, 3404–3415. [Google Scholar] [CrossRef] [PubMed]
- Shahidatul-Adha, M.; Zunaina, E.; Aini-Amalina, M.N. Evaluation of vascular endothelial growth factor (VEGF) level in the tears and serum of age-related macular degeneration patients. Sci. Rep. 2022, 12, 4423. [Google Scholar] [CrossRef] [PubMed]
- Lechner, J.; O’Leary, O.E.; Stitt, A.W. The pathology associated with diabetic retinopathy. Vision. Res. 2017, 139, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; He, M.; Congdon, N. The worldwide epidemic of diabetic retinopathy. Indian. J. Ophthalmol. 2012, 60, 428–431. [Google Scholar]
- Scherer, T.; Sakamoto, K.; Buettner, C. Brain insulin signalling in metabolic homeostasis and disease. Nat. Rev. Endocrinol. 2021, 17, 468–483. [Google Scholar] [CrossRef]
- Stoeckel, L.E.; Arvanitakis, Z.; Gandy, S.; Small, D.; Kahn, C.R.; Pascual-Leone, A.; Pawlyk, A.; Sherwin, R.; Smith, P. Complex mechanisms linking neurocognitive dysfunction to insulin resistance and other metabolic dysfunction. F1000Research 2016, 5, 353. [Google Scholar] [CrossRef]
- Umegaki, H.; Kawamura, T.; Mogi, N.; Umemura, T.; Kanai, A.; Sano, T. Glucose control levels, ischaemic brain lesions, and hyperinsulinaemia were associated with cognitive dysfunction in diabetic elderly. Age Ageing 2008, 37, 458–461. [Google Scholar] [CrossRef][Green Version]
- Exalto, L.G.; Biessels, G.J.; Karter, A.J.; Huang, E.S.; Katon, W.J.; Minkoff, J.R.; Whitmer, R.A. Risk score for prediction of 10 year dementia risk in individuals with type 2 diabetes: A cohort study. Lancet Diabetes Endocrinol. 2013, 1, 183–190. [Google Scholar] [CrossRef]
- Tolppanen, A.-M. Prediction of dementia in people with diabetes. Lancet Diabetes Endocrinol. 2013, 1, 164–165. [Google Scholar] [CrossRef]
- Bello-Chavolla, O.Y.; Aguilar-Salinas, C.A.; Avila-Funes, J.A. The type 2 diabetes-specific dementia risk score (DSDRS) is associated with frailty, cognitive and functional status amongst Mexican community-dwelling older adults. BMC Geriatr. 2020, 20, 363. [Google Scholar] [CrossRef]
- Exalto, L.G.; Biessels, G.J.; Karter, A.J.; Huang, E.S.; Quesenberry, C.P.; Whitmer, R.A. Severe Diabetic Retinal Disease and Dementia Risk in Type 2 Diabetes. J. Alzheimer’s Dis. 2014, 42, S109–S117. [Google Scholar] [CrossRef] [PubMed]
- Gasecka, A.; Siwik, D.; Gajewska, M.; Jaguszewski, M.J.; Mazurek, T.; Filipiak, K.J.; Postula, M.; Eyileten, C. Early Biomarkers of Neurodegenerative and Neurovascular Disorders in Diabetes. J. Clin. Med. 2020, 9, 2807. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, F.N.; Stokholm, L.; Pouwer, F.; Hass Rubin, K.; Peto, T.; Frydkjaer-Olsen, U.; Thykjaer, A.S.; Andersen, N.; Andresen, J.; Bek, T.; et al. Diabetic Retinopathy Predicts Risk of Alzheimer’s Disease: A Danish Registry-Based Nationwide Cohort Study. J. Alzheimer’s Dis. 2022, 86, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Zhao, X.; Yang, S.; Wang, G.; Ning, G. Association Between Diabetic Retinopathy and Cognitive Impairment: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2021, 13, 692911. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wen, Z.; Qi, C.X.; Tong, Y.; Shen, Y. Dynamic Changes of Amplitude of Low-Frequency Fluctuations in Patients With Diabetic Retinopathy. Front. Neurol. 2021, 12, 611702. [Google Scholar] [CrossRef]
- Crosby-Nwaobi, R.R.; Sivaprasad, S.; Amiel, S.; Forbes, A. The Relationship Between Diabetic Retinopathy and Cognitive Impairment. Diabetes Care 2013, 36, 3177–3186. [Google Scholar] [CrossRef]
- Martinez, B.; Peplow, P.V. MicroRNAs as biomarkers of diabetic retinopathy and disease progression. Neural Regen. Res. 2019, 14, 1858–1869. [Google Scholar]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Meneilly, G.S.; Hill, A. Alterations in glucose metabolism in patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 1993, 41, 710–714. [Google Scholar] [CrossRef]
- Pomytkin, I.; Costa-Nunes, J.P.; Kasatkin, V.; Veniaminova, E.; Demchenko, A.; Lyundup, A.; Lesch, K.-P.; Ponomarev, E.D.; Strekalova, T. Insulin receptor in the brain: Mechanisms of activation and the role in the CNS pathology and treatment. CNS Neurosci. Ther. 2018, 24, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Pansky, B.; Budd, G.C. Demonstration of insulin-specific mRNA in cultured rat retinal glial cells. Investig. Ophthalmol. Vis. Sci. 1987, 28, 1800–1810. [Google Scholar]
- Reiter, C.E.; Gardner, T.W. Functions of insulin and insulin receptor signaling in retina: Possible implications for diabetic retinopathy. Prog. Retin. Eye Res. 2003, 22, 545–562. [Google Scholar] [CrossRef]
- Freiherr, J.; Hallschmid, M.; Frey, W.H., 2nd; Brunner, Y.F.; Chapman, C.D.; Holscher, C.; Craft, S.; De Felice, F.G.; Benedict, C. Intranasal insulin as a treatment for Alzheimer’s disease: A review of basic research and clinical evidence. CNS Drugs 2013, 27, 505–514. [Google Scholar] [CrossRef]
- Schioth, H.B.; Craft, S.; Brooks, S.J.; Frey, W.H., 2nd; Benedict, C. Brain insulin signaling and Alzheimer’s disease: Current evidence and future directions. Mol. Neurobiol. 2012, 46, 4–10. [Google Scholar] [CrossRef]
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37. [Google Scholar] [CrossRef]
- Vieira, M.N.N.; Lima-Filho, R.A.S.; De Felice, F.G. Connecting Alzheimer’s disease to diabetes: Underlying mechanisms and potential therapeutic targets. Neuropharmacology 2018, 136, 160–171. [Google Scholar] [CrossRef]
- Yildirim Simsir, I.; Soyaltin, U.E.; Cetinkalp, S. Glucagon like peptide-1 (GLP-1) likes Alzheimer’s disease. Diabetes Metab. Syndr. 2018, 12, 469–475. [Google Scholar] [CrossRef]
- Perry, T.; Greig, N.H. Enhancing central nervous system endogenous GLP-1 receptor pathways for intervention in Alzheimer’s disease. Curr. Alzheimer Res. 2005, 2, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Baudouin, C.; Kolko, M.; Melik-Parsadaniantz, S.; Messmer, E.M. Inflammation in Glaucoma: From the back to the front of the eye, and beyond. Prog. Retin. Eye Res. 2021, 83, 100916. [Google Scholar] [CrossRef] [PubMed]
- Rieck, J. The pathogenesis of glaucoma in the interplay with the immune system. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2393–2409. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Van Bergen, N.J.; Kong, G.Y.; Chrysostomou, V.; Waugh, H.S.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp. Eye Res. 2011, 93, 204–212. [Google Scholar] [CrossRef]
- Osborne, N.N. Pathogenesis of Ganglion “Cell Death” in Glaucoma and Neuroprotection: Focus on Ganglion Cell Axonal Mitochondria; Elsevier: Amsterdam, The Netherlands, 2008; pp. 339–352. [Google Scholar]
- Mancino, R.; Martucci, A.; Cesareo, M.; Giannini, C.; Corasaniti, M.T.; Bagetta, G.; Nucci, C. Glaucoma and Alzheimer Disease: One Age-Related Neurodegenerative Disease of the Brain. Curr. Neuropharmacol. 2018, 16, 971–977. [Google Scholar] [CrossRef]
- Liu, Y.H.; Tian, T. Hypothesis of optineurin as a new common risk factor in normal-tension glaucoma and Alzheimer’s disease. Med. Hypotheses 2011, 77, 591–592. [Google Scholar] [CrossRef]
- Chalasani, M.L.; Balasubramanian, D.; Swarup, G. Focus on molecules: Optineurin. Exp. Eye Res. 2008, 87, 1–2. [Google Scholar] [CrossRef]
- Osawa, T.; Mizuno, Y.; Fujita, Y.; Takatama, M.; Nakazato, Y.; Okamoto, K. Optineurin in neurodegenerative diseases. Neuropathology 2011, 31, 569–574. [Google Scholar] [CrossRef]
- Nucci, C.; Martucci, A.; Cesareo, M.; Garaci, F.; Morrone, L.A.; Russo, R.; Corasaniti, M.T.; Bagetta, G.; Mancino, R. Links among glaucoma, neurodegenerative, and vascular diseases of the central nervous system. Prog. Brain Res. 2015, 221, 49–65. [Google Scholar]
- Bayer, A.U.; Ferrari, F.; Erb, C. High Occurrence Rate of Glaucoma among Patients with Alzheimer’s Disease. Eur. Neurol. 2002, 47, 165–168. [Google Scholar] [CrossRef]
- Kessing, L.V.; Lopez, A.G.; Andersen, P.K.; Kessing, S.V. No increased risk of developing Alzheimer disease in patients with glaucoma. J. Glaucoma 2007, 16, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.D.; Goldacre, R.; Goldacre, M.J. Associations between primary open angle glaucoma, Alzheimer’s disease and vascular dementia: Record linkage study. Br. J. Ophthalmol. 2015, 99, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Lv, X.; Wu, G.; Zhou, X.; Tian, H.; Qu, X.; Sun, H.; He, Y.; Zhang, Y.; Wang, C.; et al. Glaucoma Is Not Associated With Alzheimer’s Disease or Dementia: A Meta-Analysis of Cohort Studies. Front. Med. 2021, 8, 688551. [Google Scholar] [CrossRef] [PubMed]
- Wostyn, P.; Audenaert, K.; De Deyn, P.P. Alzheimer’s disease-related changes in diseases characterized by elevation of intracranial or intraocular pressure. Clin. Neurol. Neurosurg. 2008, 110, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, S.; Hara, H.; Hirata, A.; Fukushima, M.; Inomata, Y.; Tanihara, H. Vitreous fluid levels of β-amyloid(1–42) and tau in patients with retinal diseases. Jpn. J. Ophthalmol. 2005, 49, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.H.; Zou, J.Y.; Geng, W.; Wang, A.Y. Association between glaucoma and the risk of Alzheimer’s disease: A systematic review of observational studies. Acta Ophthalmol. 2019, 97, 665–671. [Google Scholar] [CrossRef]
- Moon, J.Y.; Kim, H.J.; Park, Y.H.; Park, T.K.; Park, E.C.; Kim, C.Y.; Lee, S.H. Association between Open-Angle Glaucoma and the Risks of Alzheimer’s and Parkinson’s Diseases in South Korea: A 10-year Nationwide Cohort Study. Sci. Rep. 2018, 8, 11161. [Google Scholar] [CrossRef]
- Lin, I.C.; Wang, Y.H.; Wang, T.J.; Wang, I.J.; Shen, Y.D.; Chi, N.F.; Chien, L.N. Glaucoma, Alzheimer’s disease, and Parkinson’s disease: An 8-year population-based follow-up study. PLoS ONE 2014, 9, e108938. [Google Scholar] [CrossRef]
- Pelletier, A.A.; Theoret, M.E.; Boutin, T.; Kergoat, M.J.; Massoud, F.; Latour, J.; Chayer, C.; Kergoat, H. Prevalence of glaucoma in hospitalized older adults with Alzheimer’s disease. Can. J. Neurol. Sci. 2014, 41, 206–209. [Google Scholar] [CrossRef]
- Gheorghe, A.; Mahdi, L.; Musat, O. Age-Related Macular Degeneration. Rom. J. Ophthalmol. 2015, 59, 74–77. [Google Scholar]
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Matsui, K. Parallel findings in age-related macular degeneration and Alzheimer’s disease. Prog. Retin. Eye Res. 2011, 30, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.Y.; Alekseev, O.; Li, Y.; Song, Y.; Dunaief, J.L. Iron increases APP translation and amyloid-beta production in the retina. Exp. Eye Res. 2014, 129, 31–37. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lynn, S.A.; Keeling, E.; Munday, R.; Gabha, G.; Griffiths, H.; Lotery, A.J.; Ratnayaka, J.A. The complexities underlying age-related macular degeneration: Could amyloid beta play an important role? Neural Regen. Res. 2017, 12, 538–548. [Google Scholar]
- Haan, J.; Verbraak, F.D.; Visser, P.J.; Bouwman, F.H. Retinal thickness in Alzheimer’s disease: A systematic review and meta-analysis. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2017, 6, 162–170. [Google Scholar]
- Wen, L.Y.; Wan, L.; Lai, J.N.; Chen, C.S.; Chen, J.J.; Wu, M.Y.; Hu, K.C.; Chiu, L.T.; Tien, P.T.; Lin, H.J. Increased risk of Alzheimer’s disease among patients with age-related macular degeneration: A nationwide population-based study. PLoS ONE 2021, 16, e0250440. [Google Scholar] [CrossRef]
- Palanimuthu, D.; Wu, Z.; Jansson, P.J.; Braidy, N.; Bernhardt, P.V.; Richardson, D.R.; Kalinowski, D.S. Novel chelators based on adamantane-derived semicarbazones and hydrazones that target multiple hallmarks of Alzheimer’s disease. Dalton Trans. 2018, 47, 7190–7205. [Google Scholar] [CrossRef]
- Dekens, D.W.; De Deyn, P.P.; Sap, F.; Eisel, U.L.M.; Naude, P.J.W. Iron chelators inhibit amyloid-beta-induced production of lipocalin 2 in cultured astrocytes. Neurochem. Int. 2020, 132, 104607. [Google Scholar] [CrossRef]
- Baker, M.L.; Wang, J.J.; Rogers, S.; Klein, R.; Kuller, L.H.; Larsen, E.K.; Wong, T.Y. Early age-related macular degeneration, cognitive function, and dementia: The Cardiovascular Health Study. Arch. Ophthalmol. 2009, 127, 667–673. [Google Scholar] [CrossRef]
- Chan, V.T.T.; Sun, Z.; Tang, S.; Chen, L.J.; Wong, A.; Tham, C.C.; Wong, T.Y.; Chen, C.; Ikram, M.K.; Whitson, H.E.; et al. Spectral-Domain OCT Measurements in Alzheimer’s Disease: A Systematic Review and Meta-analysis. Ophthalmology 2019, 126, 497–510. [Google Scholar] [CrossRef]
- Lopez-Cuenca, I.; de Hoz, R.; Salobrar-Garcia, E.; Elvira-Hurtado, L.; Rojas, P.; Fernandez-Albarral, J.A.; Barabash, A.; Salazar, J.J.; Ramirez, A.I.; Ramirez, J.M. Macular Thickness Decrease in Asymptomatic Subjects at High Genetic Risk of Developing Alzheimer’s Disease: An OCT Study. J. Clin. Med. 2020, 9, 1728. [Google Scholar] [CrossRef] [PubMed]
- Farzinvash, Z.; Abutorabi-Zarchi, M.; Manaviat, M.; Zare Mehrjerdi, H. Retinal Ganglion Cell Complex in Alzheimer Disease: Comparing Ganglion Cell Complex and Central Macular Thickness in Alzheimer Disease and Healthy Subjects Using Spectral Domain-Optical Coherence Tomography. Basic Clin. Neurosci. 2022, 13, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Iseri, P.K.; Altinas, O.; Tokay, T.; Yuksel, N. Relationship between cognitive impairment and retinal morphological and visual functional abnormalities in Alzheimer disease. J. Neuroophthalmol. 2006, 26, 18–24. [Google Scholar] [CrossRef]
- Polo, V.; Garcia-Martin, E.; Bambo, M.P.; Pinilla, J.; Larrosa, J.M.; Satue, M.; Otin, S.; Pablo, L.E. Reliability and validity of Cirrus and Spectralis optical coherence tomography for detecting retinal atrophy in Alzheimer’s disease. Eye 2014, 28, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.A. Neuroimaging of retinal nerve fiber layer in AD using optical coherence tomography. Neurology 2007, 69, 1060. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.Y.; Ong, Y.T.; Hilal, S.; Ikram, M.K.; Low, S.; Ong, Y.L.; Venketasubramanian, N.; Yap, P.; Seow, D.; Chen, C.L.; et al. Retinal ganglion cell analysis using high-definition optical coherence tomography in patients with mild cognitive impairment and Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.P.; Lopes, L.C.; Costa-Cunha, L.V.F.; Costa, C.F.; Pires, L.A.; Almeida, A.L.M.; Monteiro, M.L.R. Macular Thickness Measurements with Frequency Domain-OCT for Quantification of Retinal Neural Loss and its Correlation with Cognitive Impairment in Alzheimerʼs Disease. PLoS ONE 2016, 11, e0153830. [Google Scholar] [CrossRef]
- Song, A.; Johnson, N.; Ayala, A.; Thompson, A.C. Optical Coherence Tomography in Patients with Alzheimer’s Disease: What Can It Tell Us? Eye Brain 2021, 13, 1–20. [Google Scholar] [CrossRef]
- Thomson, K.L.; Yeo, J.M.; Waddell, B.; Cameron, J.R.; Pal, S. A systematic review and meta-analysis of retinal nerve fiber layer change in dementia, using optical coherence tomography. Alzheimer’s Dement. 2015, 1, 136–143. [Google Scholar] [CrossRef]
- Cunha, L.P.; Almeida, A.L.; Costa-Cunha, L.V.; Costa, C.F.; Monteiro, M.L. The role of optical coherence tomography in Alzheimer’s disease. Int. J. Retin. Vitr. 2016, 2, 24. [Google Scholar] [CrossRef]
- Cunha, J.P.; Moura-Coelho, N.; Proenca, R.P.; Dias-Santos, A.; Ferreira, J.; Louro, C.; Castanheira-Dinis, A. Alzheimer’s disease: A review of its visual system neuropathology. Optical coherence tomography-a potential role as a study tool in vivo. Graefes Arch. Clin. Exp. Ophthalmol. 2016, 254, 2079–2092. [Google Scholar] [CrossRef] [PubMed]
- Alber, J.; Goldfarb, D.; Thompson, L.I.; Arthur, E.; Hernandez, K.; Cheng, D.; DeBuc, D.C.; Cordeiro, F.; Provetti-Cunha, L.; den Haan, J.; et al. Developing retinal biomarkers for the earliest stages of Alzheimer’s disease: What we know, what we don’t, and how to move forward. Alzheimer’s Dement. 2020, 16, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Alzheimer disease and cerebrovascular pathology: An update. J. Neural Transm. 2002, 109, 813–836. [Google Scholar] [CrossRef]
- Katsimpris, A.; Karamaounas, A.; Sideri, A.M.; Katsimpris, J.; Georgalas, I.; Petrou, P. Optical coherence tomography angiography in Alzheimer’s disease: A systematic review and meta-analysis. Eye 2021, 36, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Bulut, M.; Kurtuluş, F.; Gözkaya, O.; Erol, M.K.; Cengiz, A.; Akıdan, M.; Yaman, A. Evaluation of optical coherence tomography angiographic findings in Alzheimer’s type dementia. Br. J. Ophthalmol. 2018, 102, 233–237. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, X.; Azhati, G.; Li, T.; Xu, G.; Liu, F. Retinal microvascular attenuation in mental cognitive impairment and Alzheimer’s disease by optical coherence tomography angiography. Acta Ophthalmol. 2020, 98, e781–e787. [Google Scholar] [CrossRef]
- Chua, J.; Sim, R.; Tan, B.; Wong, D.; Yao, X.; Liu, X.; Ting, D.S.W.; Schmidl, D.; Ang, M.; Garhofer, G.; et al. Optical Coherence Tomography Angiography in Diabetes and Diabetic Retinopathy. J. Clin. Med. 2020, 9, 1723. [Google Scholar] [CrossRef]
- Lee, T.H.; Lim, H.B.; Nam, K.Y.; Kim, K.; Kim, J.Y. Factors Affecting Repeatability of Assessment of the Retinal Microvasculature Using Optical Coherence Tomography Angiography in Healthy Subjects. Sci. Rep. 2019, 9, 16291. [Google Scholar] [CrossRef]
- Gomez-Ulla, F.; Cutrin, P.; Santos, P.; Fernandez, M.; Abraldes, M.; Abalo-Lojo, J.M.; Gonzalez, F. Age and gender influence on foveal avascular zone in healthy eyes. Exp. Eye Res. 2019, 189, 107856. [Google Scholar] [CrossRef]
- Hadoux, X.; Hui, F.; Lim, J.K.H.; Masters, C.L.; Pébay, A.; Chevalier, S.; Ha, J.; Loi, S.; Fowler, C.J.; Rowe, C.; et al. Non-invasive in vivo hyperspectral imaging of the retina for potential biomarker use in Alzheimer’s disease. Nat. Commun. 2019, 10, 4227. [Google Scholar] [CrossRef]
- More, S.S.; Beach, J.M.; McClelland, C.; Mokhtarzadeh, A.; Vince, R. In Vivo Assessment of Retinal Biomarkers by Hyperspectral Imaging: Early Detection of Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 4492–4501. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, S.; Van Eijgen, J.; Van Keer, K.; Jacob, J.; Moylett, S.; De Groef, L.; Vancraenendonck, T.; De Boever, P.; Stalmans, I. Hyperspectral Imaging and the Retina: Worth the Wave? Transl. Vis. Sci. Technol. 2020, 9, 9. [Google Scholar] [CrossRef]
- Lim, J.K.H.; Li, Q.-X.; Ryan, T.; Bedggood, P.; Metha, A.; Vingrys, A.J.; Bui, B.V.; Nguyen, C.T.O. Retinal hyperspectral imaging in the 5xFAD mouse model of Alzheimer’s disease. Sci. Rep. 2021, 11, 6387. [Google Scholar] [CrossRef] [PubMed]
- More, S.S.; Beach, J.M.; Vince, R. Early Detection of Amyloidopathy in Alzheimer’s Mice by Hyperspectral Endoscopy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3231–3238. [Google Scholar] [CrossRef] [PubMed]
- McGrory, S.; Cameron, J.R.; Pellegrini, E.; Warren, C.; Doubal, F.N.; Deary, I.J.; Dhillon, B.; Wardlaw, J.M.; Trucco, E.; MacGillivray, T.J. The application of retinal fundus camera imaging in dementia: A systematic review. Alzheimer’s Dement. 2017, 6, 91–107. [Google Scholar] [CrossRef]
- Cheung, C.Y.; Ong, Y.T.; Ikram, M.K.; Ong, S.Y.; Li, X.; Hilal, S.; Catindig, J.A.; Venketasubramanian, N.; Yap, P.; Seow, D.; et al. Microvascular network alterations in the retina of patients with Alzheimer’s disease. Alzheimer’s Dement. 2014, 10, 135–142. [Google Scholar] [CrossRef]
- Ong, Y.T.; Hilal, S.; Cheung, C.Y.; Xu, X.; Chen, C.; Venketasubramanian, N.; Wong, T.Y.; Ikram, M.K. Retinal vascular fractals and cognitive impairment. Dement. Geriatr. Cogn. Dis. Extra 2014, 4, 305–313. [Google Scholar] [CrossRef]
- Czako, C.; Kovacs, T.; Ungvari, Z.; Csiszar, A.; Yabluchanskiy, A.; Conley, S.; Csipo, T.; Lipecz, A.; Horvath, H.; Sandor, G.L.; et al. Retinal biomarkers for Alzheimer’s disease and vascular cognitive impairment and dementia (VCID): Implication for early diagnosis and prognosis. Geroscience 2020, 42, 1499–1525. [Google Scholar] [CrossRef]
- Fereshetian, S.; Agranat, J.S.; Siegel, N.; Ness, S.; Stein, T.D.; Subramanian, M.L. Protein and Imaging Biomarkers in the Eye for Early Detection of Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2021, 5, 375–387. [Google Scholar] [CrossRef]
- Wright, L.M.; Stein, T.D.; Jun, G.; Chung, J.; McConnell, K.; Fiorello, M.; Siegel, N.; Ness, S.; Xia, W.; Turner, K.L.; et al. Association of Cognitive Function with Amyloid-beta and Tau Proteins in the Vitreous Humor. J. Alzheimer’s Dis. 2019, 68, 1429–1438. [Google Scholar] [CrossRef]
- Tian, T.; Zhang, B.; Jia, Y.; Li, Z. Promise and challenge: The lens model as a biomarker for early diagnosis of Alzheimer’s disease. Dis. Markers 2014, 2014, 826503. [Google Scholar] [CrossRef]
- Kerbage, C.; Sadowsky, C.H.; Jennings, D.; Cagle, G.D.; Hartung, P.D. Alzheimer’s disease diagnosis by detecting exogenous fluorescent signal of ligand bound to Beta amyloid in the lens of human eye: An exploratory study. Front. Neurol. 2013, 4, 62. [Google Scholar] [CrossRef]
- Kerbage, C.; Sadowsky, C.H.; Tariot, P.N.; Agronin, M.; Alva, G.; Turner, F.D.; Nilan, D.; Cameron, A.; Cagle, G.D.; Hartung, P.D. Detection of Amyloid beta Signature in the Lens and Its Correlation in the Brain to Aid in the Diagnosis of Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Demen. 2015, 30, 738–745. [Google Scholar] [CrossRef]
- Petropoulos, I.N.; Ponirakis, G.; Khan, A.; Almuhannadi, H.; Gad, H.; Malik, R.A. Diagnosing Diabetic Neuropathy: Something Old, Something New. Diabetes Metab. J. 2018, 42, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Che, N.N.; Yang, H.Q. Potential use of corneal confocal microscopy in the diagnosis of Parkinson’s disease associated neuropathy. Transl. Neurodegener. 2020, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, I.N.; Kamran, S.; Li, Y.; Khan, A.; Ponirakis, G.; Akhtar, N.; Deleu, D.; Shuaib, A.; Malik, R.A. Corneal Confocal Microscopy: An Imaging Endpoint for Axonal Degeneration in Multiple Sclerosis. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3677–3681. [Google Scholar] [CrossRef] [PubMed]
- Dehghani, C.; Frost, S.; Jayasena, R.; Masters, C.L.; Kanagasingam, Y. Ocular Biomarkers of Alzheimer’s Disease: The Role of Anterior Eye and Potential Future Directions. Investig. Opthalmol. Vis. Sci. 2018, 59, 3554. [Google Scholar] [CrossRef]
- Fotiou, D.F.; Brozou, C.G.; Haidich, A.B.; Tsiptsios, D.; Nakou, M.; Kabitsi, A.; Giantselidis, C.; Fotiou, F. Pupil reaction to light in Alzheimer’s disease: Evaluation of pupil size changes and mobility. Aging Clin. Exp. Res. 2007, 19, 364–371. [Google Scholar] [CrossRef]
- El Haj, M.; Chapelet, G.; Moustafa, A.A.; Boutoleau-Bretonniere, C. Pupil size as an indicator of cognitive activity in mild Alzheimer’s disease. Excli J. 2022, 21, 307–316. [Google Scholar]
- Kucewicz, M.T.; Dolezal, J.; Kremen, V.; Berry, B.M.; Miller, L.R.; Magee, A.L.; Fabian, V.; Worrell, G.A. Pupil size reflects successful encoding and recall of memory in humans. Sci. Rep. 2018, 8, 4949. [Google Scholar] [CrossRef]
- Frost, S.M.; Kanagasingam, Y.; Sohrabi, H.R.; Taddei, K.; Bateman, R.; Morris, J.; Benzinger, T.; Goate, A.; Masters, C.L.; Martins, R.N. Pupil response biomarkers distinguish amyloid precursor protein mutation carriers from non-carriers. Curr. Alzheimer Res. 2013, 10, 790–796. [Google Scholar] [CrossRef]
- Hinton, D.R.; Sadun, A.A.; Blanks, J.C.; Miller, C.A. Optic-nerve degeneration in Alzheimer’s disease. N. Engl. J. Med. 1986, 315, 485–487. [Google Scholar] [CrossRef]
- Tsai, C.S.; Ritch, R.; Schwartz, B.; Lee, S.S.; Miller, N.R.; Chi, T.; Hsieh, F.Y. Optic nerve head and nerve fiber layer in Alzheimer’s disease. Arch. Ophthalmol. 1991, 109, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Bambo, M.P.; Garcia-Martin, E.; Gutierrez-Ruiz, F.; Pinilla, J.; Perez-Olivan, S.; Larrosa, J.M.; Polo, V.; Pablo, L. Analysis of optic disk color changes in Alzheimer’s disease: A potential new biomarker. Clin. Neurol. Neurosurg. 2015, 132, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, S.; Van Craenendonck, T.; Van Eijgen, J.; De Groef, L.; Bruffaerts, R.; de Jesus, D.A.; Charle, W.; Jayapala, M.; Sunaric-Megevand, G.; Standaert, A.; et al. Combination of snapshot hyperspectral retinal imaging and optical coherence tomography to identify Alzheimer’s disease patients. Alzheimers Res. Ther. 2020, 12, 144. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).