
Resistance to 2-Hydroxy-Flutamide in Prostate Cancer Cells Is Associated with the Downregulation of Phosphatidylcholine Biosynthesis and Epigenetic Modifications
 , , , and
, , , and 
Abstract
:1. Introduction
2. Results
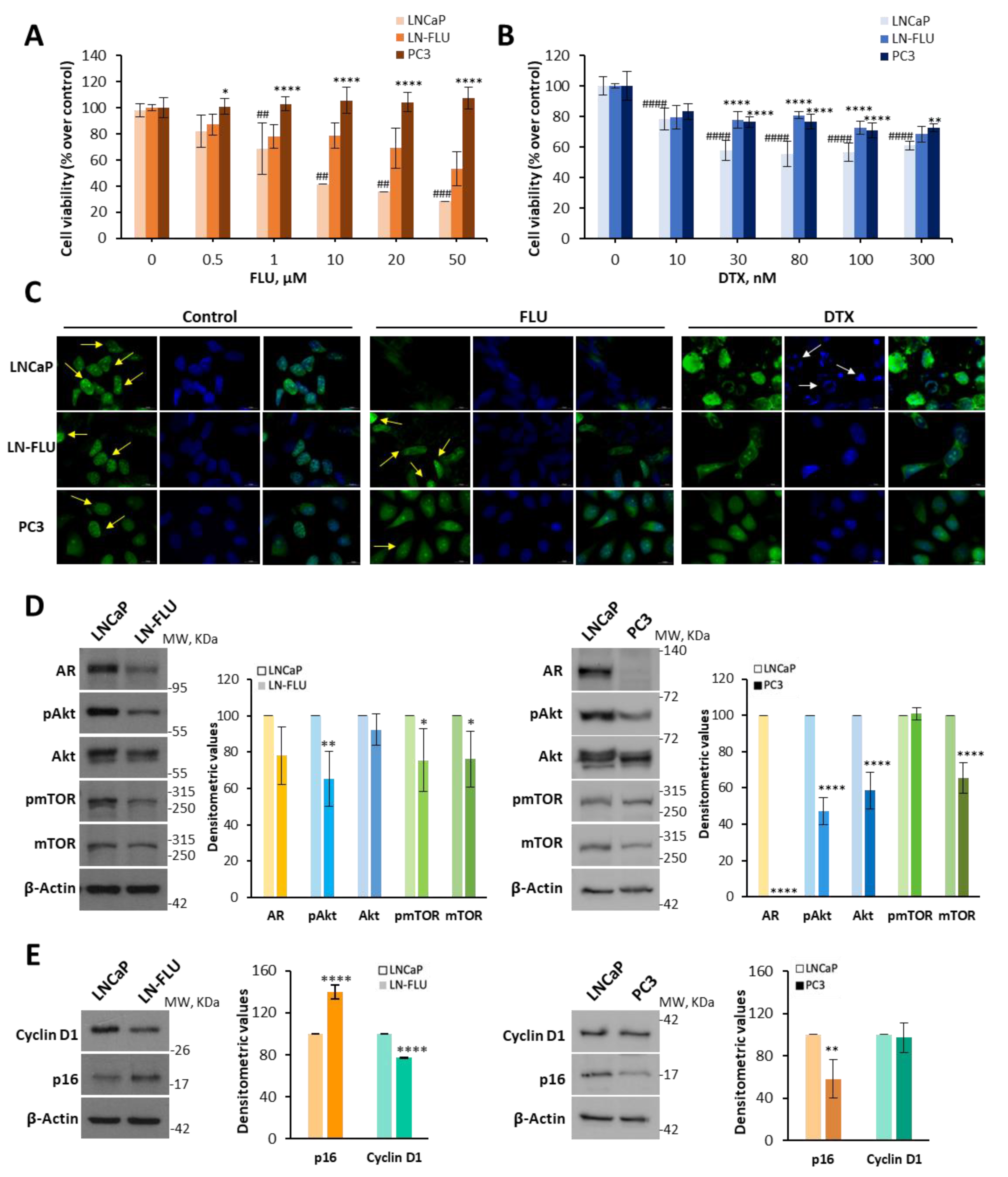
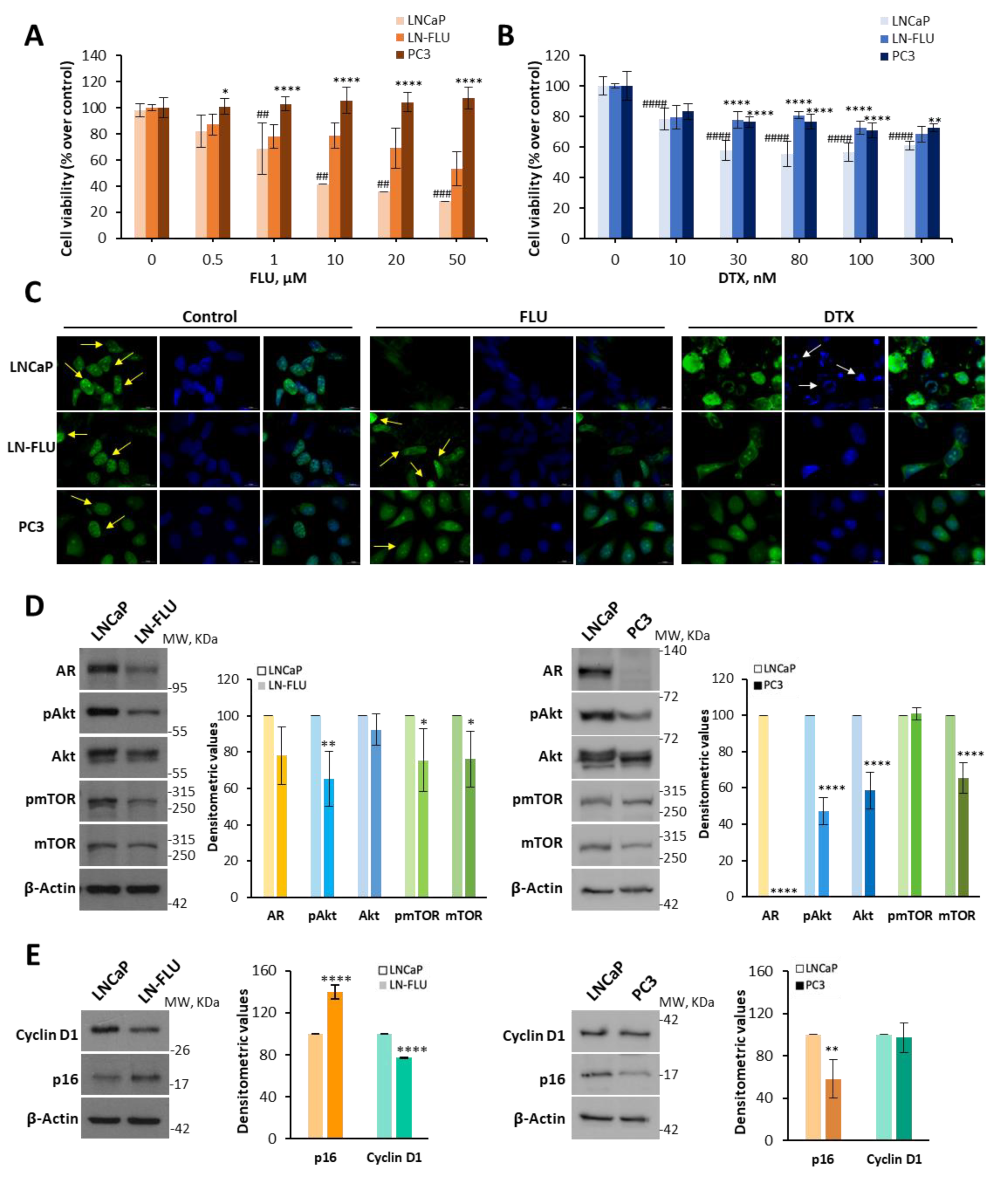
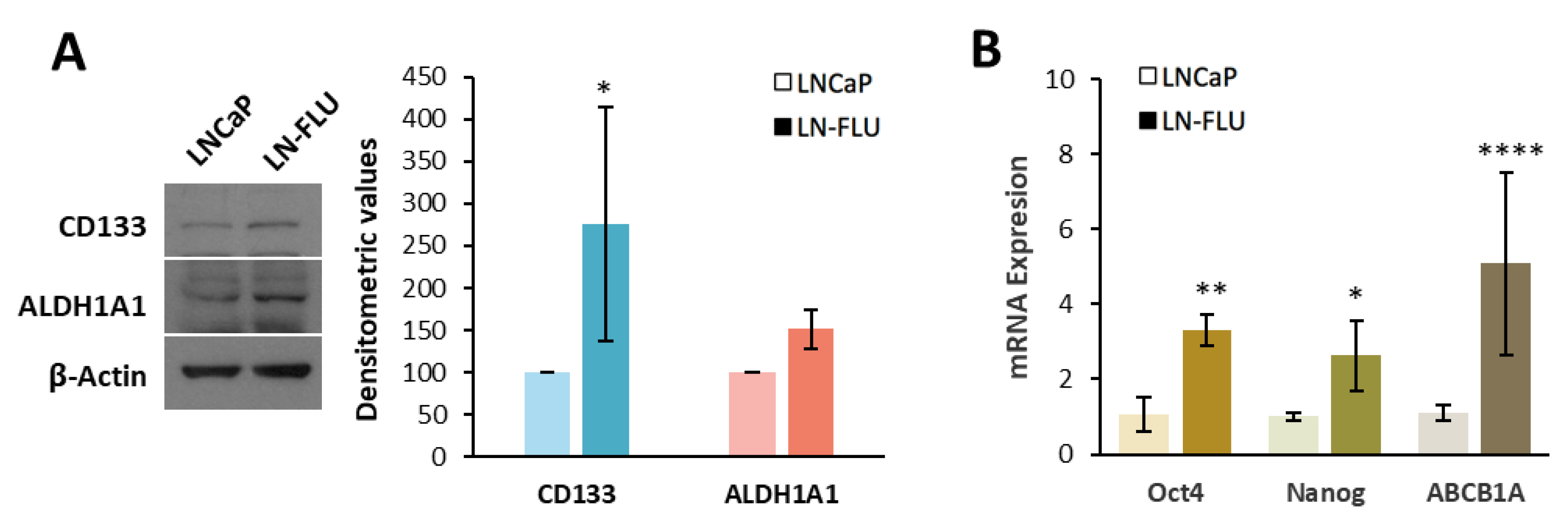
2.1. Antiandrogen-Induced Resistance in Prostate Cancer Cells: Arrest of Cell Growth and Stemness Acquirement
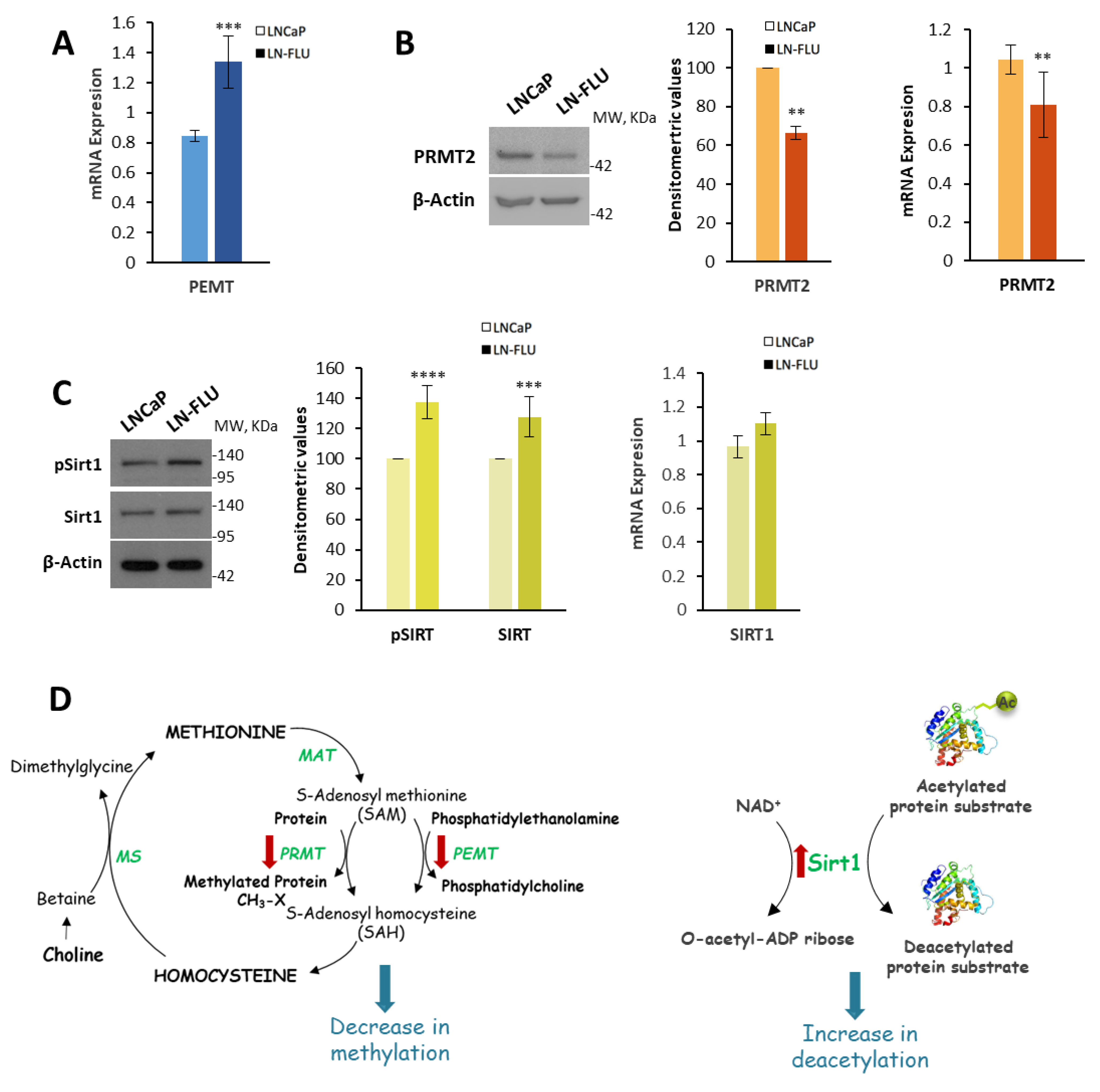
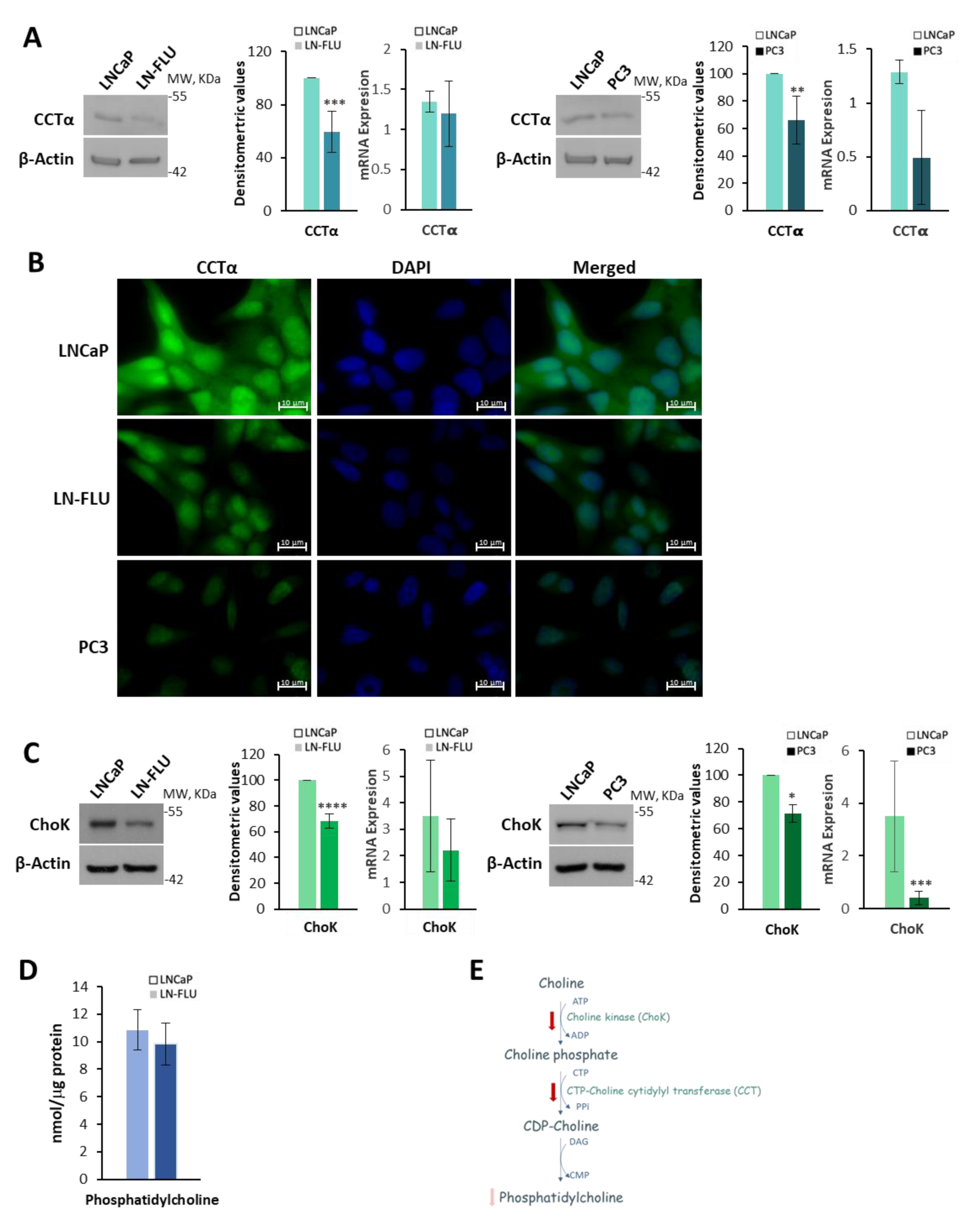
2.2. Phosphatidylcholine Biosynthesis Is Halted in LN-FLU Drug-Resistant Cells
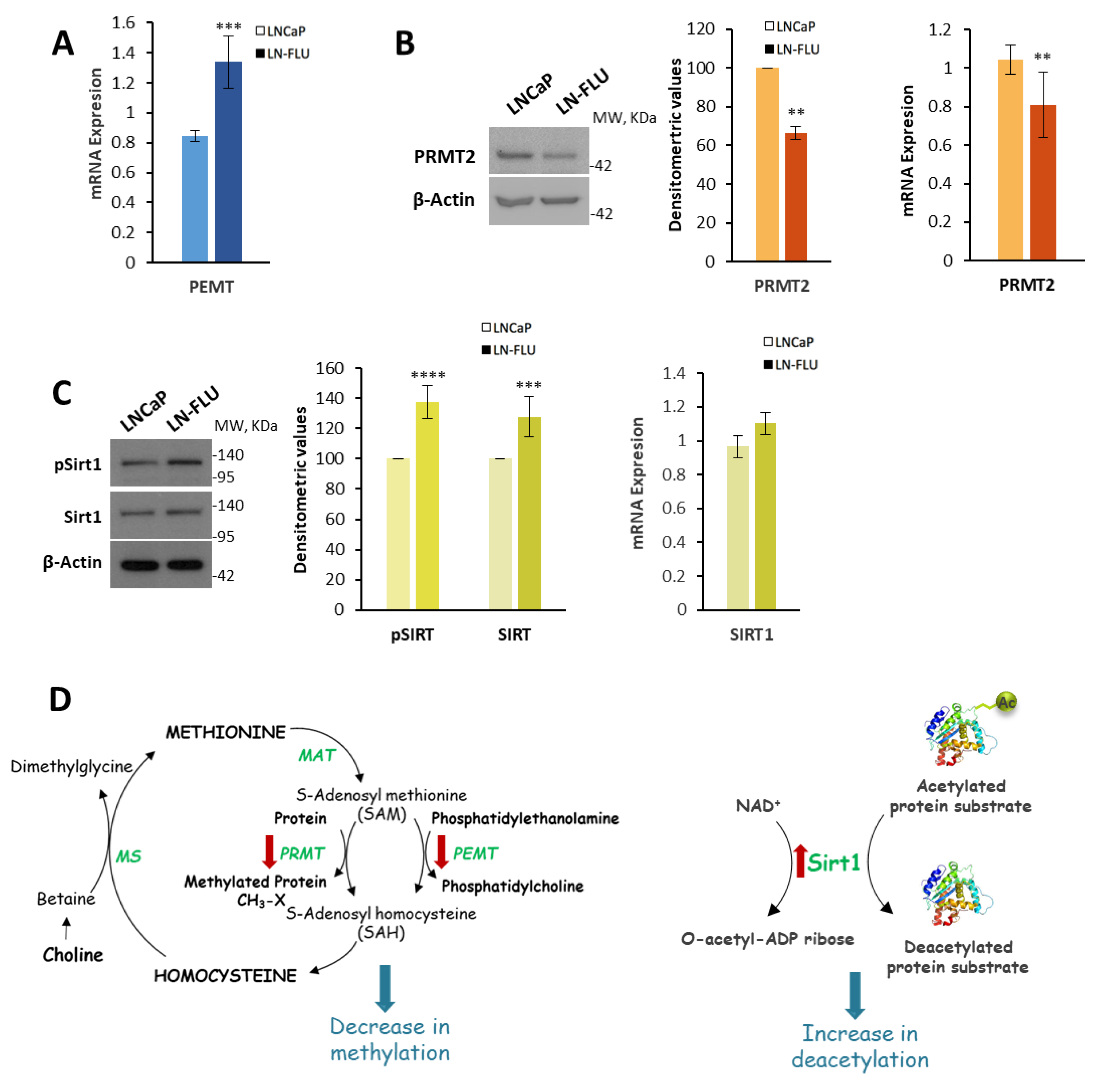
2.3. Protein Methylation and Deacetylation
2.4. Patients’ Baseline Characteristics
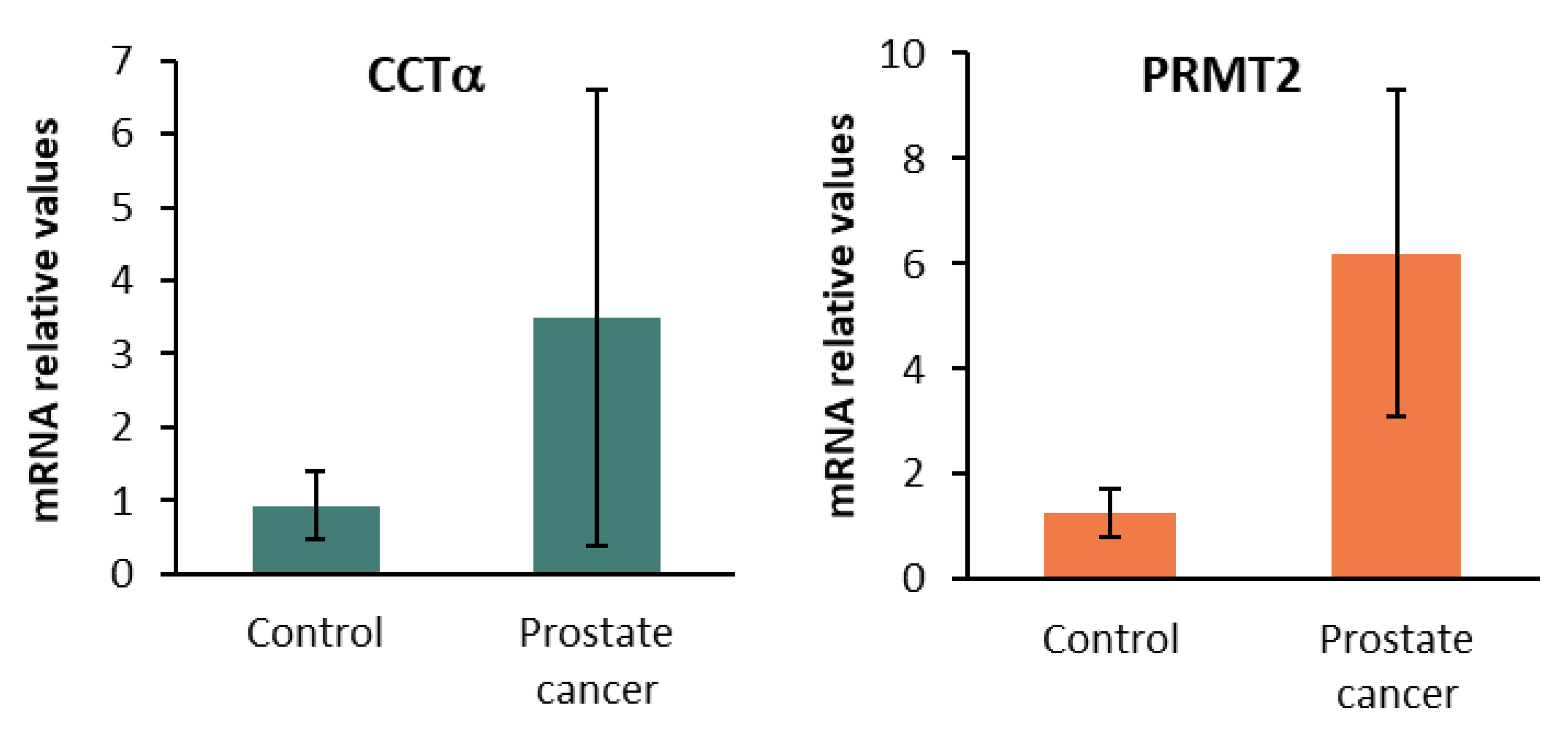
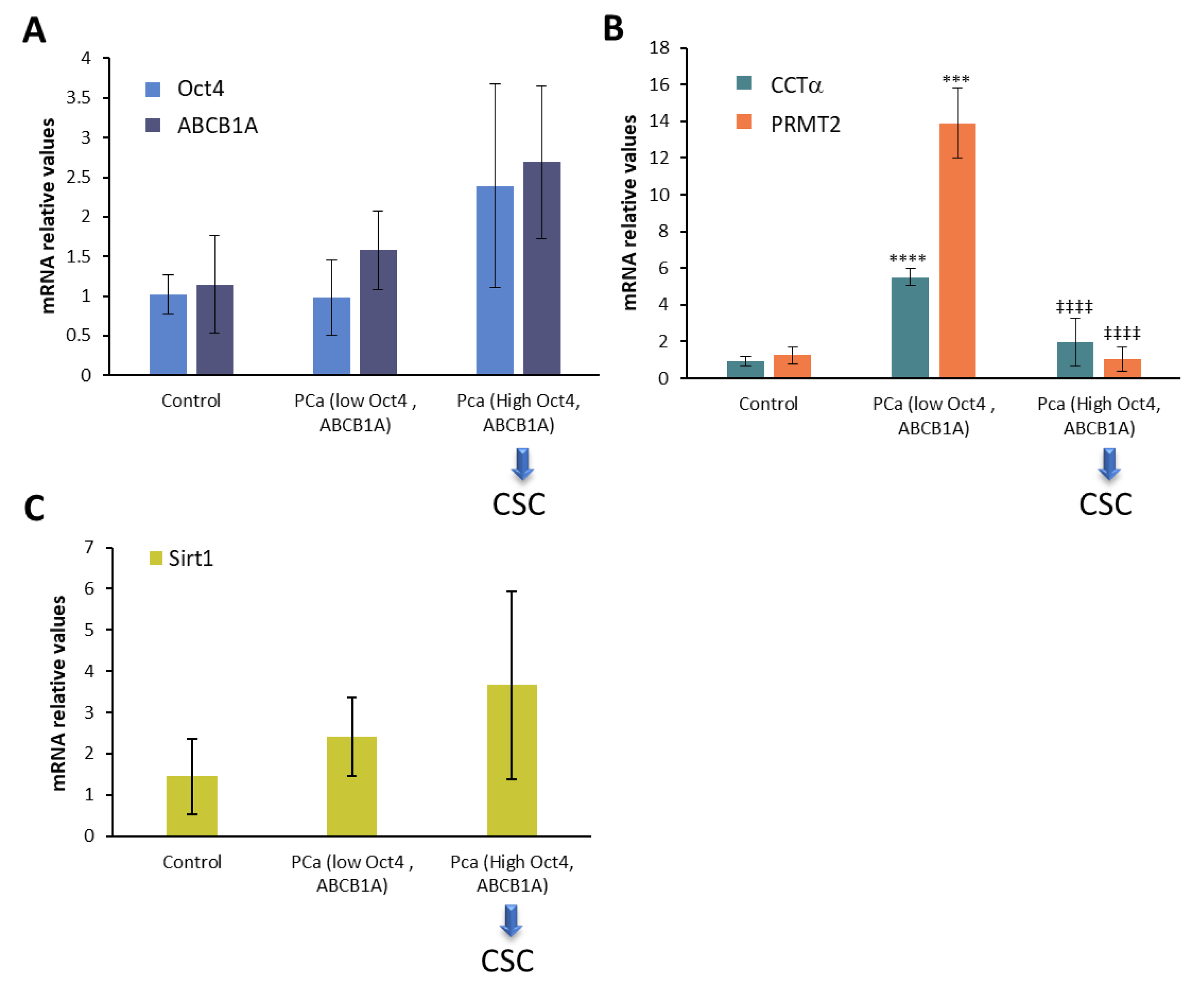
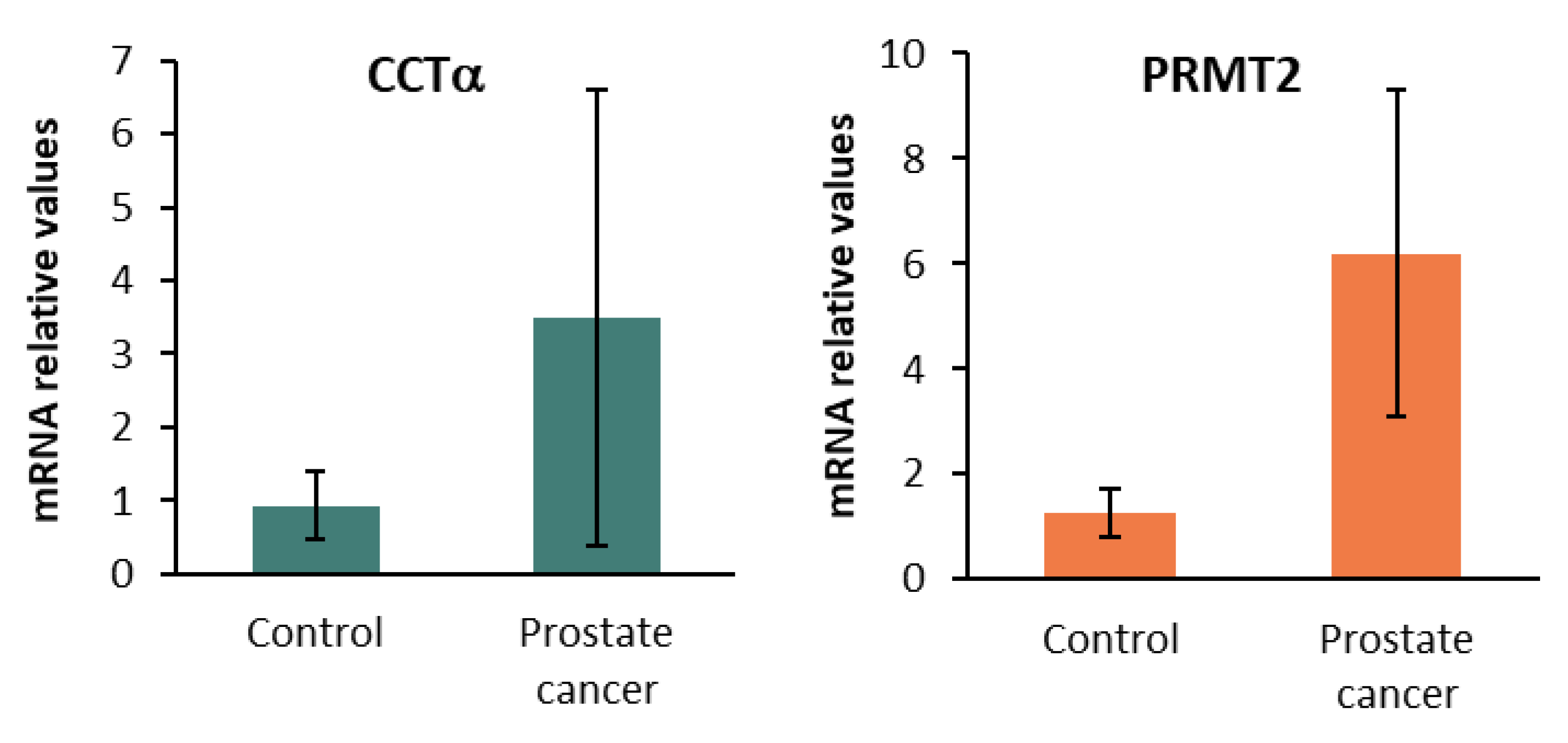
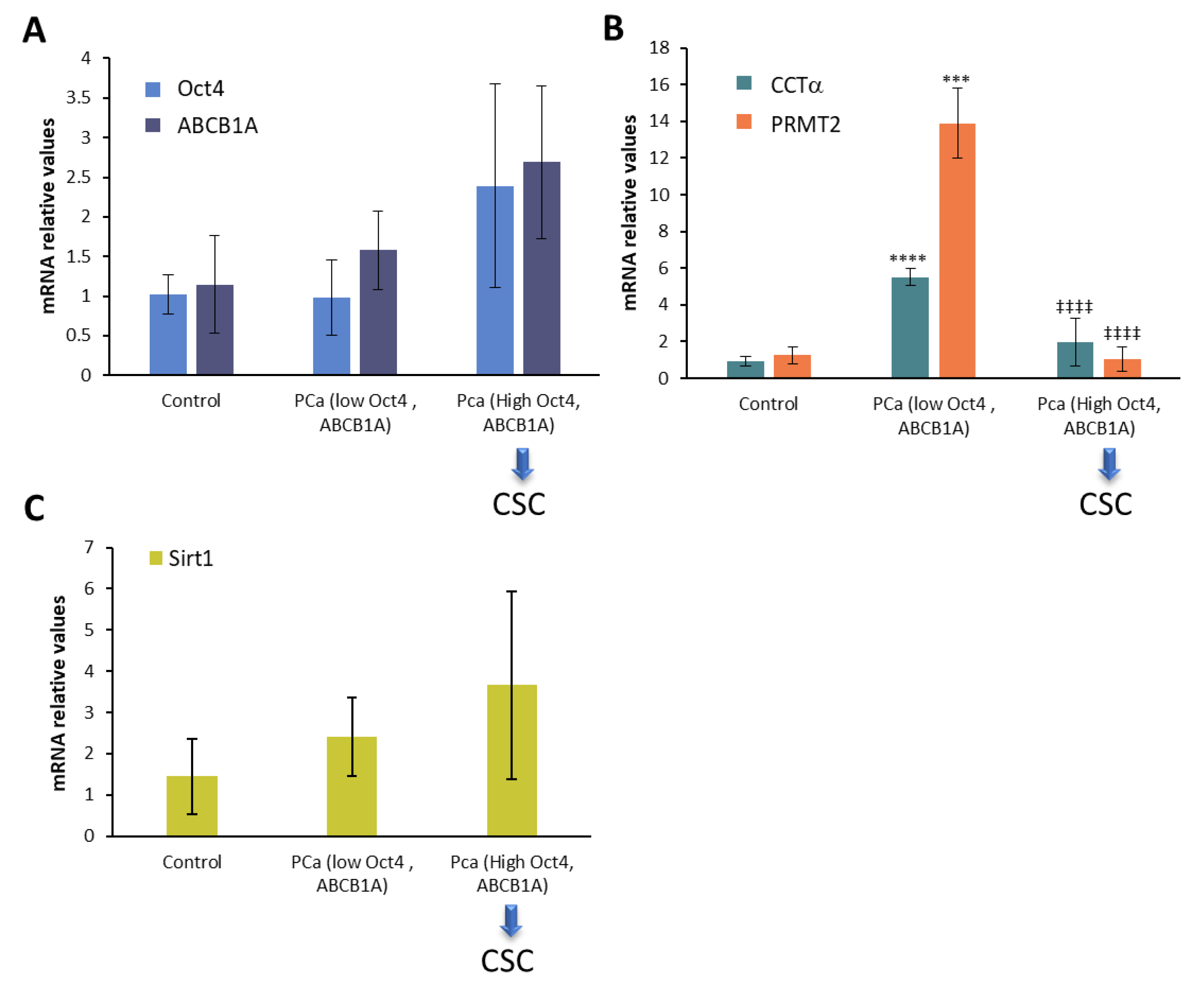
2.5. CCTα, PRMT2, and Sirt1 in Human Prostate Cancer Biopsies
3. Discussion
4. Materials and Methods
4.1. Patient Selection and Tissue Sampling
4.2. Cell Culture
4.3. Cell Viability
4.4. Cell Proliferation Assay with 5-Bromo-2′-deoxyuridine (BrdU)
4.5. Cell Proliferation Assay with Carboxyfluorescein Succinimidyl Ester (CFSE)
4.6. Western Blot
4.7. RNA Extraction and Reverse Transcription Quantitative Polymerase Chain Reaction
4.8. Fluorescence Microscopy
4.9. Analysis of Phosphatidylcholine Levels
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Morandi, A.; Indraccolo, S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 1–6. [Google Scholar] [CrossRef]
- Chen, X.; Chen, S.; Yu, D. Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites 2020, 10, 289. [Google Scholar] [CrossRef]
- Vellky, J.E.; Ricke, W.A. Development and prevalence of castration-resistant prostate cancer subtypes. Neoplasia 2020, 22, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.E.; Metzler, V.M.; Lothion-Roy, J.; Varun, D.; Woodcock, C.L.; Haigh, D.B.; Endeley, C.; Haque, M.; Toss, M.S.; Alsaleem, M.; et al. Exploring anti-androgen therapies in hormone dependent prostate cancer and new therapeutic routes for castration resistant prostate cancer. Front. Endocrinol. 2022, 13, 1006101. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.; Song, L.; Wang, B.Y.; Rezazadeh Kalebasty, A.; Uchio, E.; Zi, X. Prostate cancer immunotherapy: A review of recent advancements with novel treatment methods and efficacy. Am. J. Clin. Exp. Urol. 2022, 10, 210–233. [Google Scholar]
- Naghibi, A.F.; Daneshdoust, D.; Taha, S.R.; Abedi, S.; Dehdezi, P.A.; Zadeh, M.S.; Dokoohaki, F.; Soleymani-Goloujeh, M. Role of cancer stem cell-derived extracellular vesicles in cancer progression and metastasis. Pathol. Res. Pract. 2023, 247, 154558. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, P.P.; Verma, S.; Kumar, S.; Gupta, S. Role of prostate cancer stem-like cells in the development of antiandrogen resistance. Cancer Drug Resist. 2022, 5, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Hamburger, A.W.; Wang, L.; Khan, M.A.; Hussain, A. Androgen deprivation and stem cell markers in prostate cancers. Int. J. Clin. Exp. Pathol. 2009, 3, 128–138. [Google Scholar] [PubMed]
- Sanchez, B.G.; Bort, A.; Vara-Ciruelos, D.; Diaz-Laviada, I. Androgen Deprivation Induces Reprogramming of Prostate Cancer Cells to Stem-like Cells. Cells 2020, 9, 1441. [Google Scholar] [CrossRef] [PubMed]
- Francescangeli, F.; De Angelis, M.L.; Rossi, R.; Cuccu, A.; Giuliani, A.; De Maria, R.; Zeuner, A. Dormancy, stemness, and therapy resistance: Interconnected players in cancer evolution. Cancer Metastasis Rev. 2023, 42, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Tau, S.; Miller, T.W. The role of cancer cell bioenergetics in dormancy and drug resistance. Cancer Metastasis Rev. 2023, 42, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Van Heijster, F.H.A.; Breukels, V.; Jansen, K.; Schalken, J.A.; Heerschap, A. Carbon sources and pathways for citrate secreted by human prostate cancer cells determined by NMR tracing and metabolic modeling. Proc. Natl. Acad. Sci. USA 2022, 119, e2024357119. [Google Scholar] [CrossRef]
- Saito, R.F.; Andrade, L.N.S.; Bustos, S.O.; Chammas, R. Phosphatidylcholine-Derived Lipid Mediators: The Crosstalk Between Cancer Cells and Immune Cells. Front. Immunol. 2022, 13, 768606. [Google Scholar] [CrossRef]
- Awwad, H.M.; Geisel, J.; Obeid, R. The role of choline in prostate cancer. Clin. Biochem. 2012, 45, 1548–1553. [Google Scholar] [CrossRef]
- Beier, A.K.; Puhr, M.; Stope, M.B.; Thomas, C.; Erb, H.H.H. Metabolic changes during prostate cancer development and progression. J. Cancer Res. Clin. Oncol. 2023, 149, 2259–2270. [Google Scholar] [CrossRef]
- Hodolic, M. Imaging of Prostate Cancer Using (18)F-Choline PET/Computed Tomography. PET Clin. 2017, 12, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Abecassis, J.P.; Ghazzar, N.; Peyromaure, M.; Giraud, P. Prostate imaging: Contribution of PET PSMA and MRI. Cancer Radiother. 2020, 24, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Vera-Ramirez, L.; Hunter, K.W. Tumor cell dormancy as an adaptive cell stress response mechanism. F1000Research 2017, 6, 2134. [Google Scholar] [CrossRef]
- Gomis, R.R.; Gawrzak, S. Tumor cell dormancy. Mol. Oncol. 2017, 11, 62–78. [Google Scholar] [CrossRef]
- Linde, N.; Fluegen, G.; Aguirre-Ghiso, J.A. The Relationship Between Dormant Cancer Cells and Their Microenvironment. Adv. Cancer Res. 2016, 132, 45–71. [Google Scholar] [CrossRef]
- Yi, X.; Li, Y.; Hu, X.; Wang, F.; Liu, T. Changes in phospholipid metabolism in exosomes of hormone-sensitive and hormone-resistant prostate cancer cells. J. Cancer 2021, 12, 2893–2902. [Google Scholar] [CrossRef] [PubMed]
- Randall, E.C.; Zadra, G.; Chetta, P.; Lopez, B.G.C.; Syamala, S.; Basu, S.S.; Agar, J.N.; Loda, M.; Tempany, C.M.; Fennessy, F.M.; et al. Molecular Characterization of Prostate Cancer with Associated Gleason Score Using Mass Spectrometry Imaging. Mol. Cancer Res. 2019, 17, 1155–1165. [Google Scholar] [CrossRef]
- Li, X.; Nakayama, K.; Goto, T.; Kimura, H.; Akamatsu, S.; Hayashi, Y.; Fujita, K.; Kobayashi, T.; Shimizu, K.; Nonomura, N.; et al. High level of phosphatidylcholines/lysophosphatidylcholine ratio in urine is associated with prostate cancer. Cancer Sci. 2021, 112, 4292–4302. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Terada, N.; Inoue, T.; Kobayashi, T.; Nakayama, K.; Okada, Y.; Yoshikawa, T.; Miyazaki, Y.; Uegaki, M.; Utsunomiya, N.; et al. Decreased expression of lysophosphatidylcholine (16:0/OH) in high resolution imaging mass spectrometry independently predicts biochemical recurrence after surgical treatment for prostate cancer. Prostate 2015, 75, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Bort, A.; Sánchez, B.G.; Leon, C.; Nozal, L.; Mora-Rodriguez, J.M.; Castro, F.; Crego, A.L.; Diaz-Laviada, I. Metabolic fingerprinting of chemotherapy-resistant prostate cancer stem cells. An untargeted metabolomic approach by liquid chromatography-mass spectrometry. Front. Cell Dev. Biol. 2022, 10, 1005675. [Google Scholar] [CrossRef] [PubMed]
- Janardhan, S.; Srivani, P.; Sastry, G.N. Choline kinase: An important target for cancer. Curr. Med. Chem. 2006, 13, 1169–1186. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, C.; Yong, H.; Jiang, L.; Wang, P.; Meng, S.; Chu, S.; Li, Z.; Guo, Q.; Zheng, J.; et al. PRMT2 promotes RCC tumorigenesis and metastasis via enhancing WNT5A transcriptional expression. Cell Death Dis. 2023, 14, 322. [Google Scholar] [CrossRef]
- Grypari, I.M.; Pappa, I.; Papastergiou, T.; Zolota, V.; Bravou, V.; Melachrinou, M.; Megalooikonomou, V.; Tzelepi, V. Elucidating the role of PRMTs in prostate cancer using open access databases and a patient cohort dataset. Histol. Histopathol. 2023, 38, 287–302. [Google Scholar] [CrossRef]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Prajapati, K.S.; Kushwaha, P.P.; Shuaib, M.; Kumar Singh, A.; Kumar, S.; Gupta, S. Resistance to second generation antiandrogens in prostate cancer: Pathways and mechanisms. Cancer Drug Resist. 2020, 3, 742–761. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Shankar, E.; Kalayci, F.N.C.; Mukunda, A.; Alassfar, M.; Singh, V.; Chan, E.R.; MacLennan, G.T.; Gupta, S. Androgen Deprivation Induces Transcriptional Reprogramming in Prostate Cancer Cells to Develop Stem Cell-like Characteristics. Int. J. Mol. Sci. 2020, 21, 9568. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.E., Jr.; Kirk, J.; Ji, Y.; Tang, D.G. Tumor Dormancy and Slow-Cycling Cancer Cells. Adv. Exp. Med. Biol. 2019, 1164, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A.; Bragado, P.; Sosa, M.S. Metastasis awakening: Targeting dormant cancer. Nat. Med. 2013, 19, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Alumkal, J.J.; Sun, D.; Lu, E.; Beer, T.M.; Thomas, G.V.; Latour, E.; Aggarwal, R.; Cetnar, J.; Ryan, C.J.; Tabatabaei, S.; et al. Transcriptional profiling identifies an androgen receptor activity-low, stemness program associated with enzalutamide resistance. Proc. Natl. Acad. Sci. USA 2020, 117, 12315–12323. [Google Scholar] [CrossRef]
- Santos-de-Frutos, K.; Djouder, N. When dormancy fuels tumour relapse. Commun. Biol. 2021, 4, 747. [Google Scholar] [CrossRef]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef] [PubMed]
- Pranzini, E.; Raugei, G.; Taddei, M.L. Metabolic Features of Tumor Dormancy: Possible Therapeutic Strategies. Cancers 2022, 14, 547. [Google Scholar] [CrossRef]
- Lo Presti, C.; Yamaryo-Botte, Y.; Mondet, J.; Berthier, S.; Nutiu, D.; Botte, C.; Mossuz, P. Variation in Lipid Species Profiles among Leukemic Cells Significantly Impacts Their Sensitivity to the Drug Targeting of Lipid Metabolism and the Prognosis of AML Patients. Int. J. Mol. Sci. 2023, 24, 5988. [Google Scholar] [CrossRef]
- Yousafzai, N.A.; Jin, H.; Ullah, M.; Wang, X. Recent advances of SIRT1 and implications in chemotherapeutics resistance in cancer. Am. J. Cancer Res. 2021, 11, 5233–5248. [Google Scholar] [PubMed]
- Zietara, P.; Dziewiecka, M.; Augustyniak, M. Why Is Longevity Still a Scientific Mystery? Sirtuins-Past, Present and Future. Int. J. Mol. Sci. 2022, 24, 728. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.L.; Baris, A.M.; Fu, H.; Redon, C.E.; Pongor, L.S.; Mosavarpour, S.; Gross, J.M.; Jang, S.M.; Sebastian, R.; Utani, K.; et al. Convergence of SIRT1 and ATR signaling to modulate replication origin dormancy. Nucleic Acids Res. 2022, 50, 5111–5128. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kennedy, M.; Payne, S.; Kennedy, K.; Seewaldt, V.L.; Pizzo, S.V.; Bachelder, R.E. Model of tumor dormancy/recurrence after short-term chemotherapy. PLoS ONE 2014, 9, e98021. [Google Scholar] [CrossRef]
- Lee, S.; Mendoza, T.R.; Burner, D.N.; Muldong, M.T.; Wu, C.C.N.; Arreola-Villanueva, C.; Zuniga, A.; Greenburg, O.; Zhu, W.Y.; Murtadha, J.; et al. Novel Dormancy Mechanism of Castration Resistance in Bone Metastatic Prostate Cancer Organoids. Int. J. Mol. Sci. 2022, 23, 3203. [Google Scholar] [CrossRef] [PubMed]
- Surintrspanont, J.; Zhou, M. Prostate Pathology: What is New in the 2022 WHO Classification of Urinary and Male Genital Tumors? Pathologica 2022, 115, 41–56. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % | Mean ± SD | Median | |
|---|---|---|---|
| Age (years) | |||
| 50–59 | 12.5 | 55.5 ± 0.7 | 55.5 |
| 60–69 | 62.5 | 66.4 ± 2.9 | 68.0 |
| >70 | 25.0 | 73.5 ± 2.3 | 74.5 |
| Weight (kg) | |||
| 60–79 | 50.0 | 72.0 ± 5.3 | 74 |
| 80–99 | 31.25 | 87.8 ± 5.1 | 86 |
| >100 | 18.75 | 107.6 ± 5.8 | 110 |
| BMI (kg/m2) | |||
| 20–25 | 18.75 | 23.4 ± 0.5 | 23.7 |
| 26–30 | 50.0 | 27.6 ± 1.4 | 27.25 |
| >30 | 31.25 | 33.8 ± 3.0 | 34.1 |
| Gleason Grade | |||
| 6–7 | 26.6 | 6.5 ± 0.5 | 6.5 |
| 7–8 | 60.0 | 7.3 ± 0.5 | 7 |
| >8 | 13.4 | 9 ± 0.0 | 9 |
| PSA (ng/mL) | |||
| 1–7 | 50 | 4.9 ± 1.8 | 4.74 |
| 7–20 | 31.25 | 9.1 ± 2.2 | 8.12 |
| >20 | 18.75 | 28.4 ± 12.6 | 21.22 |
| Antibody | Dilution | Reference and Company |
|---|---|---|
| AR | 1:500 | sc-7305; Santa Cruz Biotechnology (Dallas, TX, USA) |
| pAkt (Ser 473) | 1:1000 | #4060; Cell Signaling Technology, Danvers, MA, USA |
| Akt | 1:1000 | #4691; Cell Signaling Technology, Danvers, MA, USA |
| pmTOR | 1:1000 | #2971; Cell Signaling Technology, Danvers, MA, USA |
| mTOR | 1:1000 | #2972; Cell Signaling Technology, Danvers, MA, USA |
| Cyclin D1 | 1:1000 | #RM-9104-S0; Invitrogen, Paisley, UK |
| p16 | 1:500 | sc-467; Santa Cruz Biotechnology (Dallas, TX, USA) |
| CD133 | 1:500 | #64326; Cell Signaling Technology, Danvers, MA, USA |
| ALDH1A1 | 1:500 | #12035; Cell Signaling Technology, Danvers, MA, USA |
| CCT | 1:1000 | #6931; Cell Signaling Technology, Danvers, MA, USA |
| CK | 1:1000 | #13422; Cell Signaling Technology, Danvers, MA, USA |
| PRMT2 | 1:500 | ab154154; Abcam, Cambridge, UK |
| pSIRT | 1:500 | #2314; Cell Signaling Technology, Danvers, MA, USA |
| SIRT | 1:500 | #9475; Cell Signaling Technology, Danvers, MA, USA |
| -actin | 1:5000 | A5441; Sigma, St. Louis, MO, USA |
| Rabbit (HRP) | 1:5000 | #7074S; Cell Signaling Technology, Danvers, MA, USA |
| Mouse (HRP) | 1:5000 | A-9044; Sigma, St. Louis, MO, USA |
| Gene | Forward | Reverse |
|---|---|---|
| Oct4 | 5′-GACAGGGGGAGGGGAGGAGCTAGG-3′ | 5′-CTTCCCTCCAACCAGTTGCCCCAAAC-3′ |
| Nanog | 5′-TTTGTGGGCCTGAAGAAAACT-3′ | 5′-AGGGCTGTCCTGAATAAGCAG-3′ |
| ABCB1A | 5′-TTGCTGCTTACATTCAGGTTTCA-3′ | 5′-AGCCTATCTCCTGTCGCATTA-3′ |
| PEMT | 5′-CTCTAAGCGTCACCATCCTGCT-3′ | 5′-GGTGTCCAGGCTCTCCATCCT-3′ |
| PRMT2 | 5′-GCAGTTGGACATGAGAACCGTG-3′ | 5′-AGGCTCTGGAAGTGGACGCTAA-3′ |
| Sirt1 | 5′-ACCCAGAACATAGACACGCTGGAA-3′ | 5′-TCTCCTCGTACAGCTTCACAGTCA-3′ |
| CCTα | 5′-GTGGTCATTACAGACCCTGAAAA-3′ | 5′-AACTCTTCTAACTGCCATAGCAC-3′ |
| Actin | 5′-AGAAGGATTCCTATGTGGGCG-3′ | 5′-CATGTCGTCCCAGTTGGTGAC-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mora-Rodríguez, J.M.; Sánchez, B.G.; Sebastián-Martín, A.; Díaz-Yuste, A.; Sánchez-Chapado, M.; Palacín, A.M.; Sánchez-Rodríguez, C.; Bort, A.; Díaz-Laviada, I. Resistance to 2-Hydroxy-Flutamide in Prostate Cancer Cells Is Associated with the Downregulation of Phosphatidylcholine Biosynthesis and Epigenetic Modifications. Int. J. Mol. Sci. 2023, 24, 15626. https://doi.org/10.3390/ijms242115626
Mora-Rodríguez JM, Sánchez BG, Sebastián-Martín A, Díaz-Yuste A, Sánchez-Chapado M, Palacín AM, Sánchez-Rodríguez C, Bort A, Díaz-Laviada I. Resistance to 2-Hydroxy-Flutamide in Prostate Cancer Cells Is Associated with the Downregulation of Phosphatidylcholine Biosynthesis and Epigenetic Modifications. International Journal of Molecular Sciences. 2023; 24(21):15626. https://doi.org/10.3390/ijms242115626
Chicago/Turabian StyleMora-Rodríguez, José María, Belén G. Sánchez, Alba Sebastián-Martín, Alba Díaz-Yuste, Manuel Sánchez-Chapado, Ana María Palacín, Carlos Sánchez-Rodríguez, Alicia Bort, and Inés Díaz-Laviada. 2023. "Resistance to 2-Hydroxy-Flutamide in Prostate Cancer Cells Is Associated with the Downregulation of Phosphatidylcholine Biosynthesis and Epigenetic Modifications" International Journal of Molecular Sciences 24, no. 21: 15626. https://doi.org/10.3390/ijms242115626
APA StyleMora-Rodríguez, J. M., Sánchez, B. G., Sebastián-Martín, A., Díaz-Yuste, A., Sánchez-Chapado, M., Palacín, A. M., Sánchez-Rodríguez, C., Bort, A., & Díaz-Laviada, I. (2023). Resistance to 2-Hydroxy-Flutamide in Prostate Cancer Cells Is Associated with the Downregulation of Phosphatidylcholine Biosynthesis and Epigenetic Modifications. International Journal of Molecular Sciences, 24(21), 15626. https://doi.org/10.3390/ijms242115626