
The Key Role of Astrocytes in Amyotrophic Lateral Sclerosis and Their Commitment to Glutamate Excitotoxicity


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. ALS as a Non-Cell-Autonomous Disease
2.1. Astrocyte Contribution to ALS
2.2. Microglia and Oligodendrocyte Contribution to ALS
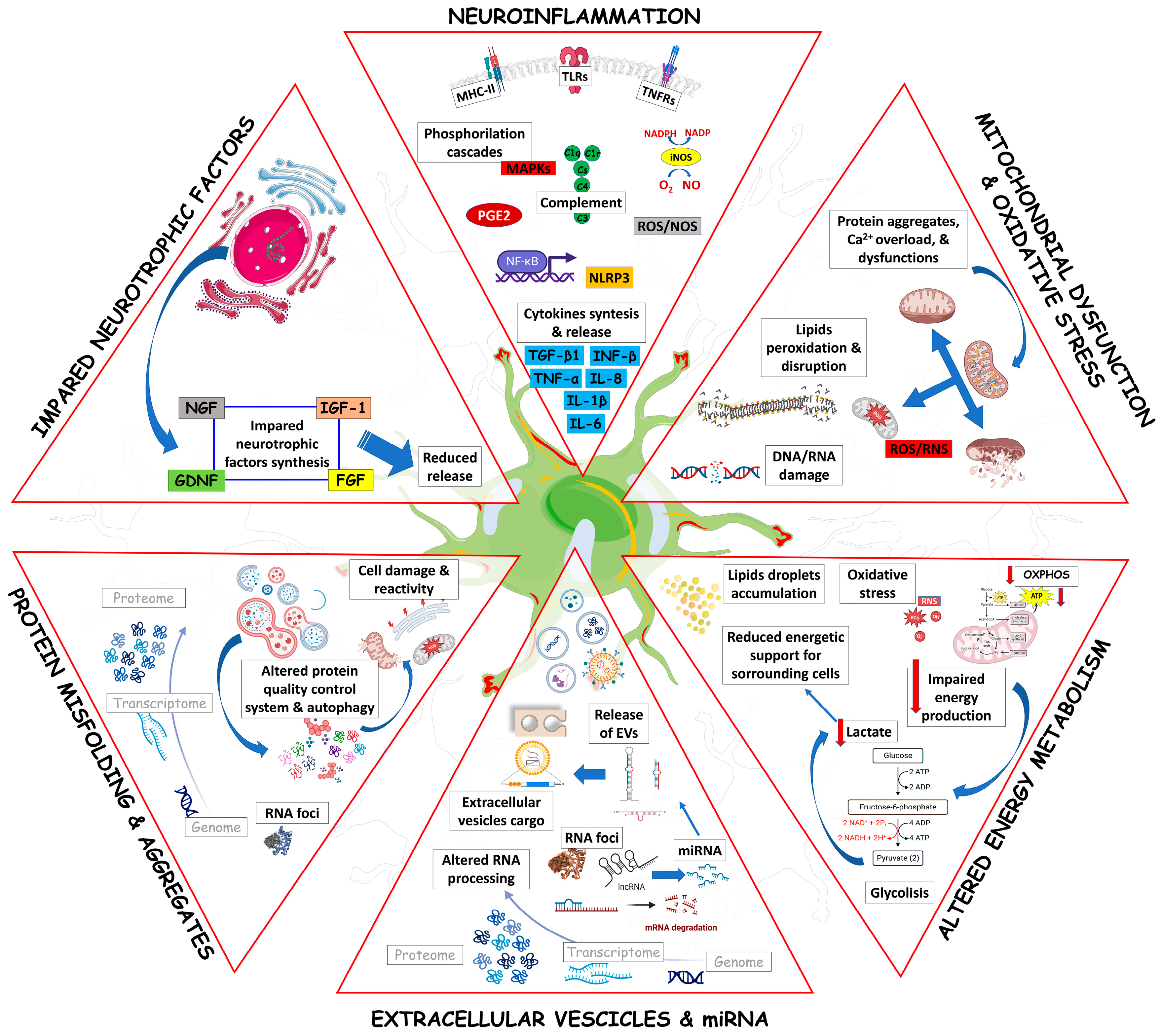
3. Astrocyte Mechanisms Fostering Neuronal Damage in ALS
3.1. Astrocytes and Neuroinflammation in ALS
3.2. Astrocytes, Mitochondrial Dysfunction, and Oxidative Stress in ALS
3.3. Astrocytes and Energy Metabolism in ALS
3.4. Astrocytes, miRNAs and Extracellular Vesicles in ALS
3.5. Astrocytes and Protein Misfolding and Autophagy in ALS
3.6. Astrocytes and Neurotrophic Factors in ALS
3.7. Astrocytes and Glutamate Excitotoxicity in ALS
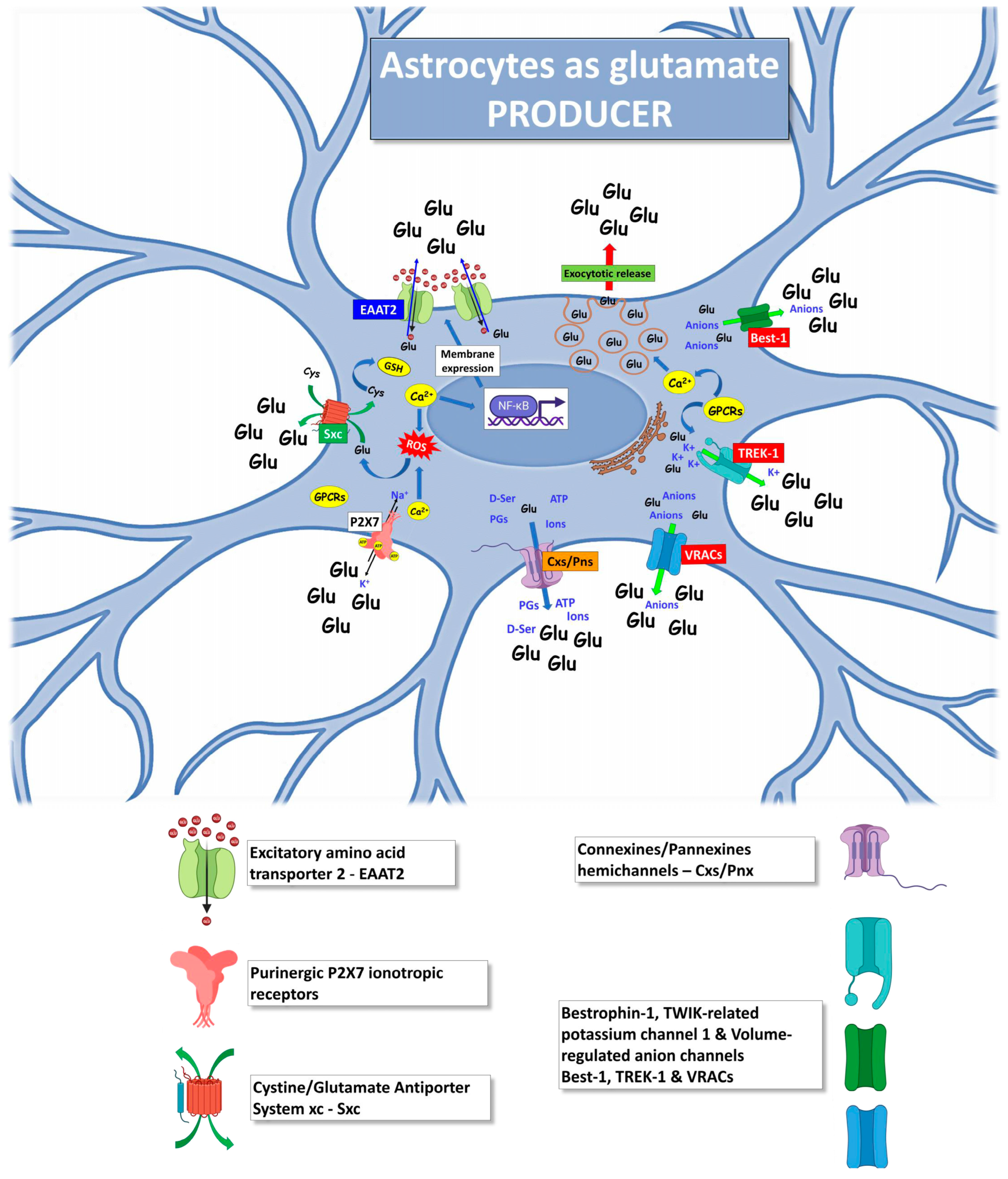
4. Astrocytes as Producers of Excessive Glutamate in ALS

4.1. Excitatory Amino Acid Transporter 2
4.2. Exocytotic Glutamate Release
4.3. Purinergic P2X7 Receptors
4.4. Cystine/Glutamate Antiporter System xc
4.5. Hemichannels
4.6. Bestrophin-1, TWIK-Related Potassium Channel 1 and Volume-Regulated Anion Channels
4.7. Other Mechanisms Triggering Astrocytic Glutamate Excitotoxicity
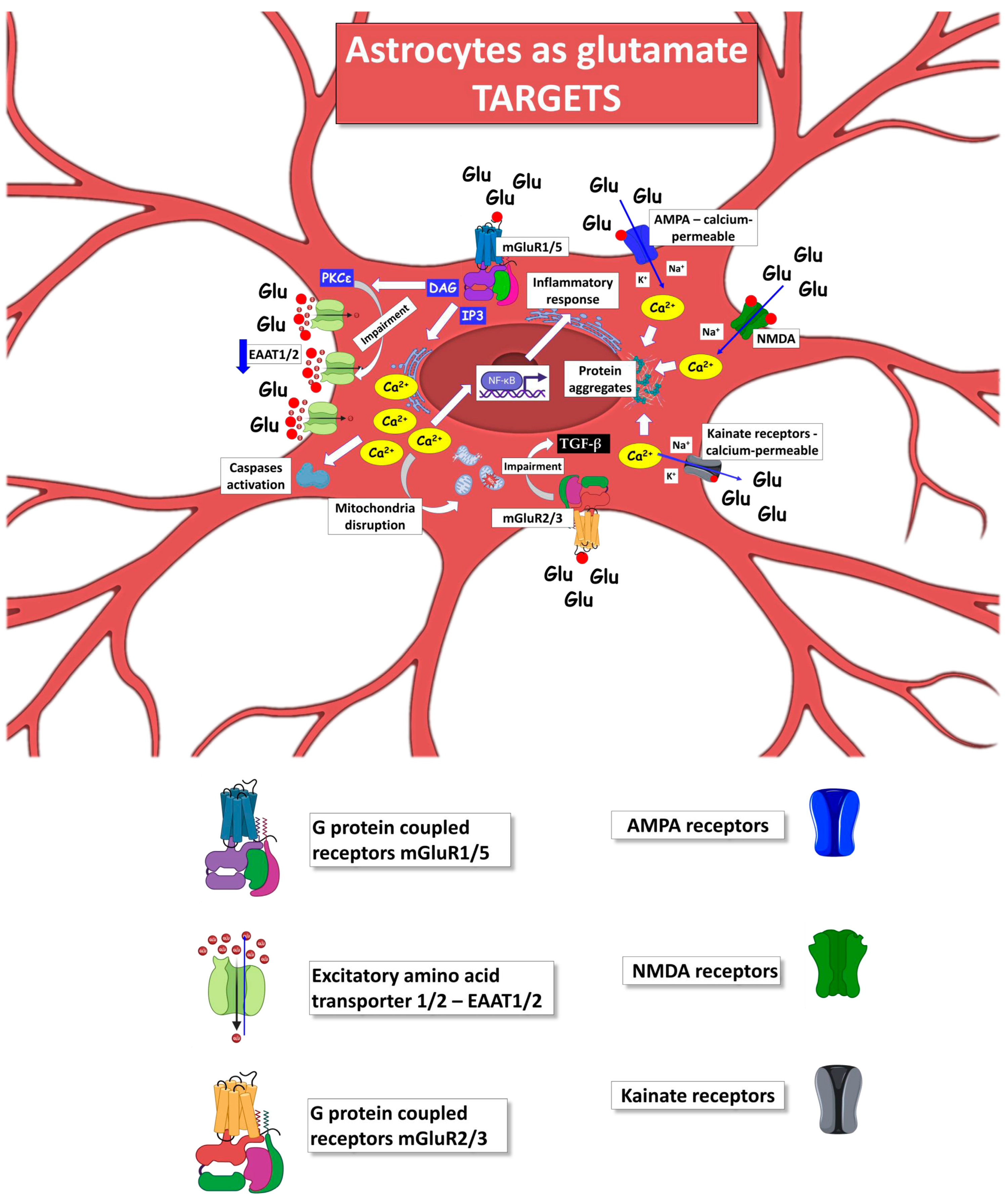
5. Astrocytes as Target of Glutamate Excitotoxicity in ALS

Metabotropic Glutamate Receptors
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, R.H.J. Amyotrophic Lateral Sclerosis: Recent Insights from Genetics and Transgenic Mice. Cell 1995, 80, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Dhasmana, S.; Dhasmana, A.; Narula, A.S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. The Panoramic View of Amyotrophic Lateral Sclerosis: A Fatal Intricate Neurological Disorder. Life Sci. 2022, 288, 120156. [Google Scholar] [CrossRef] [PubMed]
- Phukan, J.; Pender, N.P.; Hardiman, O. Cognitive Impairment in Amyotrophic Lateral Sclerosis. Lancet Neurol. 2007, 6, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17085. [Google Scholar] [CrossRef]
- Eisen, A. Amyotrophic Lateral Sclerosis: A 40-Year Personal Perspective. J. Clin. Neurosci. 2009, 16, 505–512. [Google Scholar] [CrossRef]
- Manjaly, Z.R.; Scott, K.M.; Abhinav, K.; Wijesekera, L.; Ganesalingam, J.; Goldstein, L.H.; Janssen, A.; Dougherty, A.; Willey, E.; Stanton, B.R.; et al. The Sex Ratio in Amyotrophic Lateral Sclerosis: A Population Based Study. Amyotroph. Lateral Scler. 2010, 11, 439–442. [Google Scholar] [CrossRef]
- Xu, L.; Liu, T.; Liu, L.; Yao, X.; Chen, L.; Fan, D.; Zhan, S.; Wang, S. Global Variation in Prevalence and Incidence of Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. J. Neurol. 2020, 267, 944–953. [Google Scholar] [CrossRef]
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Masson, G. Le Genetics of Amyotrophic Lateral Sclerosis: A Review. J Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current Knowledge and Recent Insights into the Genetic Basis of Amyotrophic Lateral Sclerosis. Med. Genet. 2018, 30, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Bruijn, L.I.; Wong, P.C.; Marszalek, J.R.; Vechio, J.D.; Lee, M.K.; Xu, X.-S.; Borchelt, D.R.; Sisodia, S.S.; Price, D.L. Mechanisms of Selective Motor Neuron Death in Transgenic Mouse Models of Motor Neuron Disease. Neurology 1996, 47, 54S–62S. [Google Scholar] [CrossRef] [PubMed]
- King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.K.; Vickers, J.C. Excitotoxicity in ALS: Overstimulation, or Overreaction? Exp. Neurol. 2016, 275 Pt 1, 162–171. [Google Scholar] [CrossRef]
- Morrison, B.M.; Morrison, J.H. Amyotrophic Lateral Sclerosis Associated with Mutations in Superoxide Dismutase: A Putative Mechanism of Degeneration; Elsevier: Amsterdam, The Netherlands, 1999; Volume 29. [Google Scholar]
- Tan, W.; Pasinelli, P.; Trotti, D. Role of Mitochondria in Mutant SOD1 Linked Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1295–1301. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The Role of Excitotoxicity in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 1068–1082. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Akkari, P.A.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Trojsi, F.; Lattante, S.; Ticozzi, N.; Mejzini, R.; Akkari, P.A.; et al. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as Determinants of Disease Progression in Inherited Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.; Garcia, M.L.; McAlonis-Downes, M.; Mikse, O.R.; Cleveland, D.W.; Goldstein, L.S.B. Mutant SOD1 in Cell Types Other than Motor Neurons and Oligodendrocytes Accelerates Onset of Disease in ALS Mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar] [CrossRef]
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.W.; Ostrow, L.W.; Rothstein, J.D.; Bergles, D.E. Degeneration and Impaired Regeneration of Gray Matter Oligodendrocytes in Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2013, 16, 571–579. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y.; Ryu, H. Astrocytes and Microglia as Non-Cell Autonomous Players in the Pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar] [CrossRef]
- Chen, H.; Kankel, M.W.; Su, S.C.; Han, S.W.S.; Ofengeim, D. Exploring the Genetics and Non-Cell Autonomous Mechanisms Underlying ALS/FTLD. Cell Death Differ. 2018, 25, 646–660. [Google Scholar] [CrossRef] [PubMed]
- Van Harten, A.C.M.; Phatnani, H.; Przedborski, S. Non-Cell-Autonomous Pathogenic Mechanisms in Amyotrophic Lateral Sclerosis. Trends Neurosci. 2021, 44, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W.; Llieva, H. Non-Cell Autonomous Toxicity in Neurodegenerative Disorders: ALS and Beyond Non-Cell Autonomous Toxicity in Neurodegenerativa Disorders: ALS and Beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.W.; et al. Oligodendroglia Metabolically Support Axons and Contribute to Neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Pramatarova, A.; Laganière, J.; Roussel, J.; Brisebois, K.; Rouleau, G.A. Neuron-Specific Expression of Mutant Superoxide Dismutase 1 in Transgenic Mice Does Not Lead to Motor Impairment. J. Neurosci. 2001, 21, 3369–3374. [Google Scholar] [CrossRef] [PubMed]
- Lino, M.M.; Schneider, C.; Caroni, P. Accumulation of SOD1 Mutants in Postnatal Motoneurons Does Not Cause Motoneuron Pathology or Motoneuron Disease. J. Neurosci. 2002, 22, 4825–4832. [Google Scholar] [CrossRef]
- Boillée, S.; Velde, V.C.; Cleveland, D.W.W. ALS: A Disease of Motor Neurons and Their Nonneuronal Neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef]
- Benarroch, E. What Is the Role of Oligodendrocytes in Amyotrophic Lateral Sclerosis? Neurology 2021, 97, 776–779. [Google Scholar] [CrossRef]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillée, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-Type Nonneuronal Cells Extend Survival of SOD1 Mutant Motor Neurons in ALS Mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef]
- Wang, L.; Gutmann, D.H.; Roos, R.P. Astrocyte Loss of Mutant SOD1 Delays ALS Disease Onset and Progression in G85R Transgenic Mice. Hum. Mol. Genet. 2011, 20, 286–293. [Google Scholar] [CrossRef]
- Lepore, A.C.; Rauck, B.; Dejea, C.; Pardo, A.C.; Rao, M.S.; Rothstein, J.D.; Maragakis, N.J. Focal Transplantation-Based Astrocyte Replacement Is Neuroprotective in a Model of Motor Neuron Disease. Nat. Neurosci. 2008, 11, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Papadeas, S.T.; Kraig, S.E.; O’banion, C.; Lepore, A.C.; Maragakis, N.J. Astrocytes Carrying the Superoxide Dismutase 1 (SOD1 G93A) Mutation Induce Wild-Type Motor Neuron Degeneration in Vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 17803–17808. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes Expressing ALS-Linked Mutated SOD1 Release Factors Selectively Toxic to Motor Neurons. Nat Neurosci 2007, 10, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from Familial and Sporadic ALS Patients Are Toxic to Motor Neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef]
- Rojas, F.; Cortes, N.; Abarzua, S.; Dyrda, A.; van Zundert, B. Astrocytes Expressing Mutant SOD1 and TDP43 Trigger Motoneuron Death That Is Mediated via Sodium Channels and Nitroxidative Stress. Front. Cell. Neurosci. 2014, 8, 24. [Google Scholar] [CrossRef]
- Meyer, K.; Ferraiuolo, L.; Miranda, C.J.; Likhite, S.; McElroy, S.; Renusch, S.; Ditsworth, D.; Lagier-Tourenne, C.; Smith, R.A.; Ravits, J.; et al. Direct Conversion of Patient Fibroblasts Demonstrates Non-Cell Autonomous Toxicity of Astrocytes to Motor Neurons in Familial and Sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 829–832. [Google Scholar] [CrossRef]
- Hall, C.E.; Yao, Z.; Choi, M.; Tyzack, G.E.; Serio, A.; Luisier, R.; Harley, J.; Preza, E.; Arber, C.; Crisp, S.J.; et al. Progressive Motor Neuron Pathology and the Role of Astrocytes in a Human Stem Cell Model of VCP-Related ALS. Cell Rep. 2017, 19, 1739–1749. [Google Scholar] [CrossRef]
- Zhao, C.; Devlin, A.; Chouhan, A.K.; Selvaraj, B.T.; Stavrou, M.; Burr, K.; Brivio, V.; He, X.; Mehta, A.R.; Story, D.; et al. Mutant C9orf72 Human IPSC-derived Astrocytes Cause Non-cell Autonomous Motor Neuron Pathophysiology. Glia 2020, 68, 1046–1064. [Google Scholar] [CrossRef]
- Stoklund Dittlau, K.; Van Den Bosch, L. Why Should We Care about Astrocytes in a Motor Neuron Disease? Front. Mol. Med. 2023, 3, 1047540. [Google Scholar] [CrossRef]
- Valori, C.F.; Sulmona, C.; Brambilla, L.; Rossi, D. Astrocytes: Dissecting Their Diverse Roles in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Cells 2023, 12, 1450. [Google Scholar] [CrossRef]
- Pehar, M.; Harlan, B.A.; Killoy, K.M.; Vargas, M.R. Role and Therapeutic Potential of Astrocytes in Amyotrophic Lateral Sclerosis. Curr. Pharm. Des. 2017, 23, 5010. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Zhao, W.; Liao, B.; Kano, O.; Wang, J.; Huang, A.; Appel, S.H.; Henkel, J.S. Neuroinflammation Modulates Distinct Regional and Temporal Clinical Responses in ALS Mice. Brain Behav. Immun. 2011, 25, 1025–1035. [Google Scholar] [CrossRef]
- Volonté, C.; Amadio, S.; Fabbrizio, P.; Apolloni, S. Functional Microglia Neurotransmitters in Amyotrophic Lateral Sclerosis. Semin. Cell. Dev. Biol. 2019, 94, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia Induce Motor Neuron Death via the Classical NF-κB Pathway in Amyotrophic Lateral Sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia States and Nomenclature: A Field at Its Crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Maniatis, S.; Äijö, T.; Vickovic, S.; Braine, C.; Kang, K.; Mollbrink, A.; Fagegaltier, D.; Andrusivová, Ž.; Saarenpää, S.; Saiz-Castro, G.; et al. Spatiotemporal Dynamics of Molecular Pathology in Amyotrophic Lateral Sclerosis. Science 2019, 364, 89–93. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic Supporters of Neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef]
- Sun, S.; Sun, Y.; Ling, S.-C.; Ferraiuolo, L.; McAlonis-Downes, M.; Zou, Y.; Drenner, K.; Wang, Y.; Ditsworth, D.; Tokunaga, S.; et al. Translational Profiling Identifies a Cascade of Damage Initiated in Motor Neurons and Spreading to Glia in Mutant SOD1-Mediated ALS. Proc. Natl. Acad. Sci. USA 2015, 112, E6993–E7002. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Meyer, K.; Sherwood, T.W.; Vick, J.; Likhite, S.; Frakes, A.; Miranda, C.J.; Braun, L.; Heath, P.R.; Pineda, R.; et al. Oligodendrocytes Contribute to Motor Neuron Death in ALS via SOD1-Dependent Mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, E6496–E6505. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Bento-Abreu, A.; Nonneman, A.; Haeck, W.; Staats, K.; Geelen, V.; Hersmus, N.; Küsters, B.; Van Den Bosch, L.; Van Damme, P.; et al. Oligodendrocyte Dysfunction in the Pathogenesis of Amyotrophic Lateral Sclerosis. Brain 2013, 136, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Golubczyk, D.; Malysz-Cymborska, I.; Kalkowski, L.; Janowski, M.; Coates, J.R.; Wojtkiewicz, J.; Maksymowicz, W.; Walczak, P. The Role of Glia in Canine Degenerative Myelopathy: Relevance to Human Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2019, 56, 5740–5748. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chung, A.; Na, J.E.; Lee, S.J.; Jeong, S.H.; Kim, E.; Sun, W.; Rhyu, I.J.; Park, H. Myelin Degeneration Induced by Mutant Superoxide Dismutase 1 Accumulation Promotes Amyotrophic Lateral Sclerosis. Glia 2019, 67, 23669. [Google Scholar] [CrossRef]
- Leung, G.; Sun, W.; Brookes, S.; Smith, D.; Shi, R. Potassium Channel Blocker, 4-Aminopyridine-3-Methanol, Restores Axonal Conduction in Spinal Cord of an Animal Model of Multiple Sclerosis. Exp. Neurol. 2011, 227, 232–235. [Google Scholar] [CrossRef][Green Version]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef]
- Tan, C.X.; Burrus Lane, C.J.; Eroglu, C. Role of Astrocytes in Synapse Formation and Maturation. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2021; pp. 371–407. [Google Scholar]
- Suga, M.; Kondo, T.; Inoue, H. Modeling Neurological Disorders with Human Pluripotent Stem Cell-Derived Astrocytes. Int. J. Mol. Sci. 2019, 20, 3862. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Zorec, R. Astroglial Signalling in Health and Disease. Neurosci. Lett. 2019, 689, 1–4. [Google Scholar] [CrossRef]
- Escartin, C.; Guillemaud, O.; Carrillo-de Sauvage, M.-A. Questions and (Some) Answers on Reactive Astrocytes. Glia 2019, 67, 2221–2247. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional Roles of Reactive Astrocytes in Neuroinflammation and Neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic Analysis of Reactive Astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-Y.; Huo, J. A1/A2 Astrocytes in Central Nervous System Injuries and Diseases: Angels or Devils? Neurochem. Int. 2021, 148, 105080. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Castro, B.; Gangwani, M.R.; Yu, X.; Coppola, G.; Khakh, B.S. Astrocyte Molecular Signatures in Huntington’s Disease. Sci. Transl. Med. 2019, 11, eaaw8546. [Google Scholar] [CrossRef] [PubMed]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-Associated Reactive Astrocytes Aid the Evolution of Immunosuppressive Environment in Glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Li, Z.; Noori, A.; Hyman, B.T.; Serrano-Pozo, A. Meta-Analysis of Mouse Transcriptomic Studies Supports a Context-Dependent Astrocyte Reaction in Acute CNS Injury versus Neurodegeneration. J. Neuroinflamm. 2020, 17, 227. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Nguyen, N.T.P.; Milanese, M.; Bonanno, G. Insights into Human-Induced Pluripotent Stem Cell-Derived Astrocytes in Neurodegenerative Disorders. Biomolecules 2022, 12, 344. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes Are Active Players in Cerebral Innate Immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Nardo, G.; Trolese, M.C.; Verderio, M.; Mariani, A.; De Paola, M.; Riva, N.; Dina, G.; Panini, N.; Erba, E.; Quattrini, A.; et al. Counteracting Roles of MHCI and CD8+ T Cells in the Peripheral and Central Nervous System of ALS SOD1 G93A Mice. Mol. Neurodegener. 2018, 13, 42. [Google Scholar] [CrossRef]
- Rahimifard, M.; Maqbool, F.; Moeini-Nodeh, S.; Niaz, K.; Abdollahi, M.; Braidy, N.; Nabavi, S.M.; Nabavi, S.F. Targeting the TLR4 Signaling Pathway by Polyphenols: A Novel Therapeutic Strategy for Neuroinflammation. Ageing Res. Rev. 2017, 36, 11–19. [Google Scholar] [CrossRef]
- Philips, T.; Robberecht, W. Neuroinflammation in Amyotrophic Lateral Sclerosis: Role of Glial Activation in Motor Neuron Disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Rupareliya, V.P.; Singh, A.A.; Butt, A.M.; Hariharan, A.; Kumar, H. The “Molecular Soldiers” of the CNS: Astrocytes, a Comprehensive Review on Their Roles and Molecular Signatures. Eur. J. Pharmacol. 2023, 959, 176048. [Google Scholar] [CrossRef] [PubMed]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Tripathi, P.; Rodriguez-Muela, N.; Klim, J.R.; de Boer, A.S.; Agrawal, S.; Sandoe, J.; Lopes, C.S.; Ogliari, K.S.; Williams, L.A.; Shear, M.; et al. Reactive Astrocytes Promote ALS-like Degeneration and Intracellular Protein Aggregation in Human Motor Neurons by Disrupting Autophagy through TGF-β1. Stem Cell Rep. 2017, 9, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, F.; Nyberg, S.; Giunti, D.; Torazza, C.; Parodi, B.; Bonifacino, T.; Usai, C.; Kerlero de Rosbo, N.; Milanese, M.; Uccelli, A.; et al. Micro-RNAs Shuttled by Extracellular Vesicles Secreted from Mesenchymal Stem Cells Dampen Astrocyte Pathological Activation and Support Neuroprotection in In-Vitro Models of ALS. Cells 2022, 11, 3923. [Google Scholar] [CrossRef]
- Torazza, C.; Provenzano, F.; Gallia, E.; Cerminara, M.; Balbi, M.; Bonifacino, T.; Tessitore, S.; Ravera, S.; Usai, C.; Musante, I.; et al. Genetic Downregulation of the Metabotropic Glutamate Receptor Type 5 Dampens the Reactive and Neurotoxic Phenotype of Adult ALS Astrocytes. Cells 2023, 12, 1952. [Google Scholar] [CrossRef]
- Houi, K.; Kobayashi, T.; Kato, S.; Mochio, S.; Inoue, K. Increased Plasma TGF- β 1 in Patients with Amyotrophic Lateral Sclerosis. Acta Neurol. Scand. 2002, 106, 299–301. [Google Scholar] [CrossRef]
- Endo, F.; Komine, O.; Fujimori-Tonou, N.; Katsuno, M.; Jin, S.; Watanabe, S.; Sobue, G.; Dezawa, M.; Wyss-Coray, T.; Yamanaka, K. Astrocyte-Derived TGF-β1 Accelerates Disease Progression in ALS Mice by Interfering with the Neuroprotective Functions of Microglia and T Cells. Cell Rep. 2015, 11, 592–604. [Google Scholar] [CrossRef]
- Tateishi, T.; Yamasaki, R.; Tanaka, M.; Matsushita, T.; Kikuchi, H.; Isobe, N.; Ohyagi, Y.; Kira, J. CSF Chemokine Alterations Related to the Clinical Course of Amyotrophic Lateral Sclerosis. J. Neuroimmunol. 2010, 222, 76–81. [Google Scholar] [CrossRef]
- Poloni, M.; Facchetti, D.; Mai, R.; Micheli, A.; Agnoletti, L.; Francolini, G.; Mora, G.; Camana, C.; Mazzini, L.; Bachetti, T. Circulating Levels of Tumour Necrosis Factor-α and Its Soluble Receptors Are Increased in the Blood of Patients with Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2000, 287, 211–214. [Google Scholar] [CrossRef]
- Olesen, M.N.; Wuolikainen, A.; Nilsson, A.C.; Wirenfeldt, M.; Forsberg, K.; Madsen, J.S.; Lillevang, S.T.; Brandslund, I.; Andersen, P.M.; Asgari, N. Inflammatory Profiles Relate to Survival in Subtypes of Amyotrophic Lateral Sclerosis. Neurol. Neuroimmunol. Neuroinflammation 2020, 7, e697. [Google Scholar] [CrossRef]
- Yoshihara, T.; Ishigaki, S.; Yamamoto, M.; Liang, Y.; Niwa, I.; Takeuchi, H.; Doyu, M.; Sobue, G. Differential Expression of Inflammation- and Apoptosis-Related Genes in Spinal Cords of a Mutant SOD1 Transgenic Mouse Model of Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 2002, 80, 158–167. [Google Scholar] [CrossRef]
- Brambilla, L.; Guidotti, G.; Martorana, F.; Iyer, A.M.; Aronica, E.; Valori, C.F.; Rossi, D. Disruption of the Astrocytic TNFR1-GDNF Axis Accelerates Motor Neuron Degeneration and Disease Progression in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2016, 25, ddw161. [Google Scholar] [CrossRef][Green Version]
- Tortarolo, M.; Vallarola, A.; Lidonnici, D.; Battaglia, E.; Gensano, F.; Spaltro, G.; Fiordaliso, F.; Corbelli, A.; Garetto, S.; Martini, E.; et al. Lack of TNF-Alpha Receptor Type 2 Protects Motor Neurons in a Cellular Model of Amyotrophic Lateral Sclerosis and in Mutant SOD1 Mice but Does Not Affect Disease Progression. J. Neurochem. 2015, 135, 109–124. [Google Scholar] [CrossRef]
- Dewil, M.; dela Cruz, V.F.; Van Den Bosch, L.; Robberecht, W. Inhibition of P38 Mitogen Activated Protein Kinase Activation and Mutant SOD1G93A-Induced Motor Neuron Death. Neurobiol. Dis. 2007, 26, 332–341. [Google Scholar] [CrossRef]
- Kia, A.; McAvoy, K.; Krishnamurthy, K.; Trotti, D.; Pasinelli, P. Astrocytes Expressing ALS-Linked Mutant FUS Induce Motor Neuron Death through Release of Tumor Necrosis Factor-Alpha. Glia 2018, 66, 1016–1033. [Google Scholar] [CrossRef]
- Ajmone-Cat, M.A.; Onori, A.; Toselli, C.; Stronati, E.; Morlando, M.; Bozzoni, I.; Monni, E.; Kokaia, Z.; Lupo, G.; Minghetti, L.; et al. Increased FUS Levels in Astrocytes Leads to Astrocyte and Microglia Activation and Neuronal Death. Sci. Rep. 2019, 9, 4572. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Erratum: Targeting the NLRP3 Inflammasome in Inflammatory Diseases. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef] [PubMed]
- Maran, J.J.; Adesina, M.M.; Green, C.R.; Kwakowsky, A.; Mugisho, O.O. The Central Role of the NLRP3 Inflammasome Pathway in the Pathogenesis of Age-Related Diseases in the Eye and the Brain. Ageing Res. Rev. 2023, 88, 101954. [Google Scholar] [CrossRef]
- Jose, S.; Groves, N.J.; Roper, K.E.; Gordon, R. Mechanisms of NLRP3 Activation and Pathology during Neurodegeneration. Int. J. Biochem. Cell Biol. 2022, 151, 106273. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, A.; Giacoppo, S.; Bramanti, P.; Mazzon, E. NLRP3 Inflammasome Activation in a Transgenic Amyotrophic Lateral Sclerosis Model. Inflammation 2018, 41, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Johann, S.; Heitzer, M.; Kanagaratnam, M.; Goswami, A.; Rizo, T.; Weis, J.; Troost, D.; Beyer, C. NLRP3 Inflammasome Is Expressed by Astrocytes in the SOD1 Mouse Model of ALS and in Human Sporadic ALS Patients. Glia 2015, 63, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Grottelli, S.; Costanzi, E.; Scarpelli, P.; Pigna, E.; Morozzi, G.; Mezzasoma, L.; Peirce, M.J.; Moresi, V.; Adamo, S.; et al. Peroxynitrite Activates the NLRP3 Inflammasome Cascade in SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 2350–2361. [Google Scholar] [CrossRef]
- Serrano, A.; Donno, C.; Giannetti, S.; Perić, M.; Andjus, P.; D’Ambrosi, N.; Michetti, F. The Astrocytic S100B Protein with Its Receptor RAGE Is Aberrantly Expressed in SOD1G93A Models, and Its Inhibition Decreases the Expression of Proinflammatory Genes. Mediat. Inflamm. 2017, 2017, 1626204. [Google Scholar] [CrossRef]
- Kikuchi, S.; Shinpo, K.; Ogata, A.; Tsuji, S.; Takeuchi, M.; Makita, Z.; Tashiro, K. Detection of Nε-(Carboxymethyl)Lysine (CML) and Non-CML Advanced Glycation End-Products in the Anterior Horn of Amyotrophic Lateral Sclerosis Spinal Cord. Amyotroph. Lateral Scler. 2002, 3, 63–68. [Google Scholar] [CrossRef]
- Wang, Z.; Bai, Z.; Qin, X.; Cheng, Y. Aberrations in Oxidative Stress Markers in Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Oxid. Med. Cell. Longev. 2019, 2019, 1712323. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive Species and Antioxidants. Redox Biology Is a Fundamental Theme of Aerobic Life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef]
- Yu, Y.-C.; Kuo, C.-L.; Cheng, W.-L.; Liu, C.-S.; Hsieh, M. Decreased Antioxidant Enzyme Activity and Increased Mitochondrial DNA Damage in Cellular Models of Machado-Joseph Disease. J. Neurosci. Res. 2009, 87, 1884–1891. [Google Scholar] [CrossRef]
- Halliwell, B. Biochemistry of Oxidative Stress. Biochem. Soc. Trans 2007, 35, 1147–1150. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, D.; Xiao, H. Methods of Cellular Senescence Induction Using Oxidative Stress. Methods Mol. Biol. 2013, 1048, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Jaronen, M.; Goldsteins, G.; Koistinaho, J. ER Stress and Unfolded Protein Response in Amyotrophic Lateral Sclerosis-a Controversial Role of Protein Disulphide Isomerase. Front. Cell. Neurosci. 2014, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.H.W.; Hartopp, N.; Welsh, N.J.; Mueller, S.; Glennon, E.B.; Mórotz, G.M.; Annibali, A.; Gomez-Suaga, P.; Stoica, R.; Paillusson, S.; et al. Disruption of ER−mitochondria Signalling in Fronto-Temporal Dementia and Related Amyotrophic Lateral Sclerosis. Cell Death Dis. 2018, 9, 327. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 Inhibition Reverses Axonal Transport Defects in Motor Neurons Derived from FUS-ALS Patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The Role of Astrocytes in Oxidative Stress of Central Nervous System: A Mixed Blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef]
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE Signaling Pathway: Key Mediator in Oxidative Stress and Potential Therapeutic Target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef]
- Vargas, M.R.; Pehar, M.; Cassina, P.; Beckman, J.S.; Barbeito, L. Increased Glutathione Biosynthesis by Nrf2 Activation in Astrocytes Prevents P75NTR-Dependent Motor Neuron Apoptosis. J. Neurochem. 2006, 97, 687–696. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 Activation in Astrocytes Protects against Neurodegeneration in Mouse Models of Familial Amyotrophic Lateral Sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef]
- Hoang, T.T.; Johnson, D.A.; Raines, R.T.; Johnson, J.A. Angiogenin Activates the Astrocytic Nrf2/Antioxidant-Response Element Pathway and Thereby Protects Murine Neurons from Oxidative Stress. J. Biol. Chem. 2019, 294, 15095–15103. [Google Scholar] [CrossRef]
- Greenway, M.J.; Andersen, P.M.; Russ, C.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG Mutations Segregate with Familial and “sporadic” Amyotrophic Lateral Sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef]
- Gellera, C.; Colombrita, C.; Ticozzi, N.; Castellotti, B.; Bragato, C.; Ratti, A.; Taroni, F.; Silani, V. Identification of New ANG Gene Mutations in a Large Cohort of Italian Patients with Amyotrophic Lateral Sclerosis. Neurogenetics 2008, 9, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective Effect of Nrf2/ARE Activators, CDDO Ethylamide and CDDO Trifluoroethylamide, in a Mouse Model of Amyotrophic Lateral Sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef]
- Kwak, M.-K.; Kensler, T.W. Targeting NRF2 Signaling for Cancer Chemoprevention. Toxicol. Appl. Pharmacol. 2010, 244, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human IPSC-Derived Astrocytes from ALS Patients with Mutated C9ORF72 Show Increased Oxidative Stress and Neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Rojas, F.; Gonzalez, D.; Cortes, N.; Ampuero, E.; Hernández, D.E.; Fritz, E.; Abarzua, S.; Martinez, A.; Elorza, A.A.; Alvarez, A.; et al. Reactive Oxygen Species Trigger Motoneuron Death in Non-Cell-Autonomous Models of ALS through Activation of c-Abl Signaling. Front. Cell. Neurosci. 2015, 9, 203. [Google Scholar] [CrossRef]
- Ravera, S.; Bonifacino, T.; Bartolucci, M.; Milanese, M.; Gallia, E.; Provenzano, F.; Cortese, K.; Panfoli, I.; Bonanno, G. Characterization of the Mitochondrial Aerobic Metabolism in the Pre- and Perisynaptic Districts of the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 9220–9233. [Google Scholar] [CrossRef]
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; de León, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial Dysfunction in SOD1G93A-Bearing Astrocytes Promotes Motor Neuron Degeneration: Prevention by Mitochondrial-Targeted Antioxidants. J. Neurosci. 2008, 28, 4115–4122. [Google Scholar] [CrossRef]
- Karvandi, M.S.; Sheikhzadeh Hesari, F.; Aref, A.R.; Mahdavi, M. The Neuroprotective Effects of Targeting Key Factors of Neuronal Cell Death in Neurodegenerative Diseases: The Role of ER Stress, Oxidative Stress, and Neuroinflammation. Front. Cell. Neurosci. 2023, 17, 1105247. [Google Scholar] [CrossRef]
- Carrera-Juliá, S.; Moreno, M.L.; Barrios, C.; de la Rubia Ortí, J.E.; Drehmer, E. Antioxidant Alternatives in the Treatment of Amyotrophic Lateral Sclerosis: A Comprehensive Review. Front. Physiol. 2020, 11, 63. [Google Scholar] [CrossRef]
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Bolatto, C.; Trías, E.; Gandelman, M.; Radi, R.; Barbeito, L.; Cassina, P. Modulation of Astrocytic Mitochondrial Function by Dichloroacetate Improves Survival and Motor Performance in Inherited Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e34776. [Google Scholar] [CrossRef]
- Martínez-Palma, L.; Miquel, E.; Lagos-Rodríguez, V.; Barbeito, L.; Cassina, A.; Cassina, P. Mitochondrial Modulation by Dichloroacetate Reduces Toxicity of Aberrant Glial Cells and Gliosis in the SOD1G93A Rat Model of Amyotrophic Lateral Sclerosis. Neurotherapeutics 2019, 16, 203–215. [Google Scholar] [CrossRef]
- Yoshino, H. Edaravone for the Treatment of Amyotrophic Lateral Sclerosis. Expert Rev. Neurother. 2019, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Glutamate Uptake into Astrocytes Stimulates Aerobic Glycolysis: A Mechanism Coupling Neuronal Activity to Glucose Utilization (Glutamate Rnsporter/Na+/K+-ATPase/2-Deoxyglucose/Positron-Embsson Tomography/Magnetic Resonance Imaging). Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Bouzier-Sore, A.K.; Aubert, A.; Serres, S.; Merle, M.; Costalat, R.; Magistretti, P.J. Activity-Dependent Regulation of Energy Metabolism by Astrocytes: An Update. Glia 2007, 55, 1251–1262. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular Pathways of Motor Neuron Injury in Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Madji Hounoum, B.; Mavel, S.; Coque, E.; Patin, F.; Vourc’h, P.; Marouillat, S.; Nadal-Desbarats, L.; Emond, P.; Corcia, P.; Andres, C.R.; et al. Wildtype Motoneurons, ALS-Linked SOD1 Mutation and Glutamate Profoundly Modify Astrocyte Metabolism and Lactate Shuttling. Glia 2017, 65, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Veyrat-Durebex, C.; Corcia, P.; Piver, E.; Devos, D.; Dangoumau, A.; Gouel, F.; Vourc’h, P.; Emond, P.; Laumonnier, F.; Nadal-Desbarats, L.; et al. Disruption of TCA Cycle and Glutamate Metabolism Identified by Metabolomics in an in Vitro Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2016, 53, 6910–6924. [Google Scholar] [CrossRef]
- Ravera, S.; Torazza, C.; Bonifacino, T.; Provenzano, F.; Rebosio, C.; Milanese, M.; Usai, C.; Panfoli, I.; Bonanno, G. Altered Glucose Catabolism in the Presynaptic and Perisynaptic Compartments of SOD1G93A Mouse Spinal Cord and Motor Cortex Indicates That Mitochondria Are the Site of Bioenergetic Imbalance in ALS. J. Neurochem. 2019, 151, 336–350. [Google Scholar] [CrossRef]
- Velebit, J.; Horvat, A.; Smolič, T.; Prpar Mihevc, S.; Rogelj, B.; Zorec, R.; Vardjan, N. Astrocytes with TDP-43 Inclusions Exhibit Reduced Noradrenergic CAMP and Ca2+ Signaling and Dysregulated Cell Metabolism. Sci. Rep. 2020, 10, 6003. [Google Scholar] [CrossRef]
- Yoshida, Y.; Une, F.; Utatsu, Y.; Nomoto, M.; Furukawa, Y.; Maruyama, Y.; Machigashira, N.; Matsuzaki, T.; Osame, M. Adenosine and Neopterin Levels in Cerebrospinal Fluid of Patients with Neurological Disorders. Intern. Med. 1999, 38, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, F.; Corciulo, C.; Targa, M.; Casetta, I.; Gentile, M.; Granieri, E.; Borea, P.A.; Popoli, P.; Varani, K. A2A Adenosine Receptors Are Up-Regulated in Lymphocytes from Amyotrophic Lateral Sclerosis Patients. Amyotroph Lateral Scler Front. Degener 2013, 14, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.K.; Higashimori, H.; Tolman, M.; Yang, Y. Suppression of Adenosine 2a Receptor (A2aR)-Mediated Adenosine Signaling Improves Disease Phenotypes in a Mouse Model of Amyotrophic Lateral Sclerosis. Exp. Neurol. 2015, 267, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Nomura, T.; Aihara, H.; Nishizaki, T. Adenosine Enhances Glial Glutamate Efflux via A2a Adenosine Receptors. Life Sci. 2001, 68, 1343–1350. [Google Scholar] [CrossRef]
- Boison, D.; Chen, J.-F.; Fredholm, B.B. Adenosine Signaling and Function in Glial Cells. Cell Death Differ. 2010, 17, 1071–1082. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Castelli, L.M.; Francis, L.; Woof, R.; Siskos, A.P.; Kouloura, E.; Gray, E.; Thompson, A.G.; Talbot, K.; et al. Astrocyte Adenosine Deaminase Loss Increases Motor Neuron Toxicity in Amyotrophic Lateral Sclerosis. Brain 2019, 142, 586–605. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Woof, R.; Francis, L.; Gatto, N.; Shaw, A.C.; Myszczynska, M.; Hemingway, J.; Coldicott, I.; Willcock, A.; et al. C9orf72 Expansion within Astrocytes Reduces Metabolic Flexibility in Amyotrophic Lateral Sclerosis. Brain 2019, 142, 3771–3790. [Google Scholar] [CrossRef]
- Blasco, H.; Veyrat-Durebex, C.; Bocca, C.; Patin, F.; Vourc’h, P.; Kouassi Nzoughet, J.; Lenaers, G.; Andres, C.R.; Simard, G.; Corcia, P.; et al. Lipidomics Reveals Cerebrospinal-Fluid Signatures of ALS. Sci. Rep. 2017, 7, 17652. [Google Scholar] [CrossRef]
- Trostchansky, A. Overview of Lipid Biomarkers in Amyotrophic Lateral Sclerosis (ALS). In The Role of Bioactive Lipids in Cancer, Inflammation and Related Diseases; Springer: Berlin/Heidelberg, Germany, 2019; pp. 233–241. [Google Scholar]
- Chaves-Filho, A.B.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.G.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in Lipid Metabolism of Spinal Cord Linked to Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 11642. [Google Scholar] [CrossRef]
- Jiménez-Riani, M.; Díaz-Amarilla, P.; Isasi, E.; Casanova, G.; Barbeito, L.; Olivera-Bravo, S. Ultrastructural Features of Aberrant Glial Cells Isolated from the Spinal Cord of Paralytic Rats Expressing the Amyotrophic Lateral Sclerosis-Linked SOD1G93A Mutation. Cell Tissue Res. 2017, 370, 391–401. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial Lipid Droplets and ROS Induced by Mitochondrial Defects Promote Neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletić-Savatić, M.; Bellen, H.J. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017, 26, 719–737. [Google Scholar] [CrossRef]
- Gomes, C.; Cunha, C.; Nascimento, F.; Ribeiro, J.A.; Vaz, A.R.; Brites, D. Cortical Neurotoxic Astrocytes with Early ALS Pathology and MiR-146a Deficit Replicate Gliosis Markers of Symptomatic SOD1G93A Mouse Model. Mol. Neurobiol. 2019, 56, 2137–2158. [Google Scholar] [CrossRef]
- Iyer, A.; Zurolo, E.; Prabowo, A.; Fluiter, K.; Spliet, W.G.M.; van Rijen, P.C.; Gorter, J.A.; Aronica, E. MicroRNA-146a: A Key Regulator of Astrocyte-Mediated Inflammatory Response. PLoS ONE 2012, 7, e44789. [Google Scholar] [CrossRef]
- Kang, J.; Li, Z.; Zhi, Z.; Wang, S.; Xu, G. MiR-21 Derived from the Exosomes of MSCs Regulates the Death and Differentiation of Neurons in Patients with Spinal Cord Injury. Gene Ther. 2019, 26, 491–503. [Google Scholar] [CrossRef]
- Le, M.T.N.; Xie, H.; Zhou, B.; Chia, P.H.; Rizk, P.; Um, M.; Udolph, G.; Yang, H.; Lim, B.; Lodish, H.F. MicroRNA-125b Promotes Neuronal Differentiation in Human Cells by Repressing Multiple Targets. Mol. Cell. Biol. 2009, 29, 5290–5305. [Google Scholar] [CrossRef]
- Varcianna, A.; Myszczynska, M.A.; Castelli, L.M.; O’Neill, B.; Kim, Y.; Talbot, J.; Nyberg, S.; Nyamali, I.; Heath, P.R.; Stopford, M.J.; et al. Micro-RNAs Secreted through Astrocyte-Derived Extracellular Vesicles Cause Neuronal Network Degeneration in C9orf72 ALS. EBioMedicine 2019, 40, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Saucier, D.; Wajnberg, G.; Roy, J.; Beauregard, A.-P.; Chacko, S.; Crapoulet, N.; Fournier, S.; Ghosh, A.; Lewis, S.M.; Marrero, A.; et al. Identification of a Circulating MiRNA Signature in Extracellular Vesicles Collected from Amyotrophic Lateral Sclerosis Patients. Brain Res. 2019, 1708, 100–108. [Google Scholar] [CrossRef]
- Katsu, M.; Hama, Y.; Utsumi, J.; Takashina, K.; Yasumatsu, H.; Mori, F.; Wakabayashi, K.; Shoji, M.; Sasaki, H. MicroRNA Expression Profiles of Neuron-Derived Extracellular Vesicles in Plasma from Patients with Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 708, 134176. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.M.; Fernando, S.M.; Grad, L.I.; Hill, A.F.; Turner, B.J.; Yerbury, J.J.; Cashman, N.R. Disease Mechanisms in ALS: Misfolded SOD1 Transferred Through Exosome-Dependent and Exosome-Independent Pathways. Cell. Mol. Neurobiol. 2016, 36, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Basso, M.; Pozzi, S.; Tortarolo, M.; Fiordaliso, F.; Bisighini, C.; Pasetto, L.; Spaltro, G.; Lidonnici, D.; Gensano, F.; Battaglia, E.; et al. Mutant Copper-Zinc Superoxide Dismutase (SOD1) Induces Protein Secretion Pathway Alterations and Exosome Release in Astrocytes: Implications for Disease Spreading and Motor Neuron Pathology in Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2013, 288, 15699–15711. [Google Scholar] [CrossRef]
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Greg Stacey, R.; Grad, L.I.; et al. CNS-Derived Extracellular Vesicles from Superoxide Dismutase 1 (SOD1) G93A ALS Mice Originate from Astrocytes and Neurons and Carry Misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome Secretion Is a Key Pathway for Clearance of Pathological TDP-43. Brain 2016, 139, 3187–3201. [Google Scholar] [CrossRef] [PubMed]
- Sproviero, D.; La Salvia, S.; Giannini, M.; Crippa, V.; Gagliardi, S.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Pansarasa, O.; Poletti, A.; et al. Pathological Proteins Are Transported by Extracellular Vesicles of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2018, 12, 487. [Google Scholar] [CrossRef]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Arenas, A.; Zhai, J.; Zhu, H.; Gal, J. Proteomic Analysis of FUS Interacting Proteins Provides Insights into FUS Function and Its Role in ALS. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Westergard, T.; Jensen, B.K.; Wen, X.; Cai, J.; Kropf, E.; Iacovitti, L.; Pasinelli, P.; Trotti, D. Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72 -ALS/FTD. Cell Rep. 2016, 17, 645–652. [Google Scholar] [CrossRef]
- Forsberg, K.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Glial Nuclear Aggregates of Superoxide Dismutase-1 Are Regularly Present in Patients with Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2011, 121, 623–634. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein Aggregation in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef]
- Strohm, L.; Behrends, C. Glia-Specific Autophagy Dysfunction in ALS. Semin. Cell Dev. Biol. 2020, 99, 172–182. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Quality Control in Neurons: Mitophagy and Other Selective Autophagy Mechanisms. J. Mol. Biol. 2020, 432, 240–260. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in Immunity and Inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.; Kim, S.U. Human Astrocytes: Secretome Profiles of Cytokines and Chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef] [PubMed]
- Didier, N.; Romero, I.A.; Créminon, C.; Wijkhuisen, A.; Grassi, J.; Mabondzo, A. Secretion of Interleukin-1β by Astrocytes Mediates Endothelin-1 and Tumour Necrosis Factor-α Effects on Human Brain Microvascular Endothelial Cell Permeability. J. Neurochem. 2004, 86, 246–254. [Google Scholar] [CrossRef]
- Pereira, G.J.S.; Antonioli, M.; Hirata, H.; Ureshino, R.P.; Nascimento, A.R.; Bincoletto, C.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Smaili, S.S. Glutamate Induces Autophagy via the Two-Pore Channels in Neural Cells. Oncotarget 2017, 8, 12730–12740. [Google Scholar] [CrossRef]
- Son, S.M.; Cha, M.-Y.; Choi, H.; Kang, S.; Choi, H.; Lee, M.-S.; Park, S.A.; Mook-Jung, I. Insulin-Degrading Enzyme Secretion from Astrocytes Is Mediated by an Autophagy-Based Unconventional Secretory Pathway in Alzheimer Disease. Autophagy 2016, 12, 784–800. [Google Scholar] [CrossRef]
- Son, S.M.; Kang, S.; Choi, H.; Mook-Jung, I. Statins Induce Insulin-Degrading Enzyme Secretion from Astrocytes via an Autophagy-Based Unconventional Secretory Pathway. Mol. Neurodegener. 2015, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Aguilera, M.O.; Colombo, M.I. ATP Is Released from Autophagic Vesicles to the Extracellular Space in a VAMP7-Dependent Manner. Autophagy 2012, 8, 1741–1756. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martins, I.; Ma, Y.; Kepp, O.; Galluzzi, L.; Kroemer, G. Autophagy-Dependent ATP Release from Dying Cells via Lysosomal Exocytosis. Autophagy 2013, 9, 1624–1625. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, G.; Zhou, W.; Song, A.; Xu, T.; Luo, Q.; Wang, W.; Gu, X.; Duan, S. Regulated ATP Release from Astrocytes through Lysosome Exocytosis. Nat. Cell Biol. 2007, 9, 945–953. [Google Scholar] [CrossRef]
- Martin, S.; Dudek-Peric, A.M.; Garg, A.D.; Roose, H.; Demirsoy, S.; Van Eygen, S.; Mertens, F.; Vangheluwe, P.; Vankelecom, H.; Agostinis, P. An Autophagy-Driven Pathway of ATP Secretion Supports the Aggressive Phenotype of BRAFV600E Inhibitor-Resistant Metastatic Melanoma Cells. Autophagy 2017, 13, 1512–1527. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-Linked SOD1 Mutant G85R Mediates Damage to Astrocytes and Promotes Rapidly Progressive Disease with SOD1-Containing Inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.H.; Parsadanian, A.S.; Andreeva, A.; Snider, W.D.; Elliott, J.L. Restricted Expression of G86R Cu/Zn Superoxide Dismutase in Astrocytes Results in Astrocytosis but Does Not Cause Motoneuron Degeneration. J. Neurosci. 2000, 20, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Motori, E.; Puyal, J.; Toni, N.; Ghanem, A.; Angeloni, C.; Malaguti, M.; Cantelli-Forti, G.; Berninger, B.; Conzelmann, K.-K.; Götz, M.; et al. Inflammation-Induced Alteration of Astrocyte Mitochondrial Dynamics Requires Autophagy for Mitochondrial Network Maintenance. Cell Metab. 2013, 18, 844–859. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, C.J.; Lenk, G.M.; Meisler, M.H. Defective Autophagy in Neurons and Astrocytes from Mice Deficient in PI(3,5)P2. Hum. Mol. Genet. 2009, 18, 4868–4878. [Google Scholar] [CrossRef]
- Di Malta, C.; Fryer, J.D.; Settembre, C.; Ballabio, A. Autophagy in Astrocytes. Autophagy 2012, 8, 1871–1872. [Google Scholar] [CrossRef]
- Melentijevic, I.; Toth, M.L.; Arnold, M.L.; Guasp, R.J.; Harinath, G.; Nguyen, K.C.; Taub, D.; Parker, J.A.; Neri, C.; Gabel, C.V.; et al. C. elegans Neurons Jettison Protein Aggregates and Mitochondria under Neurotoxic Stress. Nature 2017, 542, 367–371. [Google Scholar] [CrossRef]
- Davis, C.O.; Kim, K.-Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular Degradation of Axonal Mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef]
- Virgili, M.; Crochemore, C.; Peña-Altamira, E.; Contestabile, A. Regional and Temporal Alterations of ODC/Polyamine System during ALS-like Neurodegenerative Motor Syndrome in G93A Transgenic Mice. Neurochem. Int. 2006, 48, 201–207. [Google Scholar] [CrossRef]
- Ekegren, T.; Gomes-Trolin, C.; Nygren, I.; Askmark, H. Maintained Regulation of Polyamines in Spinal Cord from Patients with Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2004, 222, 49–53. [Google Scholar] [CrossRef]
- Cervelli, M.; Averna, M.; Vergani, L.; Pedrazzi, M.; Amato, S.; Fiorucci, C.; Rossi, M.N.; Maura, G.; Mariottini, P.; Cervetto, C.; et al. The Involvement of Polyamines Catabolism in the Crosstalk between Neurons and Astrocytes in Neurodegeneration. Biomedicines 2022, 10, 1756. [Google Scholar] [CrossRef]
- Bowie, D. Polyamine-Mediated Channel Block of Ionotropic Glutamate Receptors and Its Regulation by Auxiliary Proteins. J. Biol. Chem. 2018, 293, 18789–18802. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.G.; Lee, S. Polyamines and Potassium Channels: A 25-Year Romance. J. Biol. Chem. 2018, 293, 18779–18788. [Google Scholar] [CrossRef] [PubMed]
- Cervetto, C.; Averna, M.; Vergani, L.; Pedrazzi, M.; Amato, S.; Pelassa, S.; Giuliani, S.; Baldini, F.; Maura, G.; Mariottini, P.; et al. Reactive Astrocytosis in a Mouse Model of Chronic Polyamine Catabolism Activation. Biomolecules 2021, 11, 1274. [Google Scholar] [CrossRef] [PubMed]
- Skatchkov, S.N.; Bukauskas, F.F.; Benedikt, J.; Inyushin, M.; Kucheryavykh, Y.V. Intracellular Spermine Prevents Acid-Induced Uncoupling of Cx43 Gap Junction Channels. NeuroReport 2015, 26, 528–532. [Google Scholar] [CrossRef]
- Benedikt, J.; Inyushin, M.; Kucheryavykh, Y.V.; Rivera, Y.; Kucheryavykh, L.Y.; Nichols, C.G.; Eaton, M.J.; Skatchkov, S.N. Intracellular Polyamines Enhance Astrocytic Coupling. Neuroreport 2012, 23, 1021–1025. [Google Scholar] [CrossRef]
- Rossi, D. Astrocyte Physiopathology: At the Crossroads of Intercellular Networking, Inflammation and Cell Death. Prog. Neurobiol. 2015, 130, 86–120. [Google Scholar] [CrossRef]
- Ng, W.; Ng, S.-Y. Remodeling of Astrocyte Secretome in Amyotrophic Lateral Sclerosis: Uncovering Novel Targets to Combat Astrocyte-Mediated Toxicity. Transl. Neurodegener. 2022, 11, 54. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Higginbottom, A.; Heath, P.R.; Barber, S.; Greenald, D.; Kirby, J.; Shaw, P.J. Dysregulation of Astrocyte-Motoneuron Cross-Talk in Mutant Superoxide Dismutase 1-Related Amyotrophic Lateral Sclerosis. Brain 2011, 134, 2627–2641. [Google Scholar] [CrossRef]
- Tovar-y-Romo, L.B.; Ramírez-Jarquín, U.N.; Lazo-Gómez, R.; Tapia, R. Trophic Factors as Modulators of Motor Neuron Physiology and Survival: Implications for ALS Therapy. Front. Cell. Neurosci. 2014, 8, 61. [Google Scholar] [CrossRef]
- Thomsen, G.M.; Alkaslasi, M.; Vit, J.-P.; Lawless, G.; Godoy, M.; Gowing, G.; Shelest, O.; Svendsen, C.N. Systemic Injection of AAV9-GDNF Provides Modest Functional Improvements in the SOD1G93A ALS Rat but Has Adverse Side Effects. Gene Ther. 2017, 24, 245–252. [Google Scholar] [CrossRef]
- Thomsen, G.M.; Avalos, P.; Ma, A.A.; Alkaslasi, M.; Cho, N.; Wyss, L.; Vit, J.-P.; Godoy, M.; Suezaki, P.; Shelest, O.; et al. Transplantation of Neural Progenitor Cells Expressing Glial Cell Line-Derived Neurotrophic Factor into the Motor Cortex as a Strategy to Treat Amyotrophic Lateral Sclerosis. Stem Cells 2018, 36, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Milanese, M.; Zappettini, S.; Onofri, F.; Musazzi, L.; Tardito, D.; Bonifacino, T.; Messa, M.; Racagni, G.; Usai, C.; Benfenati, F.; et al. Abnormal Exocytotic Release of Glutamate in a Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurochem. 2011, 116, 1028–1042. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, T.; Musazzi, L.; Milanese, M.; Seguini, M.; Marte, A.; Gallia, E.; Cattaneo, L.; Onofri, F.; Popoli, M.; Bonanno, G. Altered Mechanisms Underlying the Abnormal Glutamate Release in Amyotrophic Lateral Sclerosis at a Pre-Symptomatic Stage of the Disease. Neurobiol. Dis. 2016, 95, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Milanese, M.; Giribaldi, F.; Melone, M.; Bonifacino, T.; Musante, I.; Carminati, E.; Rossi, P.I.A.; Vergani, L.; Voci, A.; Conti, F.; et al. Knocking down Metabotropic Glutamate Receptor 1 Improves Survival and Disease Progression in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2014, 64, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, T.; Cattaneo, L.; Gallia, E.; Puliti, A.; Melone, M.; Provenzano, F.; Bossi, S.; Musante, I.; Usai, C.; Conti, F.; et al. In-Vivo Effects of Knocking-down Metabotropic Glutamate Receptor 5 in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Neuropharmacology 2017, 123, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, T.; Rebosio, C.; Provenzano, F.; Torazza, C.; Balbi, M.; Milanese, M.; Raiteri, L.; Usai, C.; Fedele, E.; Bonanno, G. Enhanced Function and Overexpression of Metabotropic Glutamate Receptors 1 and 5 in the Spinal Cord of the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis during Disease Progression. Int. J. Mol. Sci. 2019, 20, 4552. [Google Scholar] [CrossRef]
- Bonifacino, T.; Provenzano, F.; Gallia, E.; Ravera, S.; Torazza, C.; Bossi, S.; Ferrando, S.; Puliti, A.; Van Den Bosch, L.; Bonanno, G.; et al. In-Vivo Genetic Ablation of Metabotropic Glutamate Receptor Type 5 Slows down Disease Progression in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2019, 129, 79–92. [Google Scholar] [CrossRef]
- Giribaldi, F.; Milanese, M.; Bonifacino, T.; Anna Rossi, P.I.; Di Prisco, S.; Pittaluga, A.; Tacchetti, C.; Puliti, A.; Usai, C.; Bonanno, G. Group I Metabotropic Glutamate Autoreceptors Induce Abnormal Glutamate Exocytosis in a Mouse Model of Amyotrophic Lateral Sclerosis. Neuropharmacology 2013, 66, 253–263. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective Loss of Glial Glutamate Transporter GLT-1 in Amyotrophic Lateral Sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- Trotti, D.; Aoki, M.; Pasinelli, P.; Berger, U.V.; Danbolt, N.C.; Brown, R.H.; Hediger, M.A. Amyotrophic Lateral Sclerosis-Linked Glutamate Transporter Mutant Has Impaired Glutamate Clearance Capacity. J. Biol. Chem. 2001, 276, 576–582. [Google Scholar] [CrossRef]
- Mohamed, L.A.; Markandaiah, S.S.; Bonanno, S.; Pasinelli, P.; Trotti, D. Excess Glutamate Secreted from Astrocytes Drives Upregulation of P-Glycoprotein in Endothelial Cells in Amyotrophic Lateral Sclerosis. Exp. Neurol. 2019, 316, 27–38. [Google Scholar] [CrossRef]
- Stigliani, S.; Zappettini, S.; Raiteri, L.; Passalacqua, M.; Melloni, E.; Venturi, C.; Tacchetti, C.; Diaspro, A.; Usai, C.; Bonanno, G. Glia Re-Sealed Particles Freshly Prepared from Adult Rat Brain Are Competent for Exocytotic Release of Glutamate. J. Neurochem. 2006, 96, 656–668. [Google Scholar] [CrossRef]
- Carney, K.E.; Milanese, M.; Van Nierop, P.; Li, K.W.; Oliet, S.H.R.; Smit, A.B.; Bonanno, G.; Verheijen, M.H.G. Proteomic Analysis of Gliosomes from Mouse Brain: Identification and Investigation of Glial Membrane Proteins. J. Proteome Res. 2014, 13, 5918–5927. [Google Scholar] [CrossRef] [PubMed]
- Milanese, M.; Zappettini, S.; Jacchetti, E.; Bonifacino, T.; Cervetto, C.; Usai, C.; Bonanno, G. In Vitro Activation of GAT1 Transporters Expressed in Spinal Cord Gliosomes Stimulates Glutamate Release That Is Abnormally Elevated in the SOD1/G93A(+) Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurochem. 2010, 113, 489–501. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases—What Is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Bristol, L.A.; Rothstein, J.D. Glutamate Transporter Gene Expression in Amyotrophic Lateral Sclerosis Motor Cortex. Ann. Neurol. 1996, 39, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-L.G.; Bristol, L.A.; Jin, L.; Dykes-Hoberg, M.; Crawford, T.; Clawson, L.; Rothstein, J.D. Aberrant RNA Processing in a Neurodegenerative Disease: The Cause for Absent EAAT2, a Glutamate Transporter, in Amyotrophic Lateral Sclerosis. Neuron 1998, 20, 589–602. [Google Scholar] [CrossRef]
- Rao, S.D.; Yin, H.Z.; Weiss, J.H. Disruption of Glial Glutamate Transport by Reactive Oxygen Species Produced in Motor Neurons. J. Neurosci. 2003, 23, 2627–2633. [Google Scholar] [CrossRef]
- Gegelashvili, G.; Danbolt, N.C.; Schousboe, A. Neuronal Soluble Factors Differentially Regulate the Expression of the GLT1 and GLAST Glutamate Transporters in Cultured Astroglia. J. Neurochem. 2002, 69, 2612–2615. [Google Scholar] [CrossRef]
- Trotti, D.; Volterra, A.; Lehre, K.P.; Rossi, D.; Gjesdal, O.; Racagni, G.; Danbolt, N.C. Arachidonic Acid Inhibits a Purified and Reconstituted Glutamate Transporter Directly from the Water Phase and Not via the Phospholipid Membrane. J. Biol. Chem. 1995, 270, 9890–9895. [Google Scholar] [CrossRef]
- Dumont, A.O.; Goursaud, S.; Desmet, N.; Hermans, E. Differential Regulation of Glutamate Transporter Subtypes by Pro-Inflammatory Cytokine TNF-α in Cortical Astrocytes from a Rat Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2014, 9, e97649. [Google Scholar] [CrossRef] [PubMed]
- van Damme, P.; Dewil, M.; Robberecht, W.; van den Bosch, L. Excitotoxicity and Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2005, 2, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, L.T.; Shamamandri-Markandaiah, S.; Ghosh, B.; Foran, E.; Lepore, A.C.; Pasinelli, P.; Trotti, D. Mutation of the Caspase-3 Cleavage Site in the Astroglial Glutamate Transporter EAAT2 Delays Disease Progression and Extends Lifespan in the SOD1-G93A Mouse Model of ALS. Exp. Neurol. 2017, 292, 145–153. [Google Scholar] [CrossRef]
- Yin, X.; Wang, S.; Qi, Y.; Wang, X.; Jiang, H.; Wang, T.; Yang, Y.; Wang, Y.; Zhang, C.; Feng, H. Astrocyte Elevated Gene-1 Is a Novel Regulator of Astrogliosis and Excitatory Amino Acid Transporter-2 via Interplaying with Nuclear Factor-ΚB Signaling in Astrocytes from Amyotrophic Lateral Sclerosis Mouse Model with HSOD1 G93A Mutation. Mol. Cell. Neurosci. 2018, 90, 1–11. [Google Scholar] [CrossRef]
- Ziff, O.J.; Clarke, B.E.; Taha, D.M.; Crerar, H.; Luscombe, N.M.; Patani, R. Meta-Analysis of Human and Mouse ALS Astrocytes Reveals Multi-Omic Signatures of Inflammatory Reactive States. Genome Res. 2022, 32, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Vaz, S.H.; Pinto, S.; Sebastião, A.M.; Brites, D. Astrocytes in Amyotrophic Lateral Sclerosis; Araki, T., Ed.; Exon Publications: Brisbane, Australia, 2021; ISBN 978-0-6450017-7-8. [Google Scholar]
- Malarkey, E.B.; Parpura, V. Mechanisms of Glutamate Release from Astrocytes. Neurochem. Int. 2008, 52, 142–154. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Matteoli, M.; Parpura, V.; Mothet, J.-P.; Zorec, R. Astrocytes as Secretory Cells of the Central Nervous System: Idiosyncrasies of Vesicular Secretion. EMBO J. 2016, 35, 239–257. [Google Scholar] [CrossRef]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-Mediated Astrocyte-Neuron Signalling. Nature 1994, 369, 744–747. [Google Scholar] [CrossRef]
- Bezzi, P.; Carmignoto, G.; Pasti, L.; Vesce, S.; Rossi, D.; Rizzini, B.L.; Pozzan, T.; Volterra, A. Prostaglandins Stimulate Calcium-Dependent Glutamate Release in Astrocytes. Nature 1998, 391, 281–285. [Google Scholar] [CrossRef]
- Zhang, Q.; Pangrsic, T.; Kreft, M.; Krzan, M.; Li, N.; Sul, J.-Y.; Halassa, M.; Van Bockstaele, E.; Zorec, R.; Haydon, P.G. Fusion-Related Release of Glutamate from Astrocytes. J. Biol. Chem. 2004, 279, 12724–12733. [Google Scholar] [CrossRef]
- Domercq, M.; Brambilla, L.; Pilati, E.; Marchaland, J.; Volterra, A.; Bezzi, P. P2Y1 Receptor-Evoked Glutamate Exocytosis from Astrocytes: Control by Tumor Necrosis Factor-Alpha and Prostaglandins. J. Biol. Chem. 2006, 281, 30684–30696. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, B.A.; Hochman, D.; Delay, M.J.; Weiss, S. Modulation of Intracellular Ca++ in Cultured Astrocytes by Influx through Voltage-Activated Ca++ Channels. Glia 1991, 4, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.M.; Chiu, S.Y. Differential Intracellular Calcium Responses to Glutamate in Type 1 and Type 2 Cultured Brain Astrocytes. J. Neurosci. 1991, 11, 1674–1684. [Google Scholar] [CrossRef]
- Paluzzi, S.; Alloisio, S.; Zappettini, S.; Milanese, M.; Raiteri, L.; Nobile, M.; Bonanno, G. Adult Astroglia Is Competent for Na+/Ca2+ Exchanger-Operated Exocytotic Glutamate Release Triggered by Mild Depolarization. J. Neurochem. 2007, 103, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Montana, V.; Ni, Y.; Sunjara, V.; Hua, X.; Parpura, V. Vesicular Glutamate Transporter-Dependent Glutamate Release from Astrocytes. J. Neurosci. 2004, 24, 2633–2642. [Google Scholar] [CrossRef] [PubMed]
- Milanese, M.; Bonifacino, T.; Zappettini, S.; Usai, C.; Tacchetti, C.; Nobile, M.; Bonanno, G. Glutamate Release from Astrocytic Gliosomes under Physiological and Pathological Conditions. Int. Rev. Neurobiol. 2009, 85, 295–318. [Google Scholar] [CrossRef]
- Maienschein, V.; Marxen, M.; Volknandt, W.; Zimmermann, H. A Plethora of Presynaptic Proteins Associated with ATP-Storing Organelles in Cultured Astrocytes. Glia 1999, 26, 233–244. [Google Scholar] [CrossRef]
- Bezzi, P.; Gundersen, V.; Galbete, J.L.; Seifert, G.; Steinhäuser, C.; Pilati, E.; Volterra, A. Astrocytes Contain a Vesicular Compartment That Is Competent for Regulated Exocytosis of Glutamate. Nat. Neurosci. 2004, 7, 613–620. [Google Scholar] [CrossRef]
- Crippa, D.; Schenk, U.; Francolini, M.; Rosa, P.; Verderio, C.; Zonta, M.; Pozzan, T.; Matteoli, M.; Carmignoto, G. Synaptobrevin2-Expressing Vesicles in Rat Astrocytes: Insights into Molecular Characterization, Dynamics and Exocytosis. J. Physiol. 2006, 570, 567–582. [Google Scholar] [CrossRef]
- Milanese, M.; Bonifacino, T.; Fedele, E.; Rebosio, C.; Cattaneo, L.; Benfenati, F.; Usai, C.; Bonanno, G. Exocytosis Regulates Trafficking of GABA and Glycine Heterotransporters in Spinal Cord Glutamatergic Synapses: A Mechanism for the Excessive Heterotransporter-Induced Release of Glutamate in Experimental Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2015, 74, 314–324. [Google Scholar] [CrossRef]
- Kawamata, H.; Ng, S.K.; Diaz, N.; Burstein, S.; Morel, L.; Osgood, A.; Sider, B.; Higashimori, H.; Haydon, P.G.; Manfredi, G.; et al. Abnormal Intracellular Calcium Signaling and SNARE-Dependent Exocytosis Contributes to SOD1G93A Astrocyte-Mediated Toxicity in Amyotrophic Lateral Sclerosis. J. Neurosci. 2014, 34, 2331–2348. [Google Scholar] [CrossRef] [PubMed]
- Stenovec, M.; Milošević, M.; Petrušić, V.; Potokar, M.; Stević, Z.; Prebil, M.; Kreft, M.; Trkov, S.; Andjus, P.R.; Zorec, R. Amyotrophic Lateral Sclerosis Immunoglobulins G Enhance the Mobility of Lysotracker-Labelled Vesicles in Cultured Rat Astrocytes. Acta Physiol. 2011, 203, 457–471. [Google Scholar] [CrossRef] [PubMed]
- North, R.A. Molecular Physiology of P2X Receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef] [PubMed]
- Cervetto, C.; Alloisio, S.; Frattaroli, D.; Mazzotta, M.C.; Milanese, M.; Gavazzo, P.; Passalacqua, M.; Nobile, M.; Maura, G.; Marcoli, M. The P2X7 Receptor as a Route for Non-Exocytotic Glutamate Release: Dependence on the Carboxyl Tail. J. Neurochem. 2013, 124, 821–831. [Google Scholar] [CrossRef]
- Weisman, G.A.; Camden, J.M.; Peterson, T.S.; Ajit, D.; Woods, L.T.; Erb, L. P2 Receptors for Extracellular Nucleotides in the Central Nervous System: Role of P2X7 and P2Y₂ Receptor Interactions in Neuroinflammation. Mol. Neurobiol. 2012, 46, 96–113. [Google Scholar] [CrossRef]
- Kukley, M.; Barden, J.A.; Steinhäuser, C.; Jabs, R. Distribution of P2X Receptors on Astrocytes in Juvenile Rat Hippocampus. Glia 2001, 36, 11–21. [Google Scholar] [CrossRef]
- Duan, S.; Anderson, C.M.; Keung, E.C.; Chen, Y.; Chen, Y.; Swanson, R.A. P2X7 Receptor-Mediated Release of Excitatory Amino Acids from Astrocytes. J. Neurosci. 2003, 23, 1320–1328. [Google Scholar] [CrossRef]
- Fellin, T.; Pozzan, T.; Carmignoto, G. Purinergic Receptors Mediate Two Distinct Glutamate Release Pathways in Hippocampal Astrocytes. J. Biol. Chem. 2006, 281, 4274–4284. [Google Scholar] [CrossRef]
- Cauwels, A.; Rogge, E.; Vandendriessche, B.; Shiva, S.; Brouckaert, P. Extracellular ATP Drives Systemic Inflammation, Tissue Damage and Mortality. Cell Death Dis. 2014, 5, e1102. [Google Scholar] [CrossRef]
- Panenka, W.; Jijon, H.; Herx, L.M.; Armstrong, J.N.; Feighan, D.; Wei, T.; Yong, V.W.; Ransohoff, R.M.; MacVicar, B.A. P2X7-like Receptor Activation in Astrocytes Increases Chemokine Monocyte Chemoattractant Protein-1 Expression via Mitogen-Activated Protein Kinase. J. Neurosci. 2001, 21, 7135–7142. [Google Scholar] [CrossRef]
- John, G.R.; Simpson, J.E.; Woodroofe, M.N.; Lee, S.C.; Brosnan, C.F. Extracellular Nucleotides Differentially Regulate Interleukin-1beta Signaling in Primary Human Astrocytes: Implications for Inflammatory Gene Expression. J. Neurosci. 2001, 21, 4134–4142. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, A.; Hernández, S.; Tarabal, O.; Rosselló, J.; Esquerda, J.E. Strong P2X4 Purinergic Receptor-like Immunoreactivity Is Selectively Associated with Degenerating Neurons in Transgenic Rodent Models of Amyotrophic Lateral Sclerosis. J. Comp. Neurol. 2008, 506, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-Immunoreactivities Are Increased in Activated Microglial Cells/Macrophages of Multiple Sclerosis and Amyotrophic Lateral Sclerosis Spinal Cord. BMC Neurol. 2006, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, S.; Amadio, S.; Montilli, C.; Volonté, C.; D’Ambrosi, N. Ablation of P2X7 Receptor Exacerbates Gliosis and Motoneuron Death in the SOD1-G93A Mouse Model of Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2013, 22, 4102–4116. [Google Scholar] [CrossRef] [PubMed]
- Volonté, C.; Amadio, S.; Liguori, F.; Fabbrizio, P. Duality of P2X7 Receptor in Amyotrophic Lateral Sclerosis. Front. Pharmacol. 2020, 11, 1148. [Google Scholar] [CrossRef]
- Ruiz-Ruiz, C.; Calzaferri, F.; García, A.G. P2X7 Receptor Antagonism as a Potential Therapy in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2020, 13, 93. [Google Scholar] [CrossRef]
- Cervetto, C.; Frattaroli, D.; Maura, G.; Marcoli, M. Motor Neuron Dysfunction in a Mouse Model of ALS: Gender-Dependent Effect of P2X7 Antagonism. Toxicology 2013, 311, 69–77. [Google Scholar] [CrossRef]
- Ruiz-Ruiz, C.; García-Magro, N.; Negredo, P.; Avendaño, C.; Bhattacharya, A.; Ceusters, M.; García, A.G. Chronic Administration of P2X7 Receptor Antagonist JNJ-47965567 Delays Disease Onset and Progression, and Improves Motor Performance in ALS SOD1G93A Female Mice. Dis. Model. Mech. 2020, 13, dmm045732. [Google Scholar] [CrossRef]
- Browne, S.E. When Too Much ATP Is a Bad Thing: A Pivotal Role for P2X7 Receptors in Motor Neuron Degeneration. J. Neurochem. 2013, 126, 301–304. [Google Scholar] [CrossRef]
- Gandelman, M.; Peluffo, H.; Beckman, J.S.; Cassina, P.; Barbeito, L. Extracellular ATP and the P2X7 Receptor in Astrocyte-Mediated Motor Neuron Death: Implications for Amyotrophic Lateral Sclerosis. J. Neuroinflamm. 2010, 7, 33. [Google Scholar] [CrossRef]
- Mckenzie, A.D.J.; Garrett, T.R.; Werry, E.L.; Kassiou, M. Purinergic P2X7 Receptor: A Therapeutic Target in Amyotrophic Lateral Sclerosis. ACS Chem. Neurosci. 2022, 13, 1479–1490. [Google Scholar] [CrossRef]
- Albano, R.; Liu, X.; Lobner, D. Regulation of System Xc− in the SOD1-G93A Mouse Model of ALS. Exp. Neurol. 2013, 250, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Mesci, P.; Zaïdi, S.; Lobsiger, C.S.; Millecamps, S.; Escartin, C.; Seilhean, D.; Sato, H.; Mallat, M.; Boillée, S. System XC− Is a Mediator of Microglial Function and Its Deletion Slows Symptoms in Amyotrophic Lateral Sclerosis Mice. Brain 2015, 138, 53–68. [Google Scholar] [CrossRef]
- Kazama, M.; Kato, Y.; Kakita, A.; Noguchi, N.; Urano, Y.; Masui, K.; Niida-Kawaguchi, M.; Yamamoto, T.; Watabe, K.; Kitagawa, K.; et al. Astrocytes Release Glutamate via Cystine/Glutamate Antiporter Upregulated in Response to Increased Oxidative Stress Related to Sporadic Amyotrophic Lateral Sclerosis. Neuropathology 2020, 40, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Giaume, C.; Sáez, J.C. Role of Connexin Hemichannels in Neurodegeneration. In Neurodegenerative Diseases—Processes, Prevention, Protection and Monitoring; InTechOpen: Rijeka, Croatia, 2011. [Google Scholar]
- Goldberg, G.S.; Lampe, P.D.; Nicholson, B.J. Selective Transfer of Endogenous Metabolites through Gap Junctions Composed of Different Connexins. Nat. Cell Biol. 1999, 1, 457–459. [Google Scholar] [CrossRef]
- Bennett, M.V.L.; Contreras, J.E.; Bukauskas, F.F.; Sáez, J.C. New Roles for Astrocytes: Gap Junction Hemichannels Have Something to Communicate. Trends Neurosci. 2003, 26, 610–617. [Google Scholar] [CrossRef]
- Ye, Z.-C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional Hemichannels in Astrocytes: A Novel Mechanism of Glutamate Release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [CrossRef]
- Orellana, J.A.; Stehberg, J. Hemichannels: New Roles in Astroglial Function. Front. Physiol. 2014, 5, 193. [Google Scholar] [CrossRef]
- Endong, L.; Shijie, J.; Sonobe, Y.; Di, M.; Hua, L.; Kawanokuchi, J.; Mizuno, T.; Suzumura, A. The Gap-Junction Inhibitor Carbenoxolone Suppresses the Differentiation of Th17 Cells through Inhibition of IL-23 Expression in Antigen Presenting Cells. J. Neuroimmunol. 2011, 240–241, 58–64. [Google Scholar] [CrossRef]
- Lutz, S.E.; Zhao, Y.; Gulinello, M.; Lee, S.C.; Raine, C.S.; Brosnan, C.F. Deletion of Astrocyte Connexins 43 and 30 Leads to a Dysmyelinating Phenotype and Hippocampal CA1 Vacuolation. J. Neurosci. 2009, 29, 7743–7752. [Google Scholar] [CrossRef]
- Liang, Z.; Wang, X.; Hao, Y.; Qiu, L.; Lou, Y.; Zhang, Y.; Ma, D.; Feng, J. The Multifaceted Role of Astrocyte Connexin 43 in Ischemic Stroke Through Forming Hemichannels and Gap Junctions. Front. Neurol. 2020, 11, 703. [Google Scholar] [CrossRef] [PubMed]
- Montero, T.D.; Orellana, J.A. Hemichannels: New Pathways for Gliotransmitter Release. Neuroscience 2015, 286, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.G.; Charles, A.C. Intercellular Calcium Signaling in Astrocytes via ATP Release through Connexin Hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Yang, T.; Cui, S.; Chen, G. Connexin Hemichannels in Astrocytes: Role in CNS Disorders. Front. Mol. Neurosci. 2019, 12, 23. [Google Scholar] [CrossRef]
- Cherian, P.P.; Siller-Jackson, A.J.; Gu, S.; Wang, X.; Bonewald, L.F.; Sprague, E.; Jiang, J.X. Mechanical Strain Opens Connexin 43 Hemichannels in Osteocytes: A Novel Mechanism for the Release of Prostaglandin. Mol. Biol. Cell. 2005, 16, 3100–3106. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, S.; Charles, A. Extracellular Calcium Sensing by Glial Cells: Low Extracellular Calcium Induces Intracellular Calcium Release and Intercellular Signaling. J. Neurochem. 2002, 69, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Papouin, T.; Oliet, S.H.R.; Rusakov, D.A. Long-Term Potentiation Depends on Release of d-Serine from Astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef]
- Meunier, C.; Wang, N.; Yi, C.; Dallerac, G.; Ezan, P.; Koulakoff, A.; Leybaert, L.; Giaume, C. Contribution of Astroglial Cx43 Hemichannels to the Modulation of Glutamatergic Currents by D-Serine in the Mouse Prefrontal Cortex. J. Neurosci. 2017, 37, 9064–9075. [Google Scholar] [CrossRef]
- Stehberg, J.; Moraga-Amaro, R.; Salazar, C.; Becerra, A.; Echeverría, C.; Orellana, J.A.; Bultynck, G.; Ponsaerts, R.; Leybaert, L.; Simon, F.; et al. Release of Gliotransmitters through Astroglial Connexin 43 Hemichannels Is Necessary for Fear Memory Consolidation in the Basolateral Amygdala. FASEB J. 2012, 26, 3649–3657. [Google Scholar] [CrossRef]
- Huang, X.; Su, Y.; Wang, N.; Li, H.; Li, Z.; Yin, G.; Chen, H.; Niu, J.; Yi, C. Astroglial Connexins in Neurodegenerative Diseases. Front. Mol. Neurosci. 2021, 14, 657514. [Google Scholar] [CrossRef]
- Sánchez, O.F.; Rodríguez, A.V.; Velasco-España, J.M.; Murillo, L.C.; Sutachan, J.-J.; Albarracin, S.-L. Role of Connexins 30, 36, and 43 in Brain Tumors, Neurodegenerative Diseases, and Neuroprotection. Cells 2020, 9, 846. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Masaki, K.; Yamasaki, R.; Imamura, S.; Suzuki, S.O.; Hayashi, S.; Sato, S.; Nagara, Y.; Kawamura, M.F.; Kira, J. Extensive Dysregulations of Oligodendrocytic and Astrocytic Connexins Are Associated with Disease Progression in an Amyotrophic Lateral Sclerosis Mouse Model. J. Neuroinflamm. 2014, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.F.; Gravel, M.; Kriz, J. Treatment with Minocycline after Disease Onset Alters Astrocyte Reactivity and Increases Microgliosis in SOD1 Mutant Mice. Exp. Neurol. 2011, 228, 69–79. [Google Scholar] [CrossRef]
- Almad, A.A.; Doreswamy, A.; Gross, S.K.; Richard, J.P.; Huo, Y.; Haughey, N.; Maragakis, N.J. Connexin 43 in Astrocytes Contributes to Motor Neuron Toxicity in Amyotrophic Lateral Sclerosis. Glia 2016, 64, 1154–1169. [Google Scholar] [CrossRef]
- Yamasaki, R. Novel Animal Model of Multiple Sclerosis: The Glial Connexin Gap Junction as an Environmental Tuner for Neuroinflammation. Clin. Exp. Neuroimmunol. 2020, 11, 34–40. [Google Scholar] [CrossRef]
- Fang, M.; Yamasaki, R.; Li, G.; Masaki, K.; Yamaguchi, H.; Fujita, A.; Isobe, N.; Kira, J. Connexin 30 Deficiency Attenuates Chronic but Not Acute Phases of Experimental Autoimmune Encephalomyelitis Through Induction of Neuroprotective Microglia. Front Immunol 2018, 9, 2588. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Yamasaki, R.; Ko, S.; Matsuo, E.; Kobayakawa, Y.; Masaki, K.; Matsuse, D.; Isobe, N. Connexin 30 Deficiency Ameliorates Disease Progression at the Early Phase in a Mouse Model of Amyotrophic Lateral Sclerosis by Suppressing Glial Inflammation. Int. J. Mol. Sci. 2022, 23, 16046. [Google Scholar] [CrossRef]
- Almad, A.A.; Taga, A.; Joseph, J.; Gross, S.K.; Welsh, C.; Patankar, A.; Richard, J.-P.; Rust, K.; Pokharel, A.; Plott, C.; et al. Cx43 Hemichannels Contribute to Astrocyte-Mediated Toxicity in Sporadic and Familial ALS. Proc. Natl. Acad. Sci. USA 2022, 119, e2107391119. [Google Scholar] [CrossRef]
- Lehrer, S.; Rheinstein, P.H. Insulin Docking Within the Open Hemichannel of Connexin 43 May Reduce Risk of Amyotrophic Lateral Sclerosis. Vivo 2023, 37, 539–547. [Google Scholar] [CrossRef]
- Milenkovic, A.; Brandl, C.; Milenkovic, V.M.; Jendryke, T.; Sirianant, L.; Wanitchakool, P.; Zimmermann, S.; Reiff, C.M.; Horling, F.; Schrewe, H.; et al. Bestrophin 1 Is Indispensable for Volume Regulation in Human Retinal Pigment Epithelium Cells. Proc. Natl. Acad. Sci. USA 2015, 112, E2630–E2639. [Google Scholar] [CrossRef]
- Park, H.; Han, K.-S.; Oh, S.-J.; Jo, S.; Woo, J.; Yoon, B.-E.; Lee, C.J. High Glutamate Permeability and Distal Localization of Best1 Channel in CA1 Hippocampal Astrocyte. Mol. Brain 2013, 6, 54. [Google Scholar] [CrossRef]
- Oh, S.-J.; Lee, C.J. Distribution and Function of the Bestrophin-1 (Best1) Channel in the Brain. Exp. Neurobiol. 2017, 26, 113–121. [Google Scholar] [CrossRef]
- Haroon, E.; Fleischer, C.C.; Felger, J.C.; Chen, X.; Woolwine, B.J.; Patel, T.; Hu, X.P.; Miller, A.H. Conceptual Convergence: Increased Inflammation Is Associated with Increased Basal Ganglia Glutamate in Patients with Major Depression. Mol. Psychiatry 2016, 21, 1351–1357. [Google Scholar] [CrossRef]
- Heo, C.; Chang, K.-A.; Choi, H.S.; Kim, H.-S.; Kim, S.; Liew, H.; Kim, J.A.; Yu, E.; Ma, J.; Suh, Y.-H. Effects of the Monomeric, Oligomeric, and Fibrillar Aβ42 Peptides on the Proliferation and Differentiation of Adult Neural Stem Cells from Subventricular Zone. J. Neurochem. 2007, 102, 493–500. [Google Scholar] [CrossRef]
- Djillani, A.; Mazella, J.; Heurteaux, C.; Borsotto, M. Role of TREK-1 in Health and Disease, Focus on the Central Nervous System. Front. Pharmacol. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xu, G.; Xie, M.; Zhang, X.; Schools, G.P.; Ma, L.; Kimelberg, H.K.; Chen, H. TWIK-1 and TREK-1 Are Potassium Channels Contributing Significantly to Astrocyte Passive Conductance in Rat Hippocampal Slices. J. Neurosci. 2009, 29, 8551–8564. [Google Scholar] [CrossRef] [PubMed]
- Mi Hwang, E.; Kim, E.; Yarishkin, O.; Ho Woo, D.; Han, K.-S.; Park, N.; Bae, Y.; Woo, J.; Kim, D.; Park, M.; et al. A Disulphide-Linked Heterodimer of TWIK-1 and TREK-1 Mediates Passive Conductance in Astrocytes. Nat. Commun. 2014, 5, 3227. [Google Scholar] [CrossRef] [PubMed]
- De Pittà, M.; Brunel, N. Modulation of Synaptic Plasticity by Glutamatergic Gliotransmission: A Modeling Study. Neural Plast. 2016, 2016, 7607924. [Google Scholar] [CrossRef] [PubMed]
- Fiacco, T.A.; Agulhon, C.; Taves, S.R.; Petravicz, J.; Casper, K.B.; Dong, X.; Chen, J.; McCarthy, K.D. Selective Stimulation of Astrocyte Calcium in Situ Does Not Affect Neuronal Excitatory Synaptic Activity. Neuron 2007, 54, 611–626. [Google Scholar] [CrossRef]
- Compan, V.; Baroja-Mazo, A.; López-Castejón, G.; Gomez, A.I.; Martínez, C.M.; Angosto, D.; Montero, M.T.; Herranz, A.S.; Bazán, E.; Reimers, D.; et al. Cell Volume Regulation Modulates NLRP3 Inflammasome Activation. Immunity 2012, 37, 487–500. [Google Scholar] [CrossRef]
- Shimizu, T.; Numata, T.; Okada, Y. A Role of Reactive Oxygen Species in Apoptotic Activation of Volume-Sensitive Cl− Channel. Proc. Natl. Acad. Sci. USA 2004, 101, 6770–6773. [Google Scholar] [CrossRef] [PubMed]
- Haskew-Layton, R.E.; Mongin, A.A.; Kimelberg, H.K. Hydrogen Peroxide Potentiates Volume-Sensitive Excitatory Amino Acid Release via a Mechanism Involving Ca2+/Calmodulin-Dependent Protein Kinase II. J. Biol. Chem. 2005, 280, 3548–3554. [Google Scholar] [CrossRef] [PubMed]
- Olmos, G.; Lladó, J. Tumor Necrosis Factor Alpha: A Link between Neuroinflammation and Excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-Activated Astrocyte Glutamate Release via TNFα: Amplification by Microglia Triggers Neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef]
- Tilleux, S.; Hermans, E. Neuroinflammation and Regulation of Glial Glutamate Uptake in Neurological Disorders. J. Neurosci. Res. 2007, 85, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pekarskaya, O.; Bencheikh, M.; Chao, W.; Gelbard, H.A.; Ghorpade, A.; Rothstein, J.D.; Volsky, D.J. Reduced Expression of Glutamate Transporter EAAT2 and Impaired Glutamate Transport in Human Primary Astrocytes Exposed to HIV-1 or Gp120. Virology 2003, 312, 60–73. [Google Scholar] [CrossRef]
- Sitcheran, R.; Gupta, P.; Fisher, P.B.; Baldwin, A.S. Positive and Negative Regulation of EAAT2 by NF-ΚB: A Role for N-Myc in TNFα-Controlled Repression. EMBO J. 2005, 24, 510–520. [Google Scholar] [CrossRef]
- Hu, S.; Sheng, W.S.; Ehrlich, L.C.; Peterson, P.K.; Chao, C.C. Cytokine Effects on Glutamate Uptake by Human Astrocytes. Neuroimmunomodulation 2000, 7, 153–159. [Google Scholar] [CrossRef]
- Ida, T.; Hara, M.; Nakamura, Y.; Kozaki, S.; Tsunoda, S.; Ihara, H. Cytokine-Induced Enhancement of Calcium-Dependent Glutamate Release from Astrocytes Mediated by Nitric Oxide. Neurosci. Lett. 2008, 432, 232–236. [Google Scholar] [CrossRef]
- Ohno, Y.; Kunisawa, N.; Shimizu, S. Emerging Roles of Astrocyte Kir4.1 Channels in the Pathogenesis and Treatment of Brain Diseases. Int. J. Mol. Sci. 2021, 22, 10236. [Google Scholar] [CrossRef]
- Olsen, M.L.; Sontheimer, H. Functional Implications for Kir4.1 Channels in Glial Biology: From K+ Buffering to Cell Differentiation. J. Neurochem. 2008, 107, 589–601. [Google Scholar] [CrossRef]
- Kofuji, P.; Newman, E.A. Potassium Buffering in the Central Nervous System. Neuroscience 2004, 129, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Kinboshi, M.; Shimizu, S. Inwardly Rectifying Potassium Channel Kir4.1 as a Novel Modulator of BDNF Expression in Astrocytes. Int. J. Mol. Sci. 2018, 19, 3313. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Maletzki, I.; Hülsmann, S.; Holtmann, B.; Schulz-Schaeffer, W.; Kirchhoff, F.; Bähr, M.; Neusch, C. Progressive Loss of a Glial Potassium Channel (KCNJ10) in the Spinal Cord of the SOD1 (G93A) Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurochem. 2006, 99, 900–912. [Google Scholar] [CrossRef]
- Bataveljić, D.; Nikolić, L.; Milosević, M.; Todorović, N.; Andjus, P.R. Changes in the Astrocytic Aquaporin-4 and Inwardly Rectifying Potassium Channel Expression in the Brain of the Amyotrophic Lateral Sclerosis SOD1G93A Rat Model. Glia 2012, 60, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Nwaobi, S.E.; Cuddapah, V.A.; Patterson, K.C.; Randolph, A.C.; Olsen, M.L. The Role of Glial-Specific Kir4.1 in Normal and Pathological States of the CNS. Acta Neuropathol. 2016, 132, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Peric, M.; Mitrecic, D.; Andjus, P.R. Targeting Astrocytes for Treatment in Amyotrophic Lateral Sclerosis. Curr. Pharm. Des. 2018, 23, 5037–5044. [Google Scholar] [CrossRef]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Ceprian, M.; Fulton, D. Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease. Int. J. Mol. Sci. 2019, 20, 2450. [Google Scholar] [CrossRef]
- Hösli, L.; Andrès, P.F.; Hösli, E. Depolarization of Cultured Astrocytes by Glutamate and Aspartate. Neuroscience 1979, 4, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Meeks, J.P.; Mennerick, S. Astrocyte Membrane Responses and Potassium Accumulation during Neuronal Activity. Hippocampus 2007, 17, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Zorec, R. Gliotransmission: Exocytotic Release from Astrocytes. Brain Res. Rev. 2010, 63, 83–92. [Google Scholar] [CrossRef]
- Zuo, T.; Gong, B.; Gao, Y.; Yuan, L. An in Vitro Study on the Stimulatory Effects of Extracellular Glutamate on Astrocytes. Mol. Biol. Rep. 2023, 50, 6611–6617. [Google Scholar] [CrossRef]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal Degeneration of Astrocytes in Amyotrophic Lateral Sclerosis. Cell Death Differ. 2008, 15, 1691–1700. [Google Scholar] [CrossRef]
- Biber, K.; Laurie, D.J.; Berthele, A.; Sommer, B.; Tölle, T.R.; Gebicke-Härter, P.J.; Van Calker, D.; Boddeke, H.W.G.M. Expression and Signaling of Group I Metabotropic Glutamate Receptors in Astrocytes and Microglia. J. Neurochem. 1999, 72, 1671–1680. [Google Scholar] [CrossRef]
- Nakahara, K.; Okada, M.; Nakanishi, S. The Metabotropic Glutamate Receptor MGluR5 Induces Calcium Oscillations in Cultured Astrocytes via Protein Kinase C Phosphorylation. J. Neurochem. 1997, 69, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Pasti, L.; Volterra, A.; Pozzan, T.; Carmignoto, G. Intracellular Calcium Oscillations in Astrocytes: A Highly Plastic, Bidirectional Form of Communication between Neurons and Astrocytes In Situ. J. Neurosci. 1997, 17, 7817–7830. [Google Scholar] [CrossRef]
- Miller, S.; Romano, C.; Cotman, C. Growth Factor Upregulation of a Phosphoinositide-Coupled Metabotropic Glutamate Receptor in Cortical Astrocytes. J. Neurosci. 1995, 15, 6103–6109. [Google Scholar] [CrossRef]
- Jean, Y.Y.; Lercher, L.D.; Dreyfus, C.F. Glutamate Elicits Release of BDNF from Basal Forebrain Astrocytes in a Process Dependent on Metabotropic Receptors and the PLC Pathway. Neuron Glia Biol. 2008, 4, 35–42. [Google Scholar] [CrossRef]
- Battaglia, G.; Riozzi, B.; Bucci, D.; Di Menna, L.; Molinaro, G.; Pallottino, S.; Nicoletti, F.; Bruno, V. Activation of MGlu3 Metabotropic Glutamate Receptors Enhances GDNF and GLT-1 Formation in the Spinal Cord and Rescues Motor Neurons in the SOD-1 Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2015, 74, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Molinaro, G.; Battaglia, G.; Giuffrida, M.L.; Riozzi, B.; Traficante, A.; Bruno, V.; Cannella, M.; Merlo, S.; Wang, X.; et al. Targeting Group II Metabotropic Glutamate (MGlu) Receptors for the Treatment of Psychosis Associated with Alzheimer’s Disease: Selective Activation of MGlu2 Receptors Amplifies β-Amyloid Toxicity in Cultured Neurons, Whereas Dual Activation of MGlu2 and MGlu3 Receptors Is Neuroprotective. Mol. Pharmacol. 2011, 79, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Bruno, V.; Battaglia, G.; Casabona, G.; Copani, A.; Caciagli, F.; Nicoletti, F. Neuroprotection by Glial Metabotropic Glutamate Receptors Is Mediated by Transforming Growth Factor-β. J. Neurosci. 1998, 18, 9594–9600. [Google Scholar] [CrossRef]
- Brownell, A.L.; Kuruppu, D.; Kil, K.E.; Jokivarsi, K.; Poutiainen, P.; Zhu, A.; Maxwell, M. PET Imaging Studies Show Enhanced Expression of MGluR5 and Inflammatory Response during Progressive Degeneration in ALS Mouse Model Expressing SOD1-G93A Gene. J. Neuroinflamm. 2015, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Müller Herde, A.; Schibli, R.; Weber, M.; Ametamey, S.M. Metabotropic Glutamate Receptor Subtype 5 Is Altered in LPS-Induced Murine Neuroinflammation Model and in the Brains of AD and ALS Patients. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 407–420. [Google Scholar] [CrossRef]
- Aronica, E.; Catania, M.V.; Geurts, J.; Yankaya, B.; Troost, D. Immunohistochemical Localization of Group I and II Metabotropic Glutamate Receptors in Control and Amyotrophic Lateral Sclerosis Human Spinal Cord: Upregulation in Reactive Astrocytes. Neuroscience 2001, 105, 509–520. [Google Scholar] [CrossRef]
- Bruno, V.; Battaglia, G.; Copani, A.; D’Onofrio, M.; Di Iorio, P.; De Blasi, A.; Melchiorri, D.; Flor, P.J.; Nicoletti, F. Metabotropic Glutamate Receptor Subtypes as Targets for Neuroprotective Drugs. J. Cereb. Blood Flow Metab. 2001, 21, 1013–1033. [Google Scholar] [CrossRef]
- Aronica, E.; Gorter, J.A.; Ijlst-Keizers, H.; Rozemuller, A.J.; Yankaya, B.; Leenstra, S.; Troost, D. Expression and Functional Role of MGluR3 and MGluR5 in Human Astrocytes and Glioma Cells: Opposite Regulation of Glutamate Transporter Proteins. Eur. J. Neurosci. 2003, 17, 2106–2118. [Google Scholar] [CrossRef]
- Vermeiren, C.; De Hemptinne, I.; Vanhoutte, N.; Tilleux, S.; Maloteaux, J.-M.M.; Hermans, E.; Hemptinne, I.; Vanhoutte, N.; Tilleux, S.; Maloteaux, J.-M.M.; et al. Loss of Metabotropic Glutamate Receptor-Mediated Regulation of Glutamate Transport in Chemically Activated Astrocytes in a Rat Model of Amyotrophic Lateral Sclerosis. J. Neurochem. 2006, 96, 719–731. [Google Scholar] [CrossRef]
- Vergouts, M.; Doyen, P.J.; Peeters, M.; Opsomer, R.; Hermans, E. Constitutive Downregulation Protein Kinase C Epsilon in HSOD1(G93A) Astrocytes Influences MGluR5 Signaling and the Regulation of Glutamate Uptake. Glia 2018, 66, 749–761. [Google Scholar] [CrossRef]
- Berger, J.V.; Dumont, A.O.; Focant, M.C.; Vergouts, M.; Sternotte, A.; Calas, A.-G.G.; Goursaud, S.; Hermans, E. Opposite Regulation of Metabotropic Glutamate Receptor 3 and Metabotropic Glutamate Receptor 5 by Inflammatory Stimuli in Cultured Microglia and Astrocytes. Neuroscience 2012, 205, 29–38. [Google Scholar] [CrossRef] [PubMed]
- D’Antoni, S.; Berretta, A.; Seminara, G.; Longone, P.; Giuffrida-Stella, A.M.; Battaglia, G.; Sortino, M.A.; Nicoletti, F.; Catania, M. V A Prolonged Pharmacological Blockade of Type-5 Metabotropic Glutamate Receptors Protects Cultured Spinal Cord Motor Neurons against Excitotoxic Death. Neurobiol. Dis. 2011, 42, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Viwatpinyo, K.; Chongthammakun, S. Activation of Group I Metabotropic Glutamate Receptors Leads to Brain-Derived Neurotrophic Factor Expression in Rat C6 Cells. Neurosci. Lett. 2009, 467, 127–130. [Google Scholar] [CrossRef]
- Horino-Shimizu, A.; Moriyama, K.; Mori, T.; Kohyama, K.; Nishito, Y.; Sakuma, H. Lipocalin-2 Production by Astrocytes in Response to High Concentrations of Glutamate. Brain Res. 2023, 1815, 148463. [Google Scholar] [CrossRef]
- Martorana, F.; Brambilla, L.; Valori, C.F.; Bergamaschi, C.; Roncoroni, C.; Aronica, E.; Volterra, A.; Bezzi, P.; Rossi, D. The BH4 Domain of Bcl-X(L) Rescues Astrocyte Degeneration in Amyotrophic Lateral Sclerosis by Modulating Intracellular Calcium Signals. Hum. Mol. Genet. 2012, 21, 826–840. [Google Scholar] [CrossRef]
- Budgett, R.F.; Bakker, G.; Sergeev, E.; Bennett, K.A.; Bradley, S.J. Targeting the Type 5 Metabotropic Glutamate Receptor: A Potential Therapeutic Strategy for Neurodegenerative Diseases? Front. Pharmacol. 2022, 13, 893422. [Google Scholar] [CrossRef]
- Milanese, M.; Bonifacino, T.; Torazza, C.; Provenzano, F.; Kumar, M.; Ravera, S.; Zerbo, A.R.; Frumento, G.; Balbi, M.; Nguyen, T.P.N.; et al. Blocking Glutamate MGlu(5) Receptors with the Negative Allosteric Modulator CTEP Improves Disease Course in SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Br. J. Pharmacol. 2021, 178, 3747–3764. [Google Scholar] [CrossRef]
- Paquet, M.; Ribeiro, F.M.; Guadagno, J.; Esseltine, J.L.; Ferguson, S.S.G.; Cregan, S.P. Role of Metabotropic Glutamate Receptor 5 Signaling and Homer in Oxygen Glucose Deprivation-Mediated Astrocyte Apoptosis. Mol. Brain 2013, 6, 9. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Provenzano, F.; Torazza, C.; Bonifacino, T.; Bonanno, G.; Milanese, M. The Key Role of Astrocytes in Amyotrophic Lateral Sclerosis and Their Commitment to Glutamate Excitotoxicity. Int. J. Mol. Sci. 2023, 24, 15430. https://doi.org/10.3390/ijms242015430
Provenzano F, Torazza C, Bonifacino T, Bonanno G, Milanese M. The Key Role of Astrocytes in Amyotrophic Lateral Sclerosis and Their Commitment to Glutamate Excitotoxicity. International Journal of Molecular Sciences. 2023; 24(20):15430. https://doi.org/10.3390/ijms242015430
Chicago/Turabian StyleProvenzano, Francesca, Carola Torazza, Tiziana Bonifacino, Giambattista Bonanno, and Marco Milanese. 2023. "The Key Role of Astrocytes in Amyotrophic Lateral Sclerosis and Their Commitment to Glutamate Excitotoxicity" International Journal of Molecular Sciences 24, no. 20: 15430. https://doi.org/10.3390/ijms242015430
APA StyleProvenzano, F., Torazza, C., Bonifacino, T., Bonanno, G., & Milanese, M. (2023). The Key Role of Astrocytes in Amyotrophic Lateral Sclerosis and Their Commitment to Glutamate Excitotoxicity. International Journal of Molecular Sciences, 24(20), 15430. https://doi.org/10.3390/ijms242015430