
Functional and Molecular Changes in the Prefrontal Cortex of the Chronic Mild Stress Rat Model of Depression and Modulation by Acute Ketamine

 ,
,  ,
,  , ,
, ,  ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
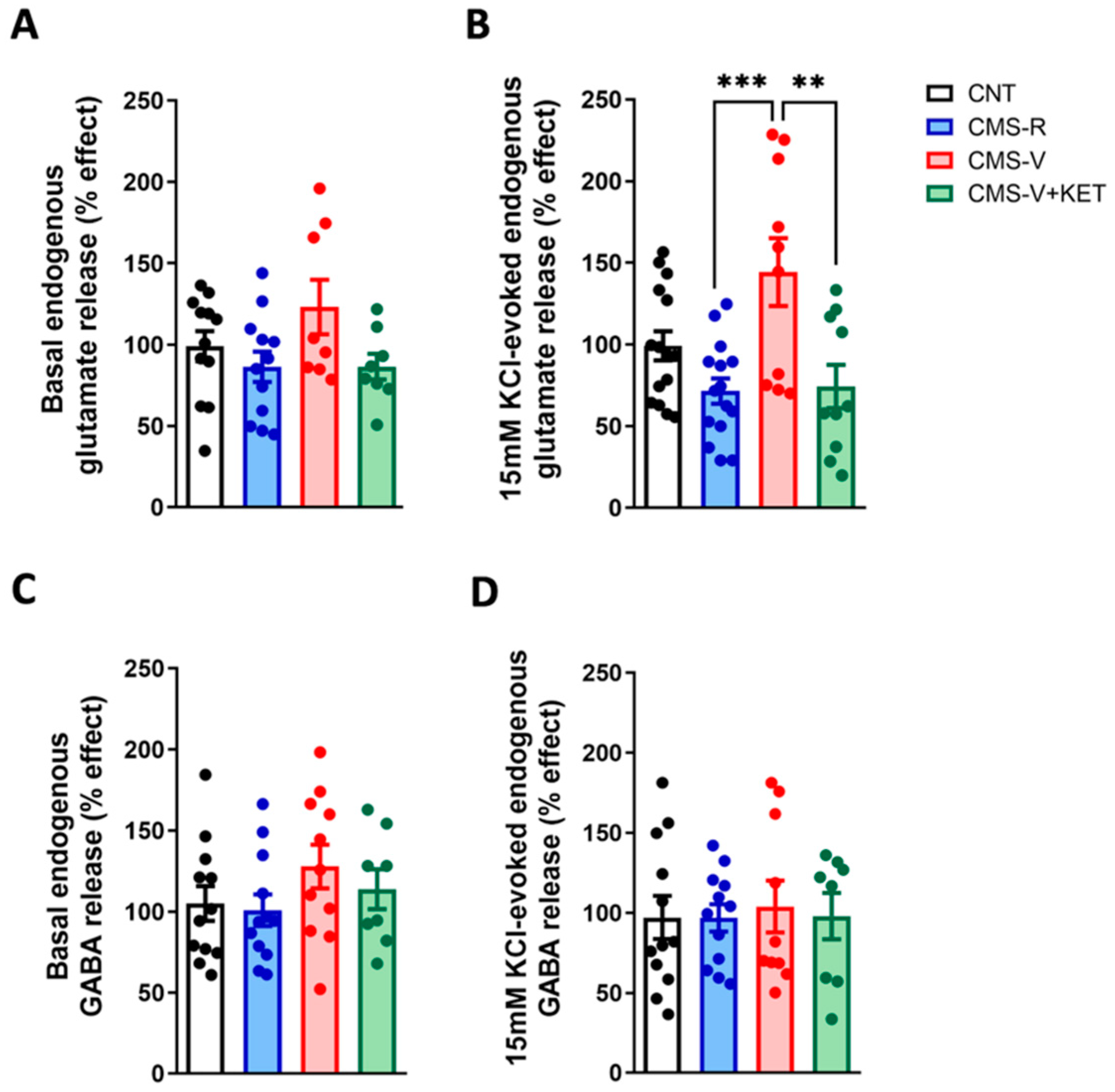
2.1. Chronic Mild Stress Increases the Activity-Dependent Presynaptic Glutamate Release in the Medial Prefrontal Cortex of Vulnerable Rats and Ketamine Rescues This Change
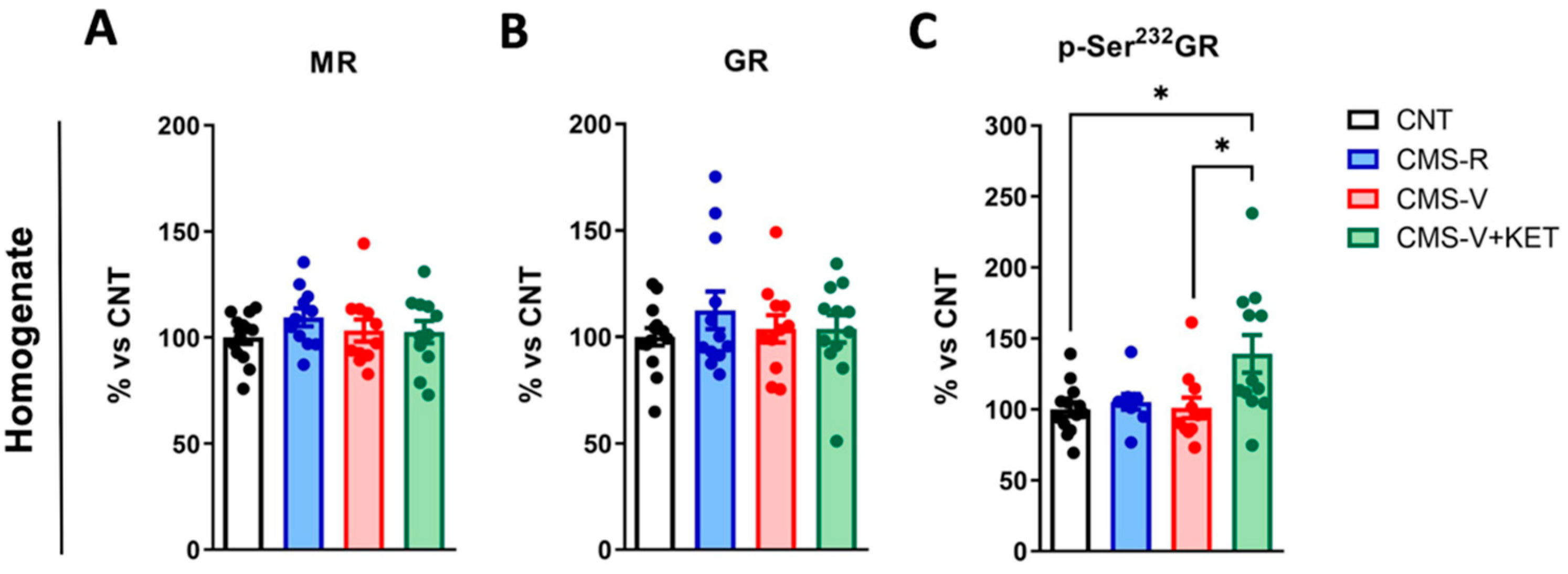
2.2. Ketamine Increases Glucocorticoid Receptor Phosphorylation in the Medial Prefrontal Cortex of Vulnerable Rats
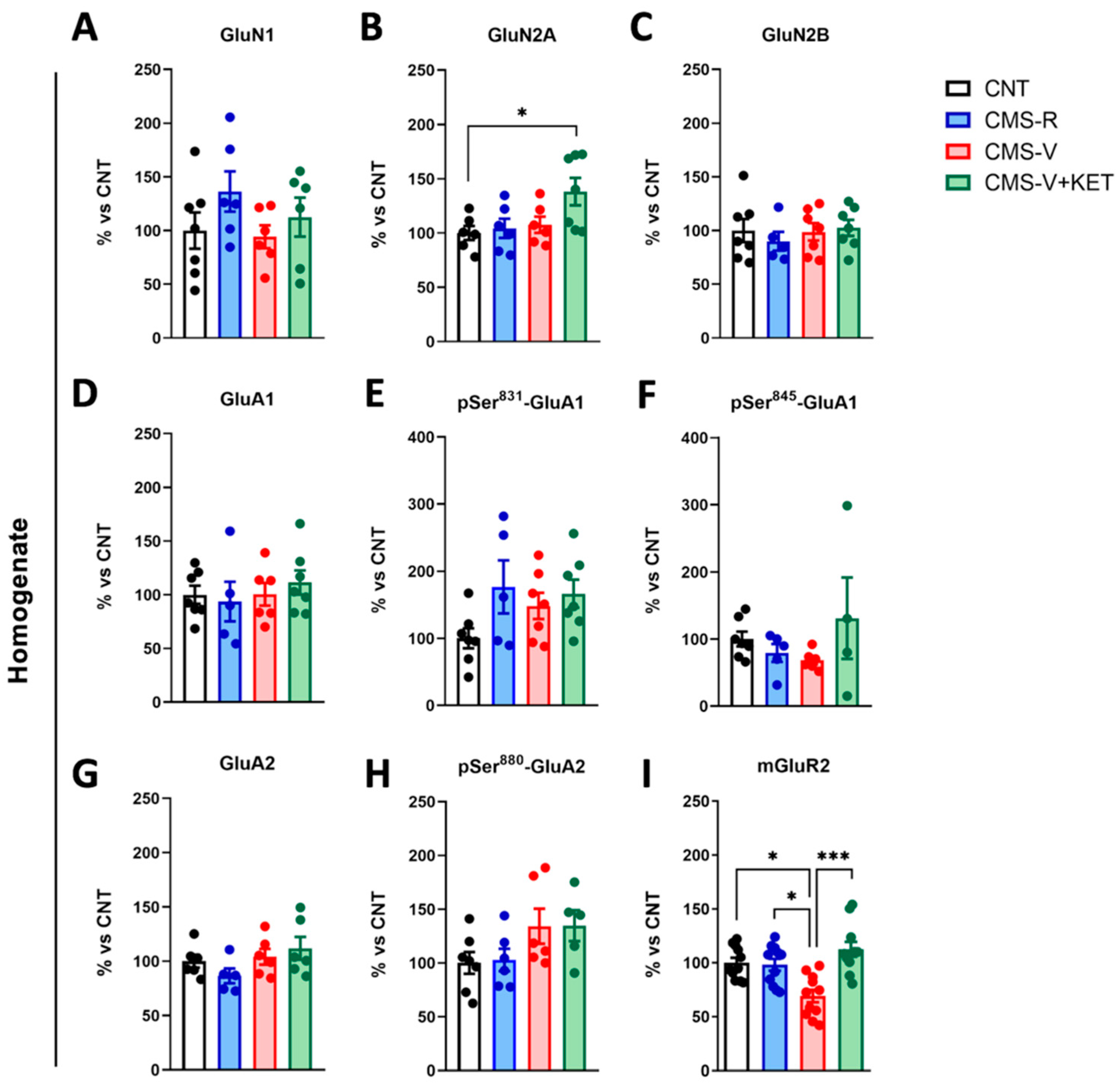
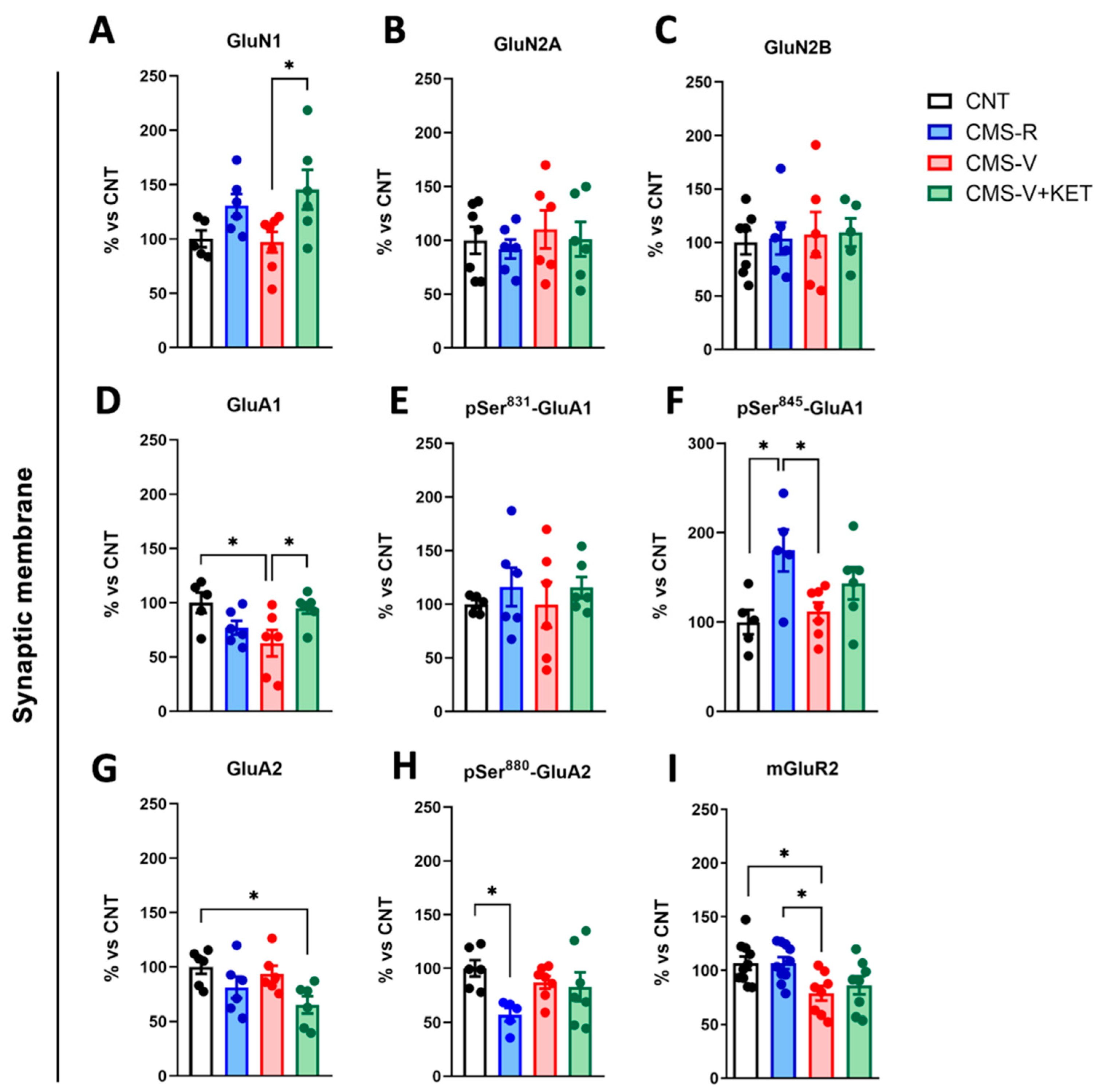
2.3. Modulation of AMPA, NMDA, and mGluR2 Glutamate Receptors Induced by Stress and Ketamine in the Medial Prefrontal Cortex of Rats
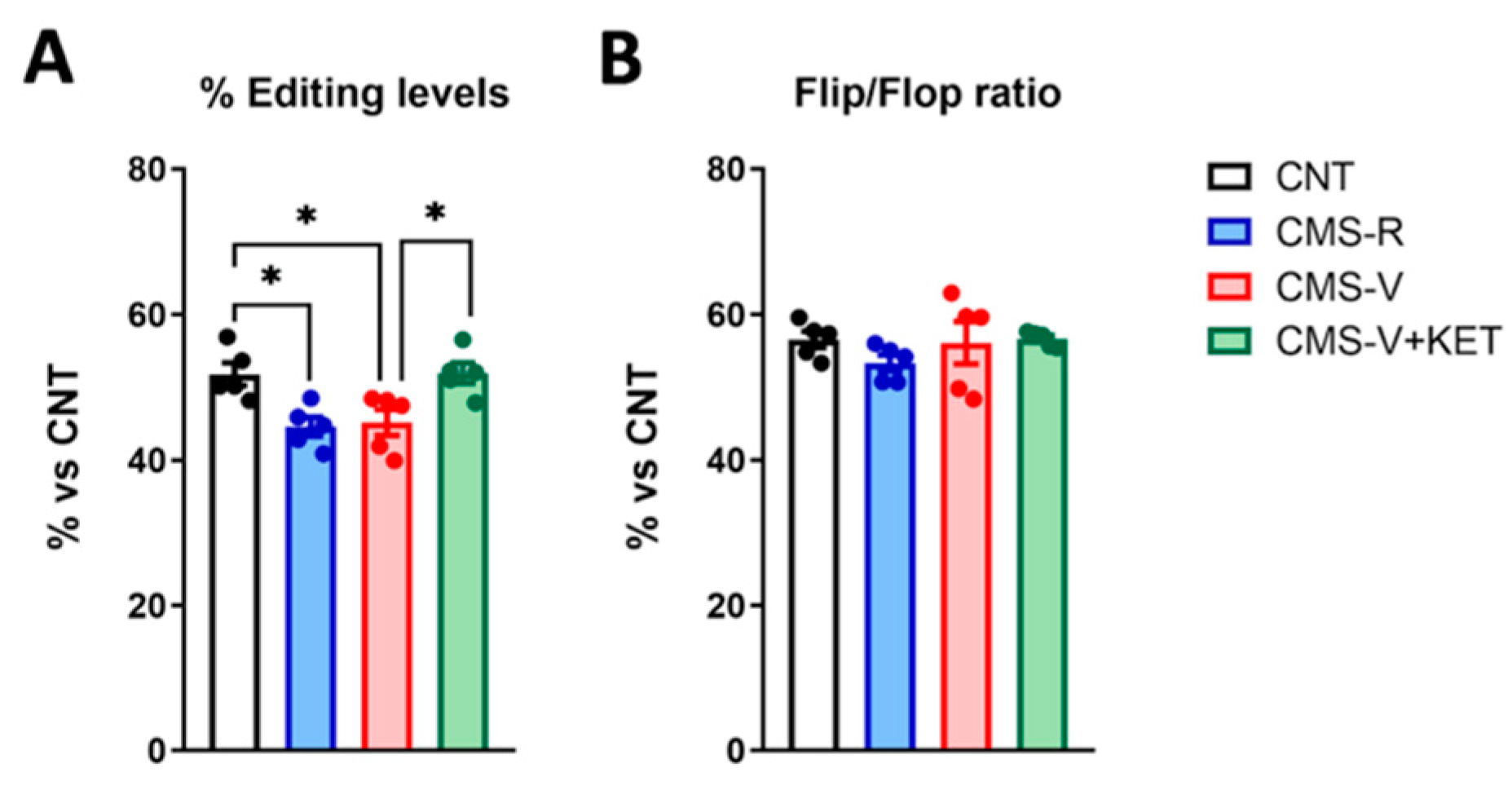
2.4. GluA2 R/G Editing but Not Flip/Flop Splicing Is Reduced in the Medial Prefrontal Cortex of Stressed Rats and Rescued by Ketamine Treatment
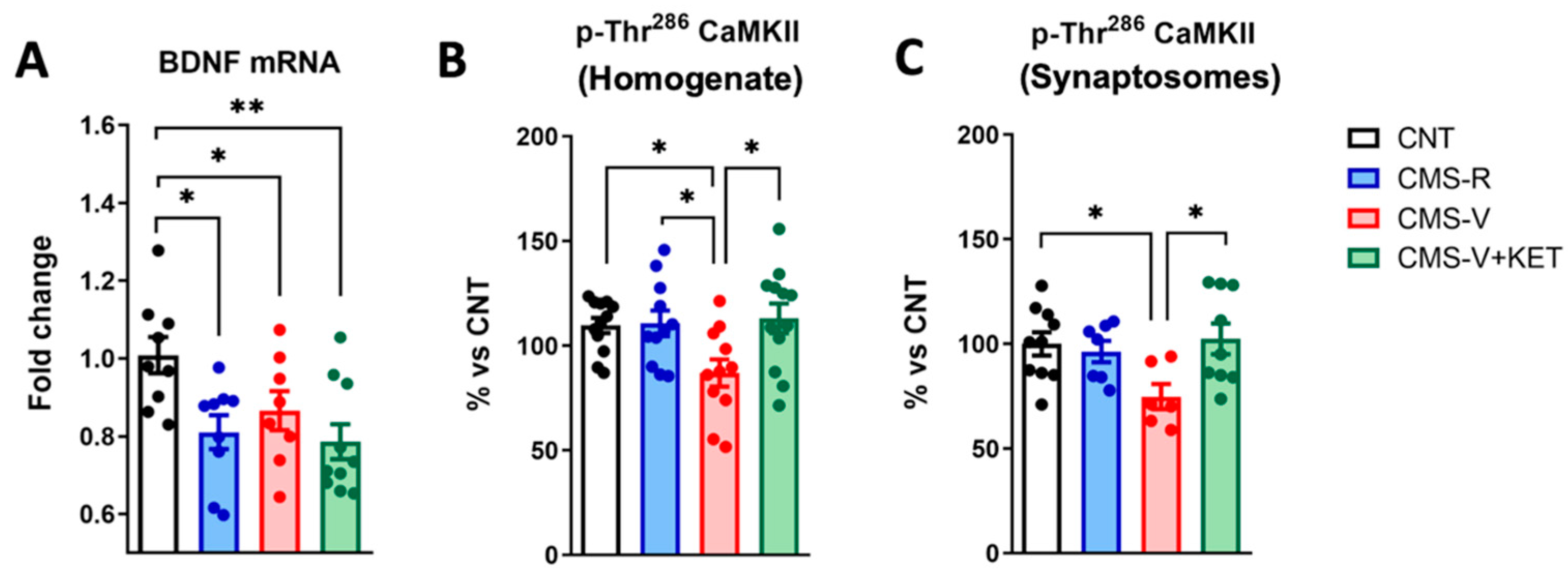
2.5. Ketamine Rescues the Stress-Dependent Reduction in CaM Kinase II Phosphorylation but Not Bdnf mRNA Expression in the Medial Prefrontal Cortex of Rats
3. Discussion
4. Materials and Methods
4.1. Animals
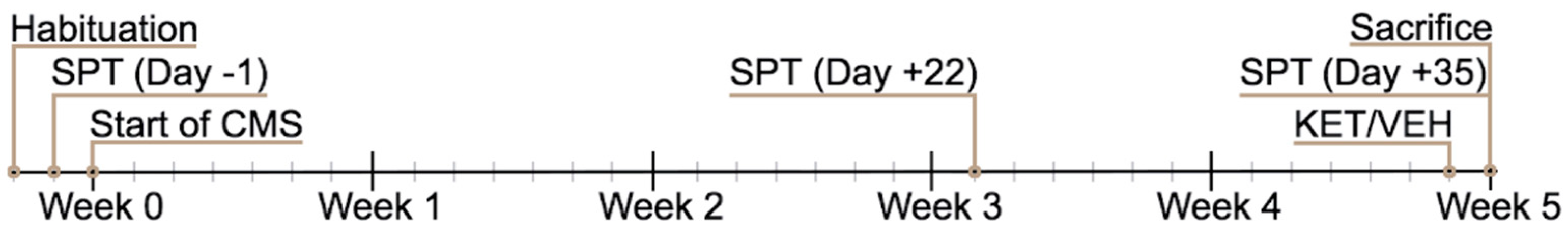
4.2. Chronic Mild Stress (CMS) Paradigm
4.3. Sucrose Preference Test
4.4. Preparation of Subcellular Fractions
4.5. Measurement of Neurotransmitter Release from Purified Synaptosomes
4.6. Western Blotting
4.7. RNA Isolation, Reverse Transcription, Real-Time PCR, RNA Editing, and Splicing Analysis
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McEwen, B.S.; Bowles, N.P.; Gray, J.D.; Hill, M.N.; Hunter, R.G.; Karatsoreos, I.N.; Nasca, C. Mechanisms of Stress in the Brain. Nat. Neurosci. 2015, 18, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Sanacora, G.; Yan, Z.; Popoli, M. The Stressed Synapse 2.0: Pathophysiological Mechanisms in Stress-Related Neuropsychiatric Disorders. Nat. Rev. Neurosci. 2022, 23, 86–103. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Nasca, C.; Gray, J.D. Stress Effects on Neuronal Structure: Hippocampus, Amygdala, and Prefrontal Cortex. Neuropsychopharmacology 2016, 41, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Han, M.-H.; Nestler, E.J. Neural Substrates of Depression and Resilience. Neurotherapeutics 2017, 14, 677–686. [Google Scholar] [CrossRef]
- Drevets, W.C.; Price, J.L.; Furey, M.L. Brain Structural and Functional Abnormalities in Mood Disorders: Implications for Neurocircuitry Models of Depression. Brain Struct. Funct. 2008, 213, 93–118. [Google Scholar] [CrossRef] [PubMed]
- Sheline, Y.I.; Liston, C.; McEwen, B.S. Parsing the Hippocampus in Depression: Chronic Stress, Hippocampal Volume, and Major Depressive Disorder. Biol. Psychiatry 2019, 85, 436–438. [Google Scholar] [CrossRef]
- Qiao, H.; Li, M.X.; Xu, C.; Chen, H.B.; An, S.C.; Ma, X.M. Dendritic Spines in Depression: What We Learned from Animal Models. Neural Plast. 2016, 2016, 8056370. [Google Scholar] [CrossRef]
- Krishnan, V.; Nestler, E.J. The Molecular Neurobiology of Depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef]
- Mrazek, D.A.; Hornberger, J.C.; Altar, C.A.; Degtiar, I. A Review of the Clinical, Economic, and Societal Burden of Treatment-Resistant Depression: 1996–2013. Psychiatr. Serv. 2014, 65, 977–987. [Google Scholar] [CrossRef]
- Johnston, J.N.; Henter, I.D.; Zarate, C.A. The Antidepressant Actions of Ketamine and Its Enantiomers. Pharmacol. Ther. 2023, 246, 108431. [Google Scholar] [CrossRef]
- Zanos, P.; Gould, T.D. Mechanisms of Ketamine Action as an Antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Kadriu, B.; Musazzi, L.; Henter, I.D.; Graves, M.; Popoli, M.; Zarate, C.A. Glutamatergic Neurotransmission: Pathway to Developing Novel Rapid-Acting Antidepressant Treatments. Int. J. Neuropsychopharmacol. 2019, 22, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Suzuki, K.; Kavalali, E.T.; Monteggia, L.M. Bridging Rapid and Sustained Antidepressant Effects of Ketamine. Trends Mol. Med. 2023, 29, 364–375. [Google Scholar] [CrossRef]
- Kim, J.; Farchione, T.; Potter, A.; Chen, Q.; Temple, R. Esketamine for Treatment-Resistant Depression—First FDA-Approved Antidepressant in a New Class. N. Engl. J. Med. 2019, 381, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tornese, P.; Sala, N.; Bonini, D.; Bonifacino, T.; La Via, L.; Milanese, M.; Treccani, G.; Seguini, M.; Ieraci, A.; Mingardi, J.; et al. Chronic Mild Stress Induces Anhedonic Behavior and Changes in Glutamate Release, BDNF Trafficking and Dendrite Morphology Only in Stress Vulnerable Rats. The Rapid Restorative Action of Ketamine. Neurobiol. Stress 2019, 10, 100160. [Google Scholar] [CrossRef]
- Elhussiny, M.E.A.; Carini, G.; Mingardi, J.; Tornese, P.; Sala, N.; Bono, F.; Fiorentini, C.; La Via, L.; Popoli, M.; Musazzi, L.; et al. Modulation by Chronic Stress and Ketamine of Ionotropic AMPA/NMDA and Metabotropic Glutamate Receptors in the Rat Hippocampus. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 104, 110033. [Google Scholar] [CrossRef]
- Mingardi, J.; La Via, L.; Tornese, P.; Carini, G.; Trontti, K.; Seguini, M.; Tardito, D.; Bono, F.; Fiorentini, C.; Elia, L.; et al. MiR-9-5p Is Involved in the Rescue of Stress-Dependent Dendritic Shortening of Hippocampal Pyramidal Neurons Induced by Acute Antidepressant Treatment with Ketamine. Neurobiol. Stress 2021, 15, 100381. [Google Scholar] [CrossRef]
- Mingardi, J.; Paoli, C.; La Via, L.; Carini, G.; Misztak, P.; Cifani, C.; Popoli, M.; Barbon, A.; Musazzi, L. Involvement of MiR-135a-5p Downregulation in Acute and Chronic Stress Response in the Prefrontal Cortex of Rats. Int. J. Mol. Sci. 2023, 24, 1552. [Google Scholar] [CrossRef]
- Arnsten, A.F.T. Stress Weakens Prefrontal Networks: Molecular Insults to Higher Cognition. Nat. Neurosci. 2015, 18, 1376–1385. [Google Scholar] [CrossRef]
- Jiang, Y.; Zou, M.; Ren, T.; Wang, Y. Are MGluR2/3 Inhibitors Potential Compounds for Novel Antidepressants? Cell. Mol. Neurobiol. 2022, 43, 1931–1940. [Google Scholar] [CrossRef]
- Musazzi, L. Targeting Metabotropic Glutamate Receptors for Rapid-Acting Antidepressant Drug Discovery. Expert Opin. Drug Discov. 2021, 16, 147–157. [Google Scholar] [CrossRef]
- Wang, J.Q.; Guo, M.-L.; Jin, D.-Z.; Xue, B.; Fibuch, E.E.; Mao, L.-M. Roles of Subunit Phosphorylation in Regulating Glutamate Receptor Function. Eur. J. Pharmacol. 2014, 728, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Diering, G.H.; Huganir, R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Filippini, A.; Bonini, D.; La Via, L.; Barbon, A. The Good and the Bad of Glutamate Receptor RNA Editing. Mol. Neurobiol. 2017, 54, 6795–6805. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor Signaling in Depression and Antidepressant Action. Biol. Psychiatry 2021, 90, 128–136. [Google Scholar] [CrossRef]
- Notaras, M.; van den Buuse, M. Neurobiology of BDNF in Fear Memory, Sensitivity to Stress, and Stress-Related Disorders. Mol. Psychiatry 2020, 25, 2251–2274. [Google Scholar] [CrossRef]
- Duman, R.S.; Deyama, S.; Fogaça, M.V. Role of BDNF in the Pathophysiology and Treatment of Depression: Activity-dependent Effects Distinguish Rapid-acting Antidepressants. Eur. J. Neurosci. 2021, 53, 126–139. [Google Scholar] [CrossRef]
- Sheng, M.; Thompson, M.A.; Greenberg, M.E. CREB: A Ca2+-Regulated Transcription Factor Phosphorylated by Calmodulin-Dependent Kinases. Science 1991, 252, 1427–1430. [Google Scholar] [CrossRef]
- Wang, Z.-W. Regulation of Synaptic Transmission by Presynaptic CaMKII and BK Channels. Mol. Neurobiol. 2008, 38, 153–166. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Riggs, L.M.; Highland, J.N.; Georgiou, P.; Pereira, E.F.R.; Albuquerque, E.X.; Thomas, C.J.; Zarate, C.A., Jr.; et al. Ketamine and Ketamine Metabolite Pharmacology: Insights into Therapeutic Mechanisms. Pharmacol. Rev. 2018, 70, 621–660. [Google Scholar] [CrossRef]
- Duman, R.S.; Sanacora, G.; Krystal, J.H. Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron 2019, 102, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.D.; Zarate, C.A.; Thompson, S.M. Molecular Pharmacology and Neurobiology of Rapid-Acting Antidepressants. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 213–236. [Google Scholar] [CrossRef] [PubMed]
- Forti, L.; Ndoj, E.; Mingardi, J.; Secchi, E.; Bonifacino, T.; Schiavon, E.; Carini, G.; La Via, L.; Russo, I.; Milanese, M.; et al. Dopamine-Dependent Ketamine Modulation of Glutamatergic Synaptic Plasticity in the Prelimbic Cortex of Adult Rats Exposed to Acute Stress. Int. J. Mol. Sci. 2023, 24, 8718. [Google Scholar] [CrossRef] [PubMed]
- Sala, N.; Paoli, C.; Bonifacino, T.; Mingardi, J.; Schiavon, E.; La Via, L.; Milanese, M.; Tornese, P.; Datusalia, A.K.; Rosa, J.; et al. Acute Ketamine Facilitates Fear Memory Extinction in a Rat Model of PTSD Along with Restoring Glutamatergic Alterations and Dendritic Atrophy in the Prefrontal Cortex. Front. Pharmacol. 2022, 13, 759626. [Google Scholar] [CrossRef] [PubMed]
- Kavalali, E.T.; Monteggia, L.M. Synaptic Mechanisms Underlying Rapid Antidepressant Action of Ketamine. Am. J. Psychiatry 2012, 169, 1150–1156. [Google Scholar] [CrossRef]
- Kim, J.-W.; Autry, A.E.; Na, E.S.; Adachi, M.; Björkholm, C.; Kavalali, E.T.; Monteggia, L.M. Sustained Effects of Rapidly Acting Antidepressants Require BDNF-Dependent MeCP2 Phosphorylation. Nat. Neurosci. 2021, 24, 1100–1109. [Google Scholar] [CrossRef]
- Ali, F.; Gerhard, D.M.; Sweasy, K.; Pothula, S.; Pittenger, C.; Duman, R.S.; Kwan, A.C. Ketamine Disinhibits Dendrites and Enhances Calcium Signals in Prefrontal Dendritic Spines. Nat. Commun. 2020, 11, 72. [Google Scholar] [CrossRef]
- Lin, P.-Y.; Ma, Z.Z.; Mahgoub, M.; Kavalali, E.T.; Monteggia, L.M. A Synaptic Locus for TrkB Signaling Underlying Ketamine Rapid Antidepressant Action. Cell Rep. 2021, 36, 109513. [Google Scholar] [CrossRef]
- Lazarevic, V.; Yang, Y.; Flais, I.; Svenningsson, P. Ketamine Decreases Neuronally Released Glutamate via Retrograde Stimulation of Presynaptic Adenosine A1 Receptors. Mol. Psychiatry 2021, 26, 7425–7435. [Google Scholar] [CrossRef]
- Lomeli, H.; Mosbacher, J.; Melcher, T.; Höger, T.; Geiger, J.R.P.; Kuner, T.; Monyer, H.; Higuchi, M.; Bach, A.; Seeburg, P.H. Control of Kinetic Properties of AMPA Receptor Channels by Nuclear RNA Editing. Science 1994, 266, 1709–1713. [Google Scholar] [CrossRef]
- Musazzi, L.; Treccani, G.; Popoli, M. Functional and Structural Remodeling of Glutamate Synapses in Prefrontal and Frontal Cortex Induced by Behavioral Stress. Front. Psychiatry 2015, 6, 60. [Google Scholar] [CrossRef]
- Monteggia, L.M.; Gideons, E.; Kavalali, E.T. The Role of Eukaryotic Elongation Factor 2 Kinase in Rapid Antidepressant Action of Ketamine. Biol. Psychiatry 2013, 73, 1199–1203. [Google Scholar] [CrossRef]
- Lu, W.; Roche, K.W. Posttranslational Regulation of AMPA Receptor Trafficking and Function. Curr. Opin. Neurobiol. 2012, 22, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Vissel, B. The Essential Role of AMPA Receptor GluR2 Subunit RNA Editing in the Normal and Diseased Brain. Front. Mol. Neurosci. 2012, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Sans, N.; Vissel, B.; Petralia, R.S.; Wang, Y.-X.; Chang, K.; Royle, G.A.; Wang, C.-Y.; O’Gorman, S.; Heinemann, S.F.; Wenthold, R.J. Aberrant Formation of Glutamate Receptor Complexes in Hippocampal Neurons of Mice Lacking the GluR2 AMPA Receptor Subunit. J. Neurosci. 2003, 23, 9367–9373. [Google Scholar] [CrossRef]
- Dick, A.L.W.; Khermesh, K.; Paul, E.; Stamp, F.; Levanon, E.Y.; Chen, A. Adenosine-to-Inosine RNA Editing Within Corticolimbic Brain Regions Is Regulated in Response to Chronic Social Defeat Stress in Mice. Front. Psychiatry 2019, 10, 277. [Google Scholar] [CrossRef]
- Cattaneo, A.; Riva, M.A. Stress-Induced Mechanisms in Mental Illness: A Role for Glucocorticoid Signalling. J. Steroid Biochem. Mol. Biol. 2016, 160, 169–174. [Google Scholar] [CrossRef]
- Anacker, C.; Zunszain, P.A.; Carvalho, L.A.; Pariante, C.M. The Glucocorticoid Receptor: Pivot of Depression and of Antidepressant Treatment? Psychoneuroendocrinology 2011, 36, 415–425. [Google Scholar] [CrossRef]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef]
- Musazzi, L.; Seguini, M.; Mallei, A.; Treccani, G.; Pelizzari, M.; Tornese, P.; Racagni, G.; Tardito, D. Time-Dependent Activation of MAPK/Erk1/2 and Akt/GSK3 Cascades: Modulation by Agomelatine. BMC Neurosci. 2014, 15, 119. [Google Scholar] [CrossRef] [PubMed]
- Treccani, G.; Musazzi, L.; Perego, C.; Milanese, M.; Nava, N.; Bonifacino, T.; Lamanna, J.; Malgaroli, A.; Drago, F.; Racagni, G.; et al. Stress and Corticosterone Increase the Readily Releasable Pool of Glutamate Vesicles in Synaptic Terminals of Prefrontal and Frontal Cortex. Mol. Psychiatry 2014, 19, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, M.; Bonanno, G.; Marchi, M.; Maura, G. Is There a Functional Linkage between Neurotransmitter Uptake Mechanisms and Presynaptic Receptors? J. Pharmacol. Exp. Ther. 1984, 231, 671–677. [Google Scholar] [PubMed]
- Bonanno, G.; Giambelli, R.; Raiteri, L.; Tiraboschi, E.; Zappettini, S.; Musazzi, L.; Raiteri, M.; Racagni, G.; Popoli, M. Chronic Antidepressants Reduce Depolarization-Evoked Glutamate Release and Protein Interactions Favoring Formation of SNARE Complex in Hippocampus. J. Neurosci. 2005, 25, 3270–3279. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, L.; Raiteri, M. Synaptosomes still viable after 25 years of superfusion. Neurochem. Res. 2000, 25, 1265–1274. [Google Scholar] [CrossRef]
- Raiteri, L.; Stigliani, S.; Zappettini, S.; Mercuri, N.B.; Raiteri, M.; Bonanno, G. Excessive and Precocious Glutamate Release in a Mouse Model of Amyotrophic Lateral Sclerosis. Neuropharmacology 2004, 46, 782–792. [Google Scholar] [CrossRef]
- Raiteri, M.; Angelini, F.; Levi, G. A Simple Apparatus for Studying the Release of Neurotransmitters from Synaptosomes. Eur. J. Pharmacol. 1974, 25, 411–414. [Google Scholar] [CrossRef]
- Raiteri, L.; Zappettini, S.; Milanese, M.; Fedele, E.; Raiteri, M.; Bonanno, G. Mechanisms of Glutamate Release Elicited in Rat Cerebrocortical Nerve Endings by ‘Pathologically’ Elevated Extraterminal K + Concentrations. J. Neurochem. 2007, 103, 952–961. [Google Scholar] [CrossRef]
- Raiteri, M.; Sala, R.; Fassio, A.; Rossetto, O.; Bonanno, G. Entrapping of Impermeant Probes of Different Size into Nonpermeabilized Synaptosomes as a Method to Study Presynaptic Mechanisms. J. Neurochem. 2001, 74, 423–431. [Google Scholar] [CrossRef]
- Musazzi, L.; Tornese, P.; Sala, N.; Popoli, M. Acute Stress Is Not Acute: Sustained Enhancement of Glutamate Release after Acute Stress Involves Readily Releasable Pool Size and Synapsin I Activation. Mol. Psychiatry 2017, 22, 1226–1227. [Google Scholar] [CrossRef]
- Orlandi, C.; La Via, L.; Bonini, D.; Mora, C.; Russo, I.; Barbon, A.; Barlati, S. AMPA Receptor Regulation at the MRNA and Protein Level in Rat Primary Cortical Cultures. PLoS ONE 2011, 6, e25350. [Google Scholar] [CrossRef]
- Bonini, D.; Filippini, A.; La Via, L.; Fiorentini, C.; Fumagalli, F.; Colombi, M.; Barbon, A. Chronic Glutamate Treatment Selectively Modulates AMPA RNA Editing and ADAR Expression and Activity in Primary Cortical Neurons. RNA Biol. 2015, 12, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, R.-J.; Dwyer, J.M.; Banasr, M.; Lee, B.; Son, H.; Li, X.-Y.; Aghajanian, G.; Duman, R.S. Glutamate N-Methyl-D-Aspartate Receptor Antagonists Rapidly Reverse Behavioral and Synaptic Deficits Caused by Chronic Stress Exposure. Biol. Psychiatry 2011, 69, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Lamanna, J.; Isotti, F.; Ferro, M.; Spadini, S.; Racchetti, G.; Musazzi, L.; Malgaroli, A. Occlusion of Dopamine-Dependent Synaptic Plasticity in the Prefrontal Cortex Mediates the Expression of Depressive-like Behavior and Is Modulated by Ketamine. Sci. Rep. 2022, 12, 11055. [Google Scholar] [CrossRef]
- Ghosal, S.; Duman, C.H.; Liu, R.-J.; Wu, M.; Terwilliger, R.; Girgenti, M.J.; Wohleb, E.; Fogaca, M.V.; Teichman, E.M.; Hare, B.; et al. Ketamine Rapidly Reverses Stress-Induced Impairments in GABAergic Transmission in the Prefrontal Cortex in Male Rodents. Neurobiol. Dis. 2020, 134, 104669. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CMS-R | CMS-V | CMS-V+KET | |
|---|---|---|---|
| Depolarization-evoked glutamate release | = | = (↑ vs. CMS-R) | = (↓ vs. CMS-V) |
| pSer232-GR (homogenate) | = | = | ↑ |
| GluN2A (homogenate) | = | = | ↑ |
| mGluR2 (homogenate) | = | ↓ | = (↑ vs. CMS-V) |
| mGluR2 (synaptic membranes) | = | ↓ | = |
| GluN1 (synaptic membranes) | = | = | = (↑ vs. CMS-V) |
| GluA1 (synaptic membranes) | = | ↓ | = (↑ vs. CMS-V) |
| pSer845-GluA1 (synaptic membranes) | ↑ | = (↓ vs. CMS-R) | = |
| GluA2 (synaptic membranes) | = | = | ↓ |
| pSer880-GluA2 (synaptic membranes) | ↓ | = | = |
| pThr286-CaM kinase II (homogenate and synaptosomes) | = | ↓ | = (↑ vs. CMS-V) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mingardi, J.; Ndoj, E.; Bonifacino, T.; Misztak, P.; Bertoli, M.; La Via, L.; Torazza, C.; Russo, I.; Milanese, M.; Bonanno, G.; et al. Functional and Molecular Changes in the Prefrontal Cortex of the Chronic Mild Stress Rat Model of Depression and Modulation by Acute Ketamine. Int. J. Mol. Sci. 2023, 24, 10814. https://doi.org/10.3390/ijms241310814
Mingardi J, Ndoj E, Bonifacino T, Misztak P, Bertoli M, La Via L, Torazza C, Russo I, Milanese M, Bonanno G, et al. Functional and Molecular Changes in the Prefrontal Cortex of the Chronic Mild Stress Rat Model of Depression and Modulation by Acute Ketamine. International Journal of Molecular Sciences. 2023; 24(13):10814. https://doi.org/10.3390/ijms241310814
Chicago/Turabian StyleMingardi, Jessica, Elona Ndoj, Tiziana Bonifacino, Paulina Misztak, Matteo Bertoli, Luca La Via, Carola Torazza, Isabella Russo, Marco Milanese, Giambattista Bonanno, and et al. 2023. "Functional and Molecular Changes in the Prefrontal Cortex of the Chronic Mild Stress Rat Model of Depression and Modulation by Acute Ketamine" International Journal of Molecular Sciences 24, no. 13: 10814. https://doi.org/10.3390/ijms241310814
APA StyleMingardi, J., Ndoj, E., Bonifacino, T., Misztak, P., Bertoli, M., La Via, L., Torazza, C., Russo, I., Milanese, M., Bonanno, G., Popoli, M., Barbon, A., & Musazzi, L. (2023). Functional and Molecular Changes in the Prefrontal Cortex of the Chronic Mild Stress Rat Model of Depression and Modulation by Acute Ketamine. International Journal of Molecular Sciences, 24(13), 10814. https://doi.org/10.3390/ijms241310814