
Neutrophils’ Contribution to Periodontitis and Periodontitis-Associated Cardiovascular Diseases
 , ,
, ,  , , ,
, , ,  ,
,  , and
, and {kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Introduction to the Biology of Neutrophils
1.2. Neutrophil Homeostasis and Activation
1.3. Neutrophil Extracellular Traps (NETs)
1.4. NETs in Diseases
1.5. Neutrophils and Trained Immunity
2. Periodontitis
3. Neutrophils, Oral Cavity, and Periodontitis
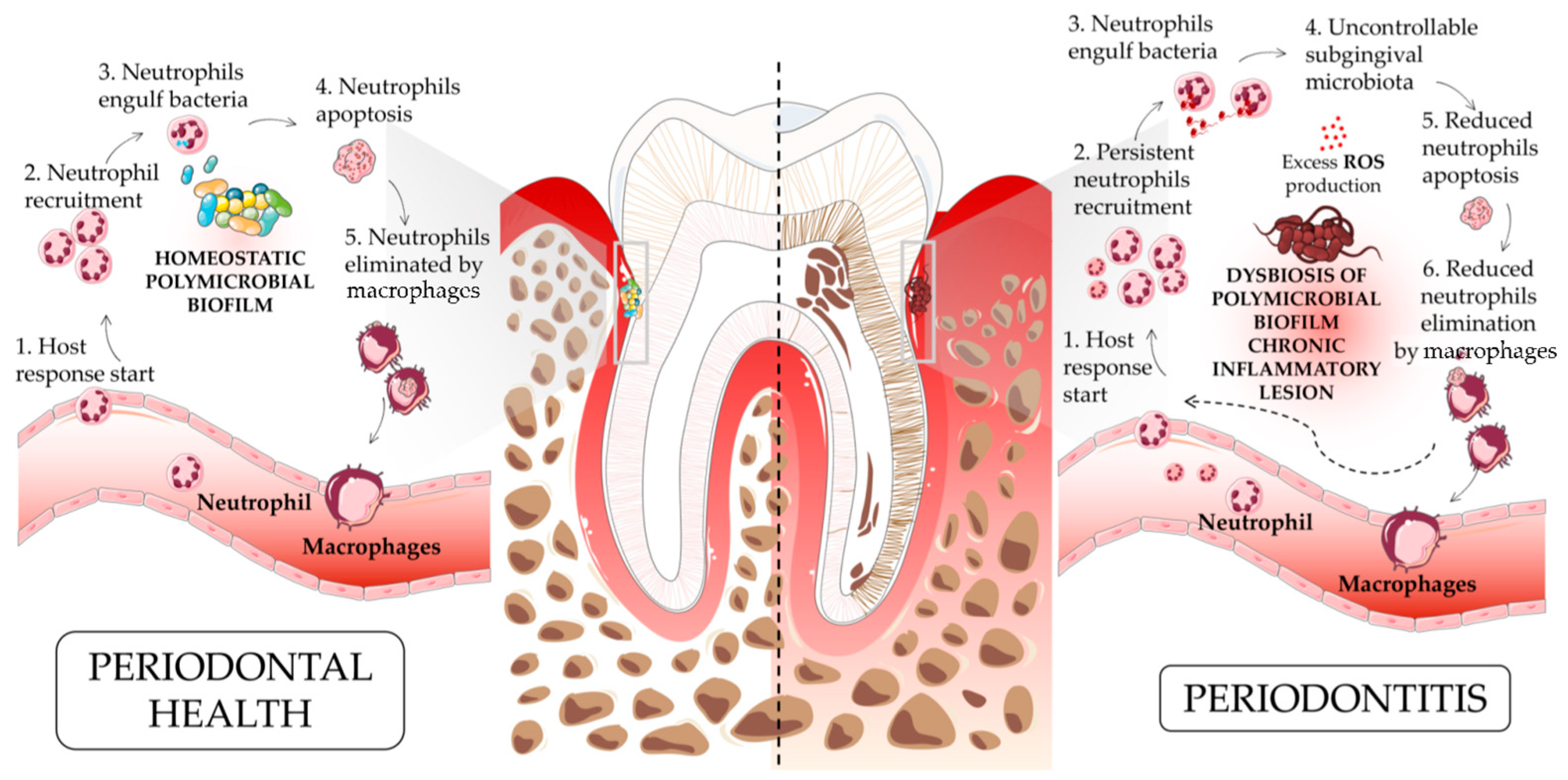
3.1. Neutrophils and Periodontitis
3.2. Neutrophil Extracellular Traps and Periodontitis
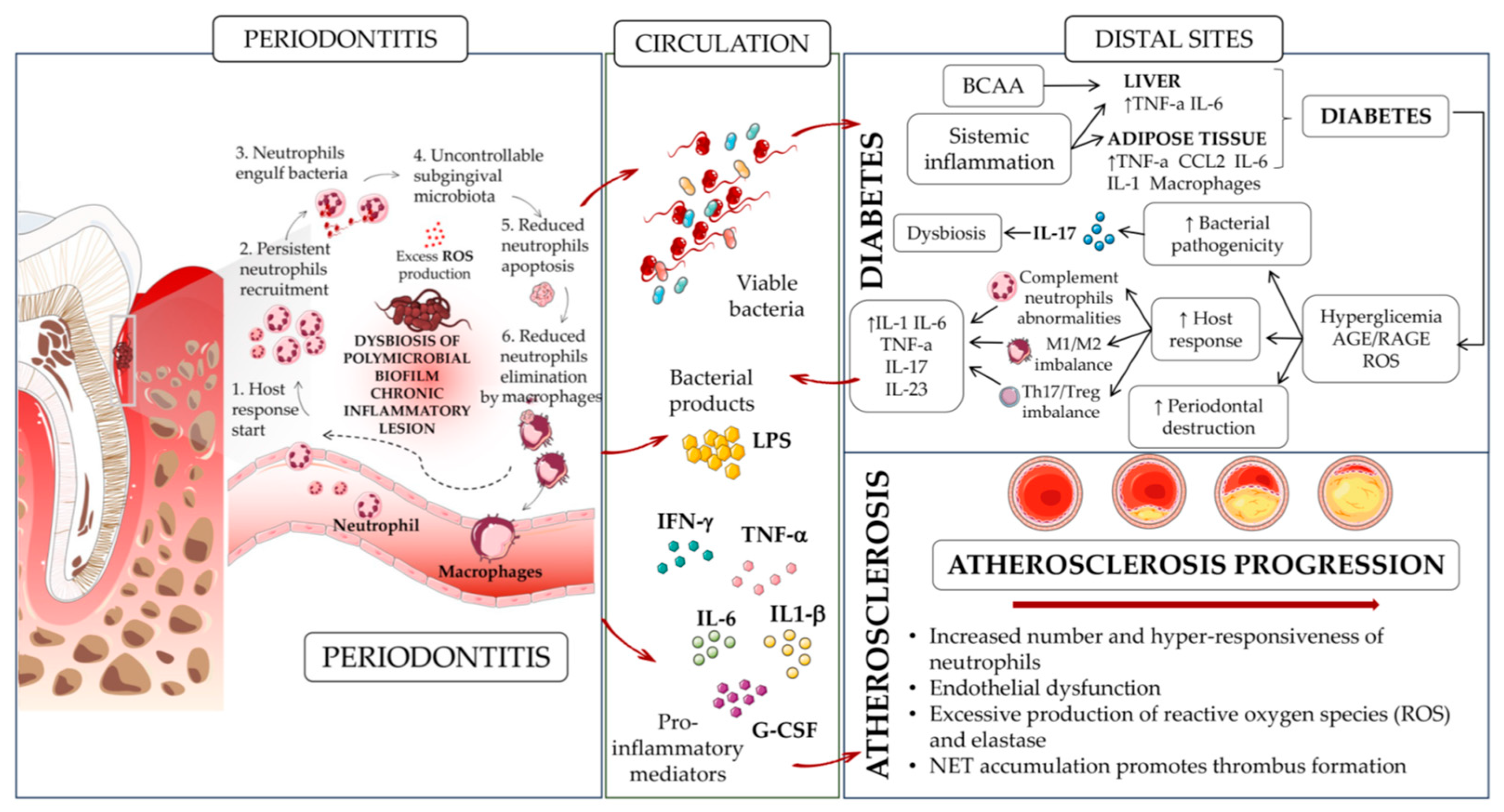
4. Neutrophils, Periodontitis, and Other Comorbidities
4.1. Diabetes
4.2. Atherosclerosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGEs | Advanced Glycation End products |
| ASCVD | AtheroSclerotic CardioVascular Disease |
| BCAA | Branched-Chain Amino Acid |
| BM | Bone Marrow |
| C/EBPα | CCAAT Enhancer-Binding Protein alpha |
| CMPs | Common Myeloid Progenitors |
| CRP | C-Reactive Protein |
| CXCL- | Chemokine (C-X-C motif) Ligand |
| CVD | Cardiovascular disease |
| fMLP | N-Formylmethionyl-leucyl-phenylalanine |
| G-CSF | Granulocyte Colony-Stimulating Factor |
| GFs | Gingival Fibroblasts |
| GMPs | Granulocyte-Monocyte Precursors |
| HFD | High-Fat Diet |
| HSC | Hematopoietic Stem Cell |
| IFN-γ | Interferon-γ |
| IL- | Interleukin- |
| LIP | Ligature-Induced Periodontitis |
| LMPs | Lymphoid and Myeloid Precursors |
| LPS | LypoPolySaccaride |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| MMPs | Matrix MetalloProteinases |
| MPO | MyeloPerOxidase |
| MPPs | MultiPotent Precursors |
| NADP-H | Nicotinamide Adenine Dinucleotide Phosphate-H |
| NE | Neutrophil Elastase |
| NETs | Neutrophil Extracellular Traps |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| PI3K/AKT | Phosphoinositide 3-Kinase Inhibitors |
| PMA | Phorbol 12-Myristate 13-Acetate |
| PRRs | Pattern Recognition Receptors |
| RAGE | Receptor for AGE |
| ROS | Reactive Oxygen Species |
| SMIRKO | Smooth Muscle Insulin Receptor Knock Out |
| TLRs | Toll-like receptors |
| TNF-α | Tumor Necrosis Factor-α |
| T2DM | Type 2 Diabetes Morbidity |
References
- Palomino-Segura, M.; Sicilia, J.; Ballesteros, I.; Hidalgo, A. Strategies of neutrophil diversification. Nat. Immunol. 2023, 24, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.G.; Ostuni, R.; Hidalgo, A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 2019, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M.; Borregaard, N. Neutrophils at work. Nat. Immunol. 2014, 15, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Trumpp, A.; Milsom, M.D. Causes and Consequences of Hematopoietic Stem Cell Heterogeneity. Cell Stem Cell 2018, 22, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.K.; Liu, F.; Iwasaki, H.; Akashi, K.; Link, D.C. Pivotal role of granulocyte colony-stimulating factor in the development of progenitors in the common myeloid pathway. Blood 2003, 102, 3562–3568. [Google Scholar] [CrossRef] [PubMed]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e368. [Google Scholar] [CrossRef]
- Hedrick, C.C.; Malanchi, I. Neutrophils in cancer: Heterogeneous and multifaceted. Nat. Rev. Immunol. 2022, 22, 173–187. [Google Scholar] [CrossRef]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef]
- Sundd, P.; Pospieszalska, M.K.; Ley, K. Neutrophil rolling at high shear: Flattening, catch bond behavior, tethers and slings. Mol. Immunol. 2013, 55, 59–69. [Google Scholar] [CrossRef]
- Filippi, M.D. Mechanism of Diapedesis: Importance of the Transcellular Route. Adv. Immunol. 2016, 129, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Mathias, J.R.; Perrin, B.J.; Liu, T.X.; Kanki, J.; Look, A.T.; Huttenlocher, A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J. Leukoc. Biol. 2006, 80, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhao, W.; Yan, M.; Mei, H. Neutrophil reverse migration. J. Inflamm. 2022, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.; Rieber, N.; Radsak, M.; von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015, 11, e1004651. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Knackstedt, S.L.; Abu Abed, U.; Deigendesch, N.; Harbort, C.J.; Caffrey, B.E.; Brinkmann, V.; Heppner, F.L.; Hinds, P.W.; Zychlinsky, A. Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev. Cell 2017, 43, 449–462.e445. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Price, C.P.; Newall, R.G.; Boyd, J.C. Use of protein:creatinine ratio measurements on random urine samples for prediction of significant proteinuria: A systematic review. Clin. Chem. 2005, 51, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Amini, P.; Stojkov, D.; Felser, A.; Jackson, C.B.; Courage, C.; Schaller, A.; Gelman, L.; Soriano, M.E.; Nuoffer, J.M.; Scorrano, L.; et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat. Commun. 2018, 9, 2958. [Google Scholar] [CrossRef]
- Cheng, O.Z.; Palaniyar, N. NET balancing: A problem in inflammatory lung diseases. Front. Immunol. 2013, 4, 1. [Google Scholar] [CrossRef]
- Wang, M.; Ishikawa, T.; Lai, Y.; Nallapothula, D.; Singh, R.R. Diverse Roles of NETosis in the Pathogenesis of Lupus. Front. Immunol. 2022, 13, 895216. [Google Scholar] [CrossRef]
- Song, W.; Ye, J.; Pan, N.; Tan, C.; Herrmann, M. Neutrophil Extracellular Traps Tied to Rheumatoid Arthritis: Points to Ponder. Front. Immunol. 2020, 11, 578129. [Google Scholar] [CrossRef] [PubMed]
- Denorme, F.; Portier, I.; Rustad, J.L.; Cody, M.J.; de Araujo, C.V.; Hoki, C.; Alexander, M.D.; Grandhi, R.; Dyer, M.R.; Neal, M.D.; et al. Neutrophil extracellular traps regulate ischemic stroke brain injury. J. Clin. Investig. 2022, 132, e154225. [Google Scholar] [CrossRef] [PubMed]
- Millrud, C.R.; Kagedal, A.; Kumlien Georen, S.; Winqvist, O.; Uddman, R.; Razavi, R.; Munck-Wikland, E.; Cardell, L.O. NET-producing CD16(high) CD62L(dim) neutrophils migrate to tumor sites and predict improved survival in patients with HNSCC. Int. J. Cancer 2017, 140, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, H.; Tan, S.; Dong, Q.; Fan, X.; Wang, Y.; Zhang, H.; He, J. The role of neutrophil extracellular traps in cancer progression, metastasis and therapy. Exp. Hematol. Oncol. 2022, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- De Meo, M.L.; Spicer, J.D. The role of neutrophil extracellular traps in cancer progression and metastasis. Semin. Immunol. 2021, 57, 101595. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Kuttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, 4227. [Google Scholar] [CrossRef]
- Hirschfeld, J.; Dommisch, H.; Skora, P.; Horvath, G.; Latz, E.; Hoerauf, A.; Waller, T.; Kawai, T.; Jepsen, S.; Deschner, J.; et al. Neutrophil extracellular trap formation in supragingival biofilms. Int. J. Med. Microbiol. 2015, 305, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, J.; White, P.C.; Milward, M.R.; Cooper, P.R.; Chapple, I.L.C. Modulation of Neutrophil Extracellular Trap and Reactive Oxygen Species Release by Periodontal Bacteria. Infect. Immun. 2017, 85, e00297-17. [Google Scholar] [CrossRef]
- Silva, L.M.; Doyle, A.D.; Greenwell-Wild, T.; Dutzan, N.; Tran, C.L.; Abusleme, L.; Juang, L.J.; Leung, J.; Chun, E.M.; Lum, A.G.; et al. Fibrin is a critical regulator of neutrophil effector function at the oral mucosal barrier. Science 2021, 374, eabl5450. [Google Scholar] [CrossRef]
- Kim, T.S.; Silva, L.M.; Theofilou, V.I.; Greenwell-Wild, T.; Li, L.; Williams, D.W.; Ikeuchi, T.; Brenchley, L.; Genomics, N.N.; Computational Biology Core; et al. Neutrophil extracellular traps and extracellular histones potentiate IL-17 inflammation in periodontitis. J. Exp. Med. 2023, 220, e20221751. [Google Scholar] [CrossRef]
- Papayannopoulos, V. NET histones inflame periodontitis. J. Exp. Med. 2023, 220, e20230783. [Google Scholar] [CrossRef] [PubMed]
- Kalafati, L.; Hatzioannou, A.; Hajishengallis, G.; Chavakis, T. The role of neutrophils in trained immunity. Immunol. Rev. 2023, 314, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Quintin, J.; van der Meer, J.W. Trained immunity: A memory for innate host defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Joosten, L.A.; Ifrim, D.C.; Saeed, S.; Jacobs, C.; van Loenhout, J.; de Jong, D.; Stunnenberg, H.G.; et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 17537–17542. [Google Scholar] [CrossRef] [PubMed]
- Kalafati, L.; Kourtzelis, I.; Schulte-Schrepping, J.; Li, X.; Hatzioannou, A.; Grinenko, T.; Hagag, E.; Sinha, A.; Has, C.; Dietz, S.; et al. Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell 2020, 183, 771–785.e712. [Google Scholar] [CrossRef] [PubMed]
- Moorlag, S.; Rodriguez-Rosales, Y.A.; Gillard, J.; Fanucchi, S.; Theunissen, K.; Novakovic, B.; de Bont, C.M.; Negishi, Y.; Fok, E.T.; Kalafati, L.; et al. BCG Vaccination Induces Long-Term Functional Reprogramming of Human Neutrophils. Cell Rep. 2020, 33, 108387. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Li, X.; Divaris, K.; Chavakis, T. Maladaptive trained immunity and clonal hematopoiesis as potential mechanistic links between periodontitis and inflammatory comorbidities. Periodontology 2000 2022, 89, 215–230. [Google Scholar] [CrossRef]
- Ishai, A.; Osborne, M.T.; El Kholy, K.; Takx, R.A.P.; Ali, A.; Yuan, N.; Hsue, P.; Van Dyke, T.E.; Tawakol, A. Periodontal Disease Associates With Arterial Inflammation Via Potentiation of a Hematopoietic-Arterial Axis. JACC Cardiovasc. Imaging 2019, 12, 2271–2273. [Google Scholar] [CrossRef]
- Fine, N.; Chadwick, J.W.; Sun, C.; Parbhakar, K.K.; Khoury, N.; Barbour, A.; Goldberg, M.; Tenenbaum, H.C.; Glogauer, M. Periodontal Inflammation Primes the Systemic Innate Immune Response. J. Dent. Res. 2021, 100, 318–325. [Google Scholar] [CrossRef]
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal diseases. Nat. Rev. Dis. Primers 2017, 3, 17038. [Google Scholar] [CrossRef]
- Schatzle, M.; Loe, H.; Lang, N.P.; Burgin, W.; Anerud, A.; Boysen, H. The clinical course of chronic periodontitis. J. Clin. Periodontol. 2004, 31, 1122–1127. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T.; Lambris, J.D. Current understanding of periodontal disease pathogenesis and targets for host-modulation therapy. Periodontology 2000 2020, 84, 14–34. [Google Scholar] [CrossRef] [PubMed]
- Gorr, S.U.; Abdolhosseini, M. Antimicrobial peptides and periodontal disease. J. Clin. Periodontol. 2011, 38 (Suppl. S11), 126–141. [Google Scholar] [CrossRef] [PubMed]
- Genco, R.J.; Sanz, M. Clinical and public health implications of periodontal and systemic diseases: An overview. Periodontology 2000 2020, 83, 7–13. [Google Scholar] [CrossRef]
- Potempa, J.; Mydel, P.; Koziel, J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 606–620. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef]
- Acharya, C.; Sahingur, S.E.; Bajaj, J.S. Microbiota, cirrhosis, and the emerging oral-gut-liver axis. JCI Insight 2017, 2, e94416. [Google Scholar] [CrossRef] [PubMed]
- Schenkein, H.A.; Papapanou, P.N.; Genco, R.; Sanz, M. Mechanisms underlying the association between periodontitis and atherosclerotic disease. Periodontology 2000 2020, 83, 90–106. [Google Scholar] [CrossRef]
- Irani, S.; Barati, I.; Badiei, M. Periodontitis and oral cancer—current concepts of the etiopathogenesis. Oncol. Rev. 2020, 14, 465. [Google Scholar] [CrossRef]
- Migliorati, C.A. Periodontal diseases and cancer. Lancet Oncol. 2008, 9, 510–512. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.; Bassani, B.; Tremolati, M.; Gini, E.; Farronato, G.; Bruno, A. Natural Killer Cells in the Orchestration of Chronic Inflammatory Diseases. J. Immunol. Res. 2017, 2017, 4218254. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, M.; Hernandez-Lemus, E. Periodontal Inflammation and Systemic Diseases: An Overview. Front. Physiol. 2021, 12, 709438. [Google Scholar] [CrossRef] [PubMed]
- Cecoro, G.; Annunziata, M.; Iuorio, M.T.; Nastri, L.; Guida, L. Periodontitis, Low-Grade Inflammation and Systemic Health: A Scoping Review. Medicina 2020, 56, 272. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Marco Del Castillo, A.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and cardiovascular diseases: Consensus report. J. Clin. Periodontol. 2020, 47, 268–288. [Google Scholar] [CrossRef]
- Priyamvara, A.; Dey, A.K.; Bandyopadhyay, D.; Katikineni, V.; Zaghlol, R.; Basyal, B.; Barssoum, K.; Amarin, R.; Bhatt, D.L.; Lavie, C.J. Periodontal Inflammation and the Risk of Cardiovascular Disease. Curr. Atheroscler. Rep. 2020, 22, 28. [Google Scholar] [CrossRef]
- Curtis, M.A.; Zenobia, C.; Darveau, R.P. The relationship of the oral microbiotia to periodontal health and disease. Cell Host Microbe 2011, 10, 302–306. [Google Scholar] [CrossRef]
- Dutzan, N.; Konkel, J.E.; Greenwell-Wild, T.; Moutsopoulos, N.M. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol. 2016, 9, 1163–1172. [Google Scholar] [CrossRef]
- Suzuki, T.; Sugita, N.; Yoshie, H.; Hara, K. Presence of activated eosinophils, high IgE and sCD23 titers in gingival crevicular fluid of patients with adult periodontitis. J. Periodontal. Res. 1995, 30, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Barros, S.P.; Williams, R.; Offenbacher, S.; Morelli, T. Gingival crevicular fluid as a source of biomarkers for periodontitis. Periodontology 2000 2016, 70, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Botelho, J.; Machado, V.; Hussain, S.B.; Zehra, S.A.; Proenca, L.; Orlandi, M.; Mendes, J.J.; D’Aiuto, F. Periodontitis and circulating blood cell profiles: A systematic review and meta-analysis. Exp. Hematol. 2021, 93, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sansores-Espana, L.D.; Melgar-Rodriguez, S.; Vernal, R.; Carrillo-Avila, B.A.; Martinez-Aguilar, V.M.; Diaz-Zuniga, J. Neutrophil N1 and N2 Subsets and Their Possible Association with Periodontitis: A Scoping Review. Int. J. Mol. Sci. 2022, 23, 12068. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.B.; Wright, H.J.; Roberts, A.; Cooper, P.R.; Chapple, I.L. Hyperactivity and reactivity of peripheral blood neutrophils in chronic periodontitis. Clin. Exp. Immunol. 2007, 147, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Dominguez-Andres, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, E.; Sanz, J.; Dunn, J.L.; Khan, N.; Mendonca, L.E.; Pacis, A.; Tzelepis, F.; Pernet, E.; Dumaine, A.; Grenier, J.C.; et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 2018, 172, 176–190.e119. [Google Scholar] [CrossRef] [PubMed]
- Welsh, C.; Welsh, P.; Mark, P.B.; Celis-Morales, C.A.; Lewsey, J.; Gray, S.R.; Lyall, D.M.; Iliodromiti, S.; Gill, J.M.R.; Pell, J.; et al. Association of Total and Differential Leukocyte Counts With Cardiovascular Disease and Mortality in the UK Biobank. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1415–1423. [Google Scholar] [CrossRef]
- Lassale, C.; Curtis, A.; Abete, I.; van der Schouw, Y.T.; Verschuren, W.M.M.; Lu, Y.; Bueno-de-Mesquita, H.B.A. Elements of the complete blood count associated with cardiovascular disease incidence: Findings from the EPIC-NL cohort study. Sci. Rep. 2018, 8, 3290. [Google Scholar] [CrossRef]
- Christ, A.; Gunther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Bassler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e114. [Google Scholar] [CrossRef] [PubMed]
- Filep, J.G. Targeting Neutrophils for Promoting the Resolution of Inflammation. Front. Immunol. 2022, 13, 866747. [Google Scholar] [CrossRef]
- Delgado-Rizo, V.; Martinez-Guzman, M.A.; Iniguez-Gutierrez, L.; Garcia-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Cooper, P.R.; Palmer, L.J.; Chapple, I.L. Neutrophil extracellular traps as a new paradigm in innate immunity: Friend or foe? Periodontology 2000 2013, 63, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed]
- Arazna, M.; Pruchniak, M.P.; Demkow, U. Neutrophil extracellular traps in bacterial infections: Strategies for escaping from killing. Respir. Physiol. Neurobiol. 2013, 187, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Berends, E.T.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; von Kockritz-Blickwede, M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Beiter, K.; Wartha, F.; Albiger, B.; Normark, S.; Zychlinsky, A.; Henriques-Normark, B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 2006, 16, 401–407. [Google Scholar] [CrossRef]
- Palmer, L.J.; Chapple, I.L.; Wright, H.J.; Roberts, A.; Cooper, P.R. Extracellular deoxyribonuclease production by periodontal bacteria. J. Periodontal. Res. 2012, 47, 439–445. [Google Scholar] [CrossRef]
- Bryzek, D.; Ciaston, I.; Dobosz, E.; Gasiorek, A.; Makarska, A.; Sarna, M.; Eick, S.; Puklo, M.; Lech, M.; Potempa, B.; et al. Triggering NETosis via protease-activated receptor (PAR)-2 signaling as a mechanism of hijacking neutrophils function for pathogen benefits. PLoS Pathog. 2019, 15, e1007773. [Google Scholar] [CrossRef]
- Jayaprakash, K.; Demirel, I.; Khalaf, H.; Bengtsson, T. The role of phagocytosis, oxidative burst and neutrophil extracellular traps in the interaction between neutrophils and the periodontal pathogen Porphyromonas gingivalis. Mol. Oral Microbiol. 2015, 30, 361–375. [Google Scholar] [CrossRef]
- Doke, M.; Fukamachi, H.; Morisaki, H.; Arimoto, T.; Kataoka, H.; Kuwata, H. Nucleases from Prevotella intermedia can degrade neutrophil extracellular traps. Mol. Oral Microbiol. 2017, 32, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Hu, Q.; Ling, Q.; Yao, X.; Liu, M.; Chen, J.; Yan, Z.; Dai, Q. Periodontal disease is associated with the risk of cardiovascular disease independent of sex: A meta-analysis. Front. Cardiovasc. Med. 2023, 10, 1114927. [Google Scholar] [CrossRef] [PubMed]
- Geerts, S.O.; Nys, M.; De, M.P.; Charpentier, J.; Albert, A.; Legrand, V.; Rompen, E.H. Systemic release of endotoxins induced by gentle mastication: Association with periodontitis severity. J. Periodontol. 2002, 73, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Forner, L.; Larsen, T.; Kilian, M.; Holmstrup, P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J. Clin. Periodontol. 2006, 33, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Preshaw, P.M.; Bissett, S.M. Periodontitis and diabetes. Br. Dent. J. 2019, 227, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Luong, A.; Tawfik, A.N.; Islamoglu, H.; Gobriel, H.S.; Ali, N.; Ansari, P.; Shah, R.; Hung, T.; Patel, T.; Henson, B.; et al. Periodontitis and diabetes mellitus co-morbidity: A molecular dialogue. J. Oral Biosci. 2021, 63, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Interconnection of periodontal disease and comorbidities: Evidence, mechanisms, and implications. Periodontology 2000 2022, 89, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Mealey, B.L.; Ocampo, G.L. Diabetes mellitus and periodontal disease. Periodontology 2000 2007, 44, 127–153. [Google Scholar] [CrossRef]
- Tsai, C.; Hayes, C.; Taylor, G.W. Glycemic control of type 2 diabetes and severe periodontal disease in the US adult population. Community Dent. Oral Epidemiol. 2002, 30, 182–192. [Google Scholar] [CrossRef]
- Kitamoto, S.; Nagao-Kitamoto, H.; Jiao, Y.; Gillilland, M.G., 3rd; Hayashi, A.; Imai, J.; Sugihara, K.; Miyoshi, M.; Brazil, J.C.; Kuffa, P.; et al. The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell 2020, 182, 447–462.e414. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.C.; DalBo, S.; Striechen, T.M.; Farias, J.M.; Olchanheski, L.R., Jr.; Mendes, R.T.; Vellosa, J.C.; Favero, G.M.; Sordi, R.; Assreuy, J.; et al. Experimental periodontitis promotes transient vascular inflammation and endothelial dysfunction. Arch. Oral Biol. 2013, 58, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, C.; Haley, M.J.; Lemarchand, E.; Smith, C.J.; Allan, S.M.; Konkel, J.E.; Lawrence, C.B. Ligature-induced periodontitis induces systemic inflammation but does not alter acute outcome after stroke in mice. Int. J. Stroke 2020, 15, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Blasco-Baque, V.; Garidou, L.; Pomie, C.; Escoula, Q.; Loubieres, P.; Le Gall-David, S.; Lemaitre, M.; Nicolas, S.; Klopp, P.; Waget, A.; et al. Periodontitis induced by Porphyromonas gingivalis drives periodontal microbiota dysbiosis and insulin resistance via an impaired adaptive immune response. Gut 2017, 66, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Liu, C.; Zheng, X.; Jia, X.; Peng, X.; Yang, R.; Zhou, X.; Xu, X. Porphyromonas gingivalis Induces Insulin Resistance by Increasing BCAA Levels in Mice. J. Dent. Res. 2020, 99, 839–846. [Google Scholar] [CrossRef] [PubMed]
- White, P.J.; Newgard, C.B. Branched-chain amino acids in disease. Science 2019, 363, 582–583. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: A novel pathway for inflammatory cell recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef]
- Lalla, E.; Lamster, I.B.; Feit, M.; Huang, L.; Spessot, A.; Qu, W.; Kislinger, T.; Lu, Y.; Stern, D.M.; Schmidt, A.M. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J. Clin. Investig. 2000, 105, 1117–1124. [Google Scholar] [CrossRef]
- Pihlstrom, B.L.; Michalowicz, B.S.; Johnson, N.W. Periodontal diseases. Lancet 2005, 366, 1809–1820. [Google Scholar] [CrossRef]
- Gursoy, U.K.; Marakoglu, I.; Oztop, A.Y. Relationship between neutrophil functions and severity of periodontitis in obese and/or type 2 diabetic chronic periodontitis patients. Quintessence Int. 2008, 39, 485–489. [Google Scholar]
- Swoboda, L.; Held, J. Impaired wound healing in diabetes. J. Wound Care 2022, 31, 882–885. [Google Scholar] [CrossRef]
- Shinjo, T.; Onizuka, S.; Zaitsu, Y.; Ishikado, A.; Park, K.; Li, Q.; Yokomizo, H.; Zeze, T.; Sato, K.; St-Louis, R.; et al. Dysregulation of CXCL1 expression and neutrophil recruitment in insulin resistance and diabetes-related periodontitis in male mice. Diabetes 2023, 72, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Machado, V.; Botelho, J.; Escalda, C.; Hussain, S.B.; Luthra, S.; Mascarenhas, P.; Orlandi, M.; Mendes, J.J.; D’Aiuto, F. Serum C-Reactive Protein and Periodontitis: A Systematic Review and Meta-Analysis. Front. Immunol. 2021, 12, 706432. [Google Scholar] [CrossRef] [PubMed]
- Munoz Aguilera, E.; Suvan, J.; Buti, J.; Czesnikiewicz-Guzik, M.; Barbosa Ribeiro, A.; Orlandi, M.; Guzik, T.J.; Hingorani, A.D.; Nart, J.; D’Aiuto, F. Periodontitis is associated with hypertension: A systematic review and meta-analysis. Cardiovasc. Res. 2020, 116, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Munoz Aguilera, E.; Suvan, J.; Orlandi, M.; Miro Catalina, Q.; Nart, J.; D’Aiuto, F. Association Between Periodontitis and Blood Pressure Highlighted in Systemically Healthy Individuals: Results From a Nested Case-Control Study. Hypertension 2021, 77, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Chavakis, T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat. Rev. Immunol. 2021, 21, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Leira, Y.; Rodriguez-Yanez, M.; Arias, S.; Lopez-Dequidt, I.; Campos, F.; Sobrino, T.; D’Aiuto, F.; Castillo, J.; Blanco, J. Periodontitis as a risk indicator and predictor of poor outcome for lacunar infarct. J. Clin. Periodontol. 2019, 46, 20–30. [Google Scholar] [CrossRef]
- Chavakis, T.; Wielockx, B.; Hajishengallis, G. Inflammatory Modulation of Hematopoiesis: Linking Trained Immunity and Clonal Hematopoiesis with Chronic Disorders. Annu. Rev. Physiol. 2022, 84, 183–207. [Google Scholar] [CrossRef]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.; Li, Y.; Popa, C.D.; Ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146.e139. [Google Scholar] [CrossRef]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161.e112. [Google Scholar] [CrossRef]
- King, K.Y.; Goodell, M.A. Inflammatory modulation of HSCs: Viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 2011, 11, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Yu, X.; Saha, G.; Kalafati, L.; Ioannidis, C.; Mitroulis, I.; Netea, M.G.; Chavakis, T.; Hajishengallis, G. Maladaptive innate immune training of myelopoiesis links inflammatory comorbidities. Cell 2022, 185, 1709–1727.e1718. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Passegue, E. TNF-alpha Coordinates Hematopoietic Stem Cell Survival and Myeloid Regeneration. Cell Stem Cell 2019, 25, 357–372.e357. [Google Scholar] [CrossRef] [PubMed]
- Schurch, C.M.; Riether, C.; Ochsenbein, A.F. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell 2014, 14, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Matatall, K.A.; Jeong, M.; Chen, S.; Sun, D.; Chen, F.; Mo, Q.; Kimmel, M.; King, K.Y. Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Rep. 2016, 17, 2584–2595. [Google Scholar] [CrossRef] [PubMed]
- Christopher, M.J.; Rao, M.; Liu, F.; Woloszynek, J.R.; Link, D.C. Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J. Exp. Med. 2011, 208, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Herault, A.; Binnewies, M.; Leong, S.; Calero-Nieto, F.J.; Zhang, S.Y.; Kang, Y.A.; Wang, X.; Pietras, E.M.; Chu, S.H.; Barry-Holson, K.; et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 2017, 544, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Del Castillo, A.M.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and Cardiovascular Diseases. Consensus Report. Glob. Heart 2020, 15, 1. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bassani, B.; Cucchiara, M.; Butera, A.; Kayali, O.; Chiesa, A.; Palano, M.T.; Olmeo, F.; Gallazzi, M.; Dellavia, C.P.B.; Mortara, L.; et al. Neutrophils’ Contribution to Periodontitis and Periodontitis-Associated Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 24, 15370. https://doi.org/10.3390/ijms242015370
Bassani B, Cucchiara M, Butera A, Kayali O, Chiesa A, Palano MT, Olmeo F, Gallazzi M, Dellavia CPB, Mortara L, et al. Neutrophils’ Contribution to Periodontitis and Periodontitis-Associated Cardiovascular Diseases. International Journal of Molecular Sciences. 2023; 24(20):15370. https://doi.org/10.3390/ijms242015370
Chicago/Turabian StyleBassani, Barbara, Martina Cucchiara, Andrea Butera, Omar Kayali, Alessandro Chiesa, Maria Teresa Palano, Francesca Olmeo, Matteo Gallazzi, Claudia Paola Bruna Dellavia, Lorenzo Mortara, and et al. 2023. "Neutrophils’ Contribution to Periodontitis and Periodontitis-Associated Cardiovascular Diseases" International Journal of Molecular Sciences 24, no. 20: 15370. https://doi.org/10.3390/ijms242015370
APA StyleBassani, B., Cucchiara, M., Butera, A., Kayali, O., Chiesa, A., Palano, M. T., Olmeo, F., Gallazzi, M., Dellavia, C. P. B., Mortara, L., Parisi, L., & Bruno, A. (2023). Neutrophils’ Contribution to Periodontitis and Periodontitis-Associated Cardiovascular Diseases. International Journal of Molecular Sciences, 24(20), 15370. https://doi.org/10.3390/ijms242015370