
Mitochondrial and Proteasome Dysfunction Occurs in the Hearts of Mice Treated with Triazine Herbicide Prometryn



Abstract
:1. Introduction
2. Results
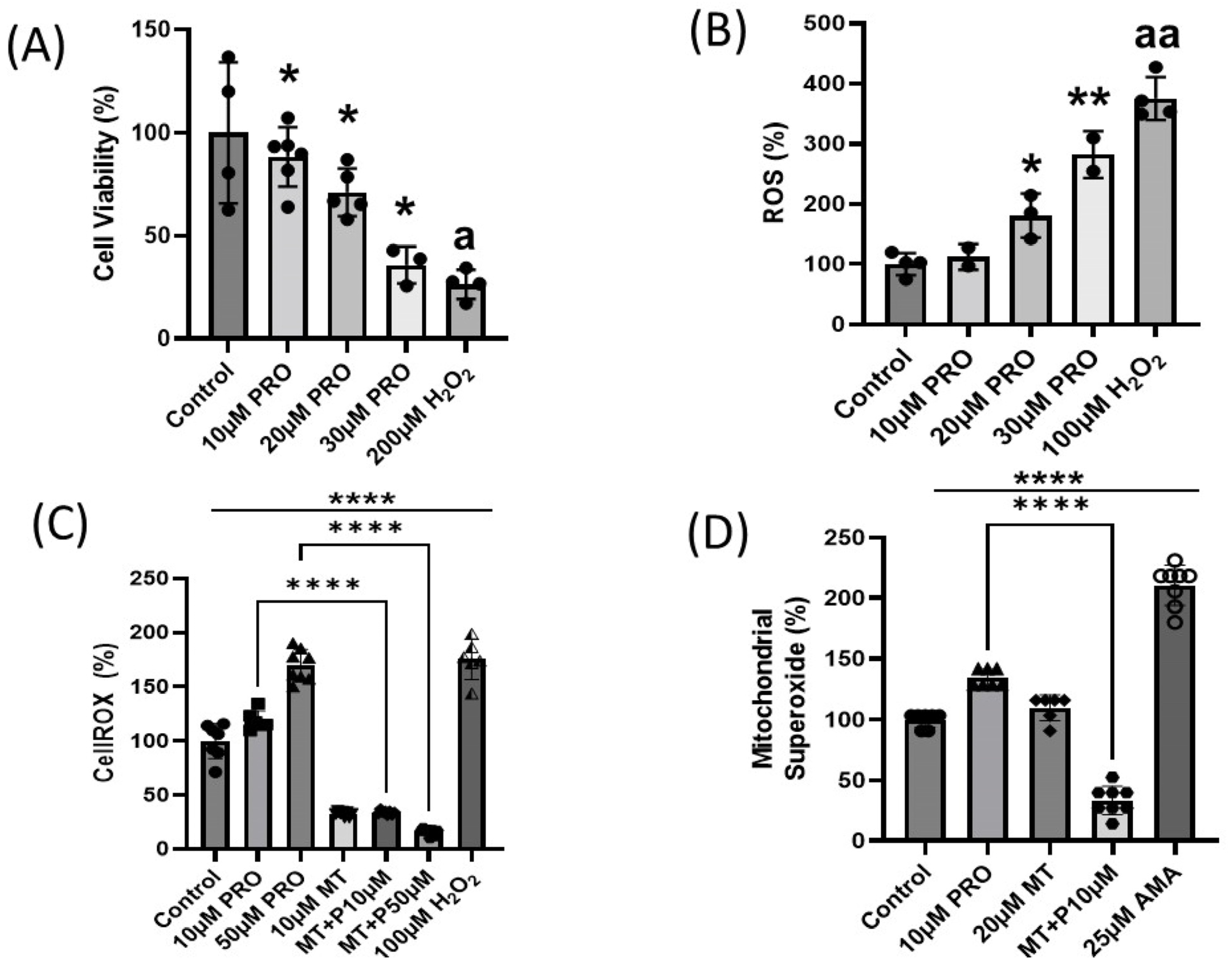
2.1. Effects of Prometryn on H9c2 Cardiac Cells
2.1.1. Prometryn Treatment Reduces Cell Viability in H9c2 Cells
2.1.2. Prometryn Treatment Elevated Oxidative Stress and Mitochondrial Superoxide
2.2. Identification of the Differentially Expressed Proteins in Prometryn Treated Mice
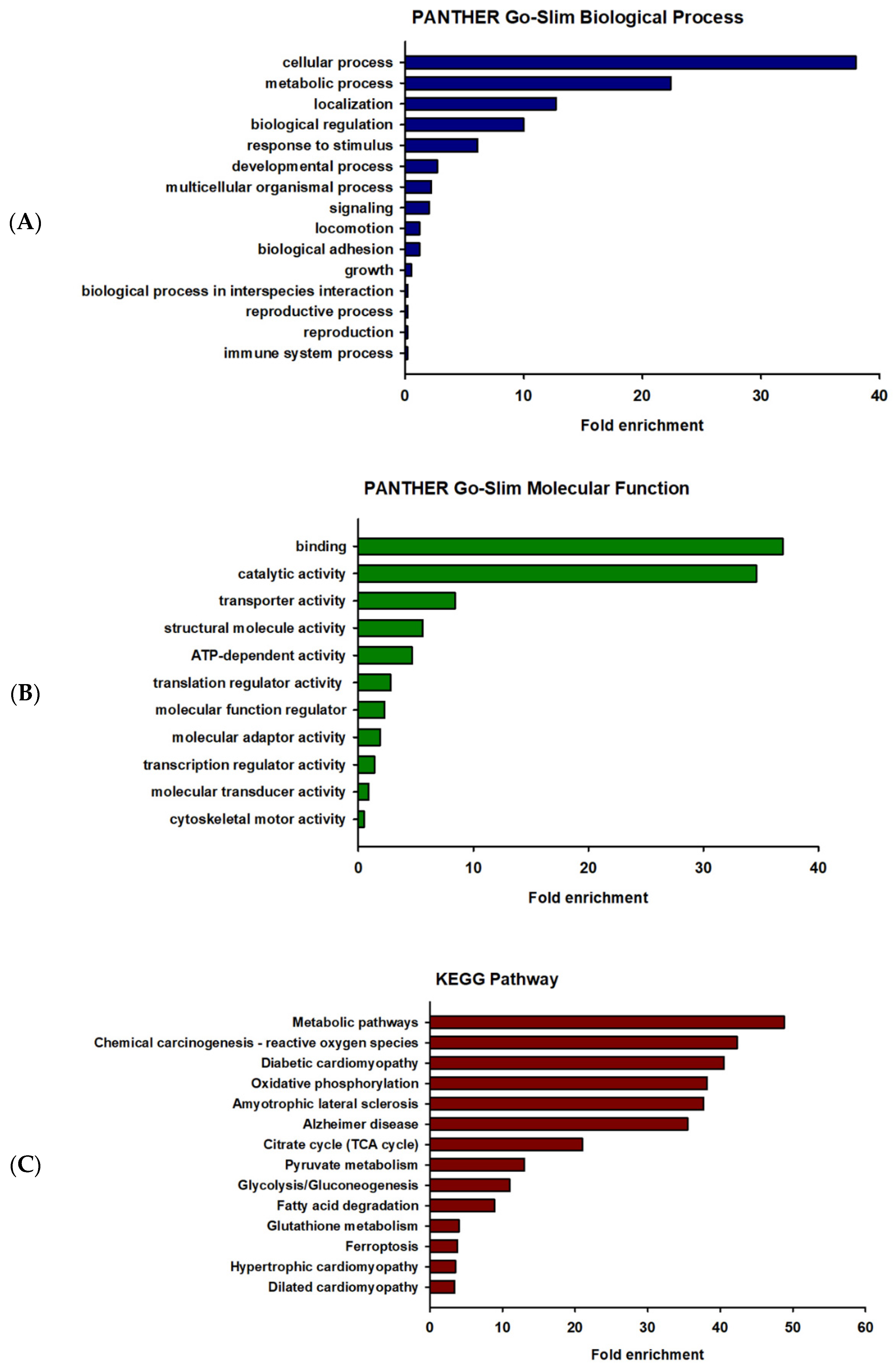
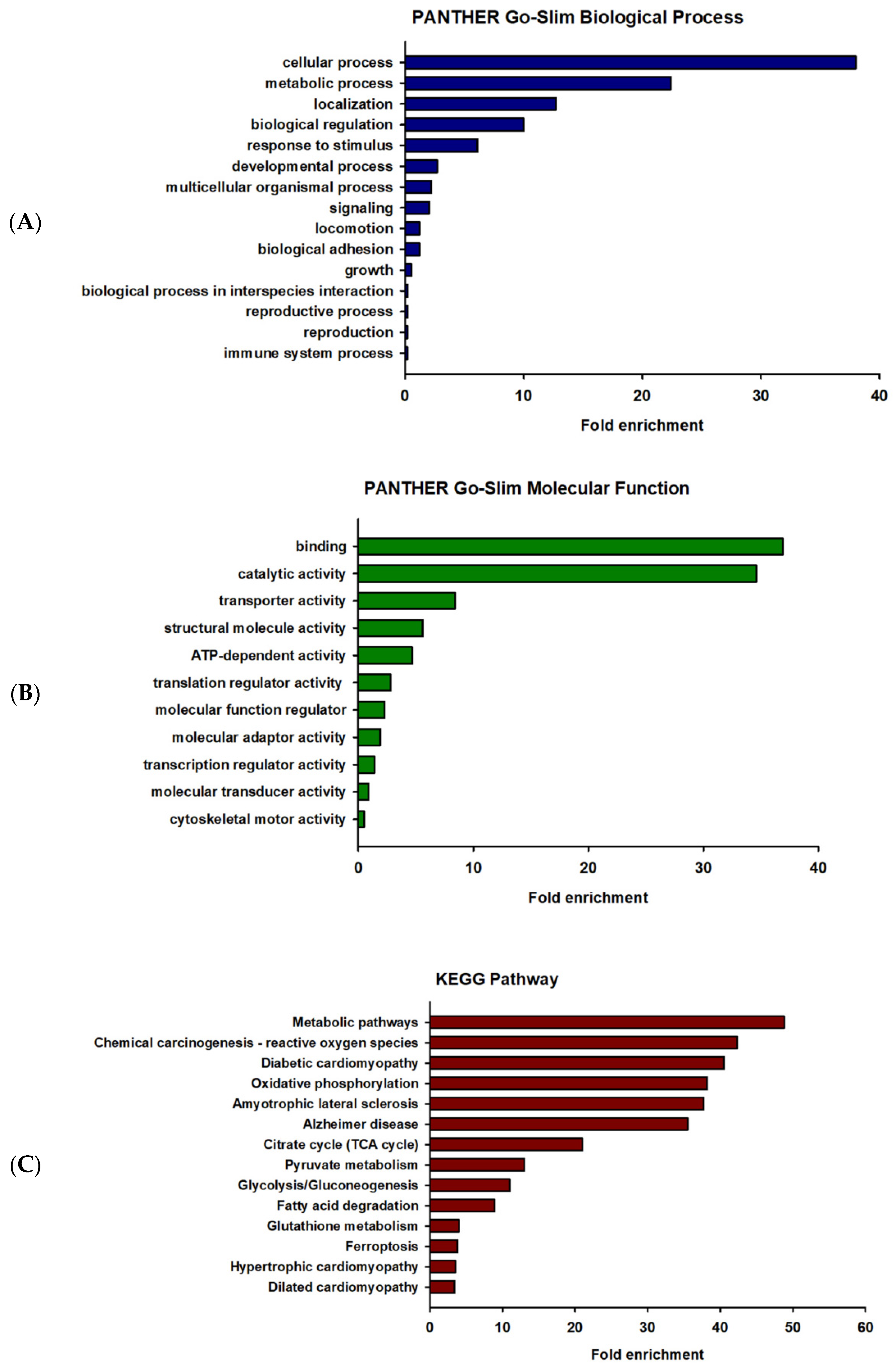
2.3. Gene Ontology (GO) Enrichment and Pathway Analysis of the Differentially Expressed Proteins
2.4. Prometryn Increased Antioxidant Defense System Proteins
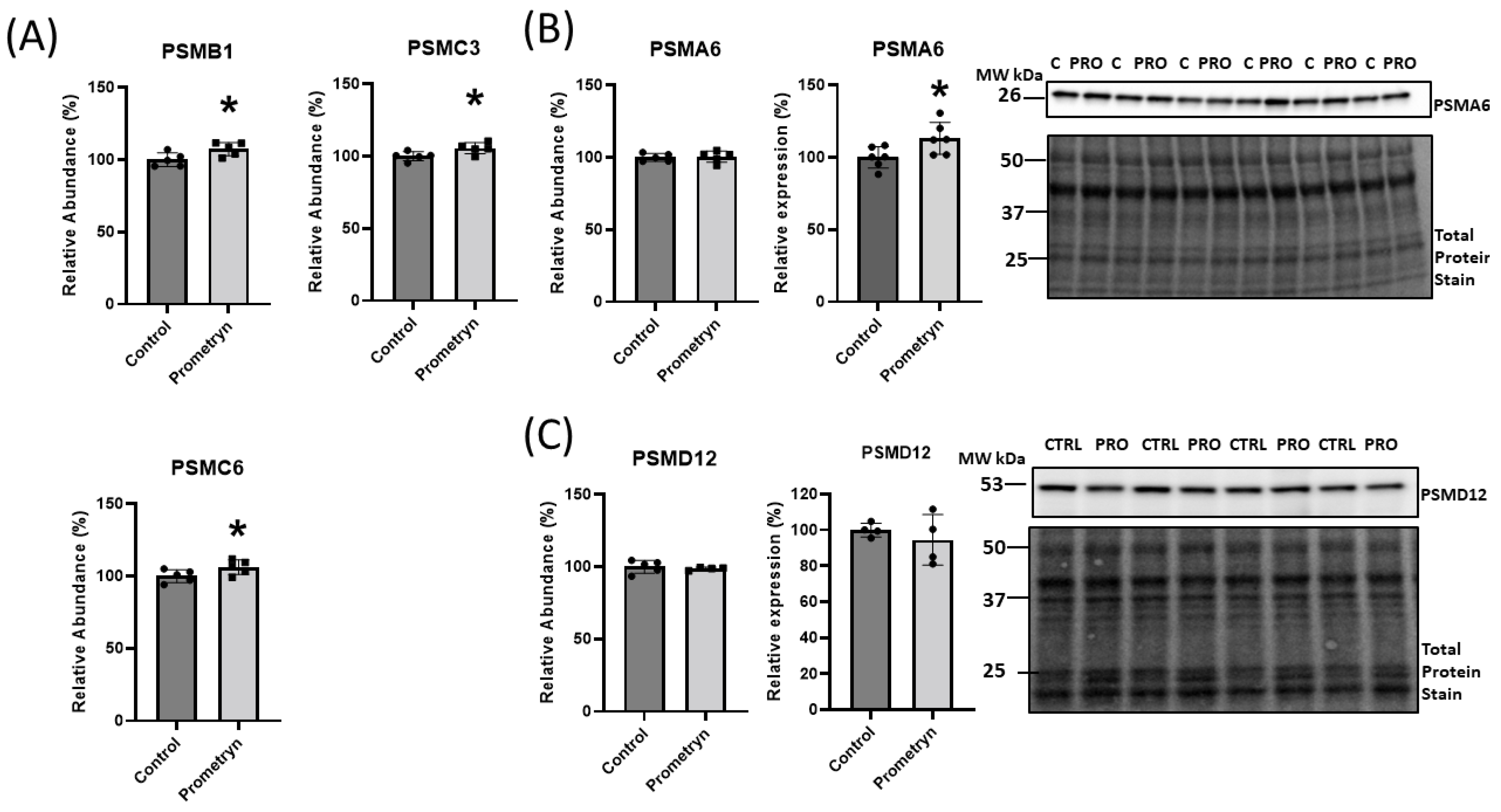
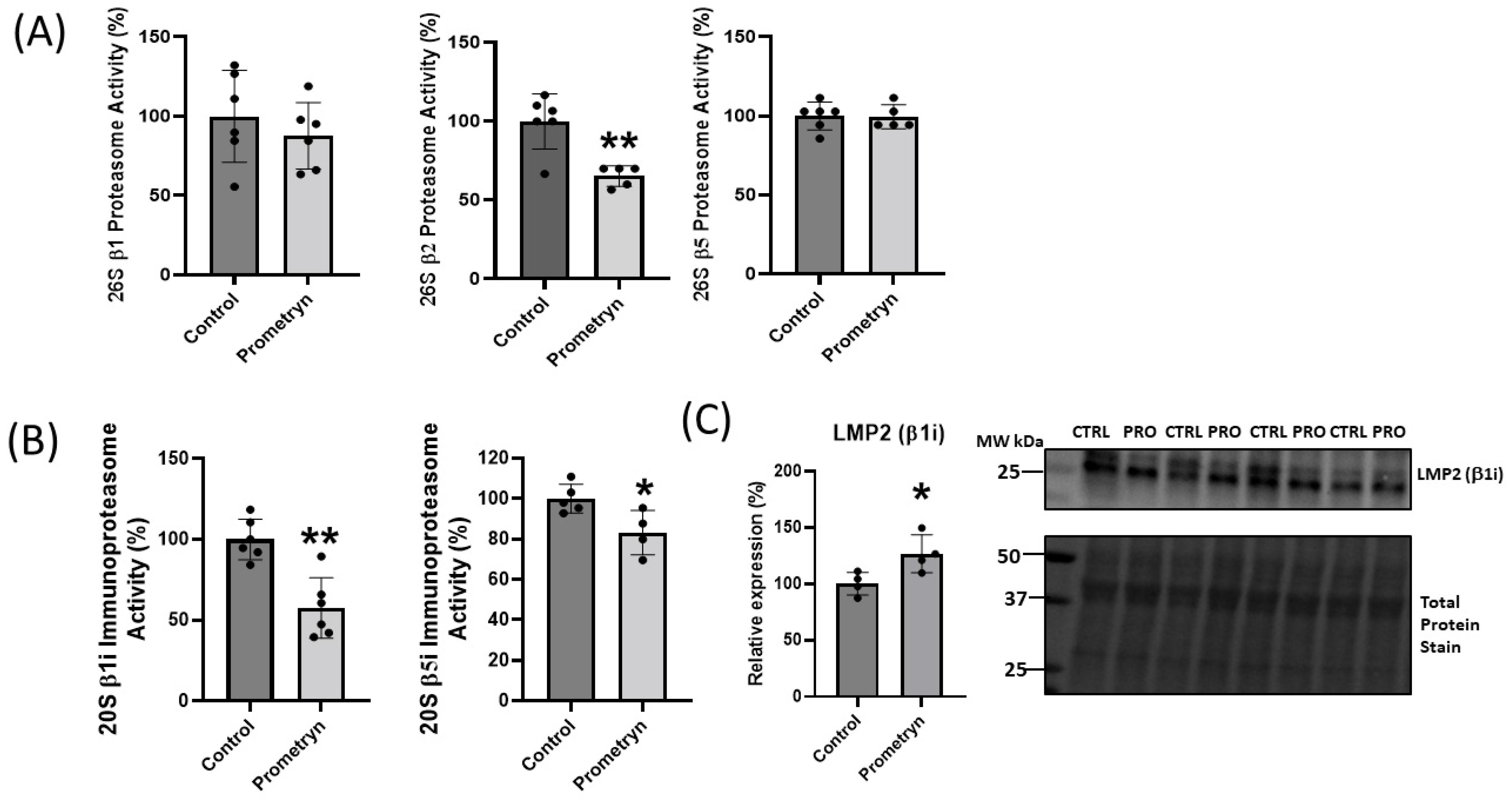
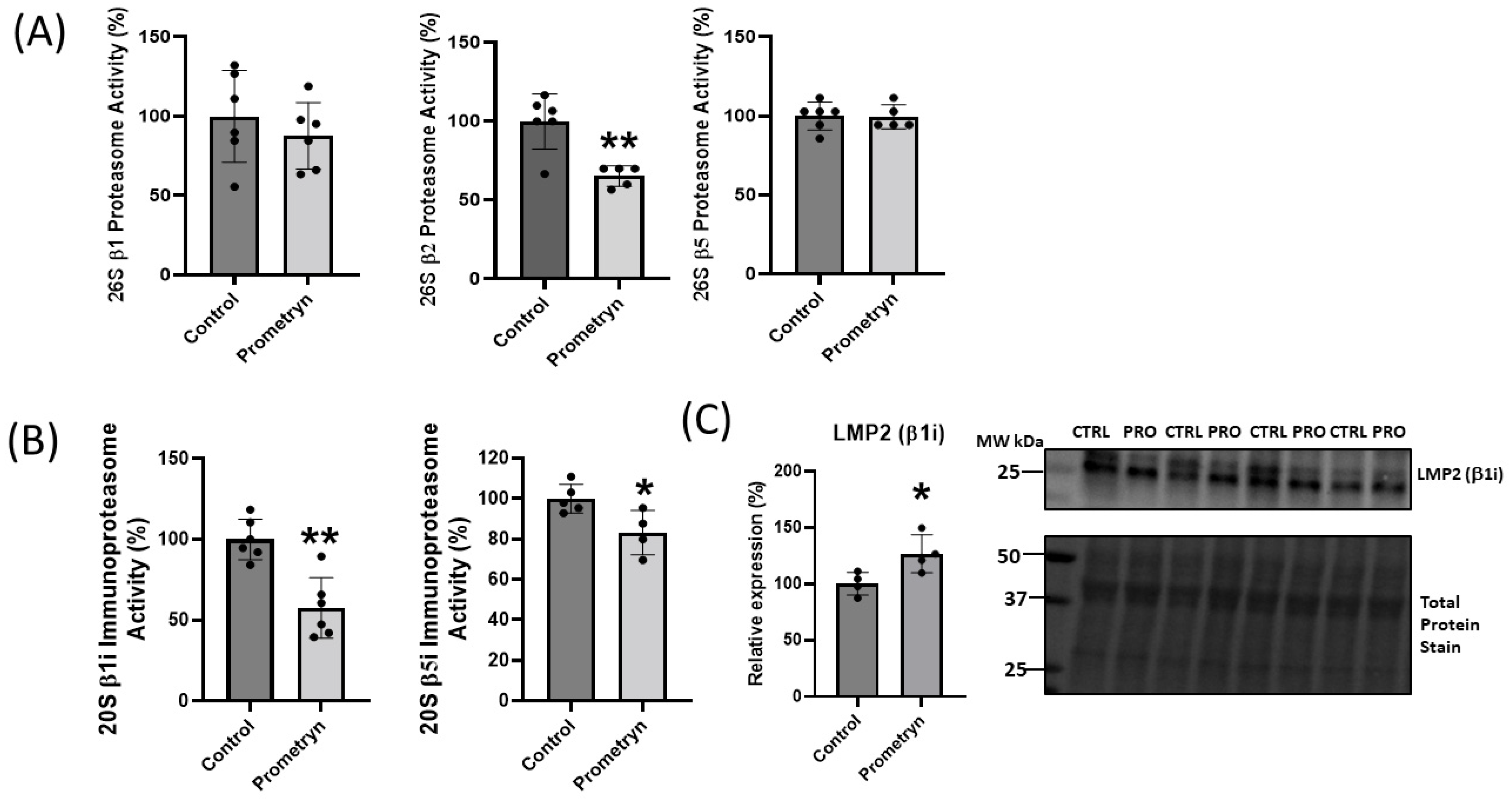
2.5. Prometryn Treatment Induced Proteasomal Dysfunction
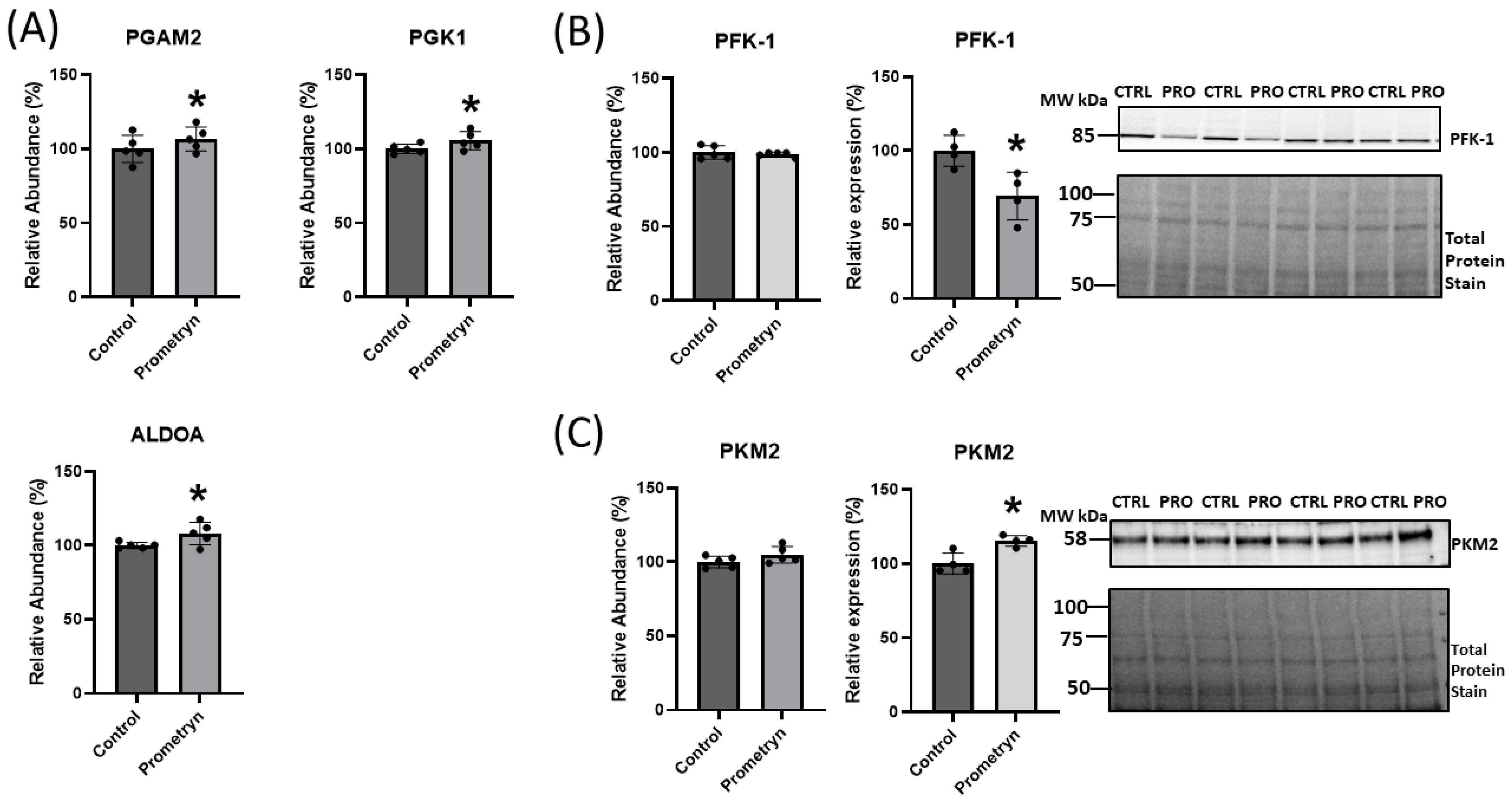
2.6. Prometryn Affected Glycolytic Pathway in Male Mice Heart
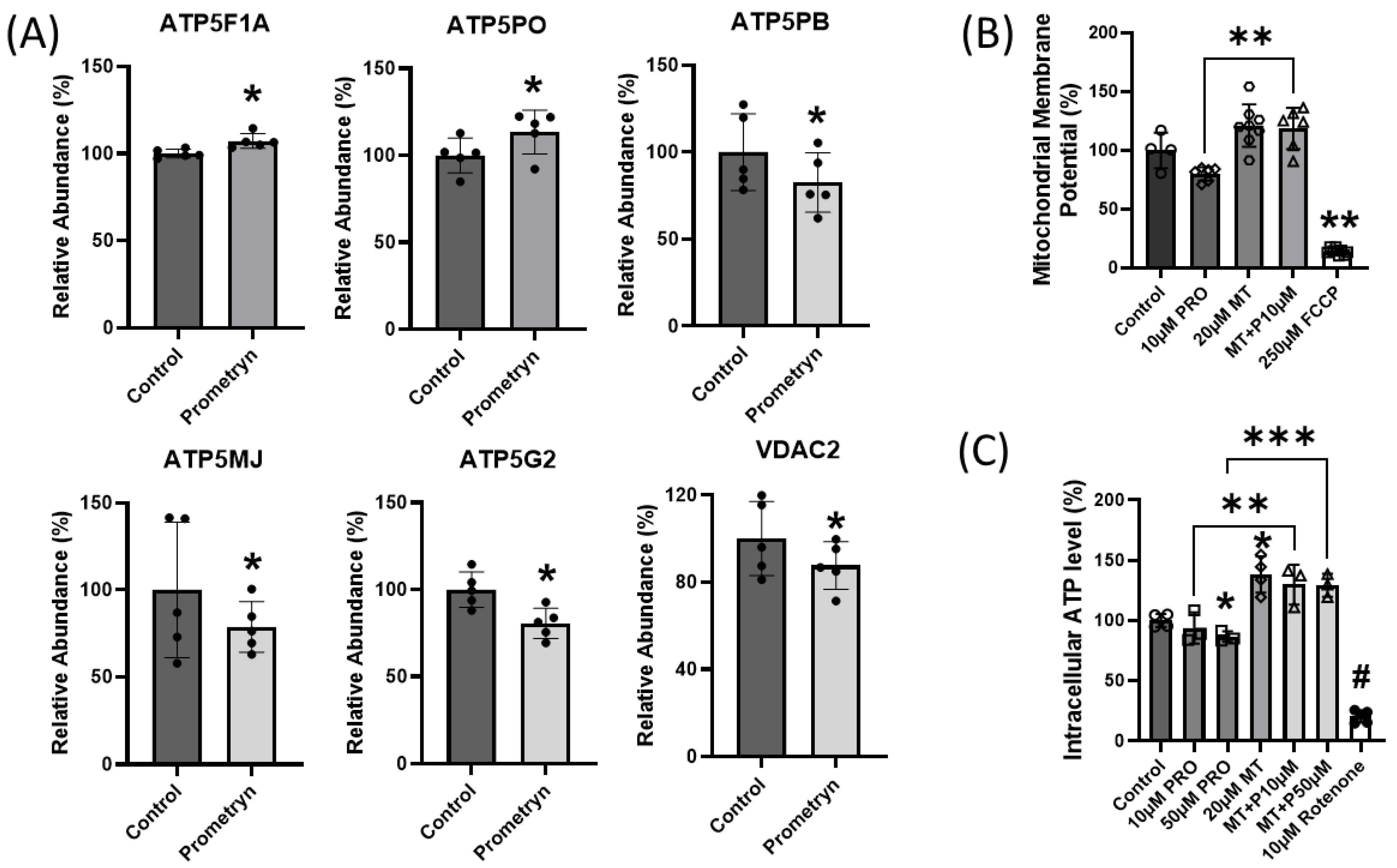
2.7. Mitochondrial Function Impaired in Prometryn-Treated Male Mice Heart
2.8. Prometryn Affects Fatty Acid Metabolism Pathway in Male Mice Heart
2.9. Prometryn Affects Calcium Signaling Pathways
3. Discussion
3.1. Proteins Involved in Antioxidant Defense System
3.2. Proteins Involved in Degradation
3.3. Proteins Involved in Energy Metabolism
3.4. Proteins Involved in Fatty Acid Metabolism Pathway
3.5. Other Pathway Proteins
3.6. Other Potential Pathways
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability Assay
4.3. Mitochondrial Membrane Potential
4.4. Intracellular ATP Level Detection
4.5. Intracellular Reactive Oxygen Species (ROS) Detection
4.6. Superoxide Anion Scavenging Assay
4.7. Animal Studies
4.8. LC-MS/MS Using 10–Plex TMT Proteomics
4.8.1. 26S Proteasome Activity Assay
4.8.2. Immunoproteasome Activity Assay
4.8.3. Immunoblotting
Sample Preparation
Electrophoresis and Western Blotting
Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sule, R.O.; Condon, L.; Gomes, A.V. A Common Feature of Pesticides: Oxidative Stress—The Role of Oxidative Stress in Pesticide-Induced Toxicity. Oxidative Med. Cell. Longev. 2022, 2022, 5563759. [Google Scholar] [CrossRef] [PubMed]
- Syafrudin, M.; Kristanti, R.A.; Yuniarto, A.; Hadibarata, T.; Rhee, J.; Al-Onazi, W.A.; Algarni, T.S.; Almarri, A.H.; Al-Mohaimeed, A.M. Pesticides in Drinking Water-A Review. Int. J. Environ. Res. Public Health 2021, 18, 468. [Google Scholar] [CrossRef] [PubMed]
- Tudi, M.; Daniel Ruan, H.; Wang, L.; Lyu, J.; Sadler, R.; Connell, D.; Chu, C.; Phung, D.T. Agriculture Development, Pesticide Application and Its Impact on the Environment. Int. J. Environ. Res. Public Health 2021, 18, 1112. [Google Scholar] [CrossRef]
- Sarka, K.; Lucie, K. Triazine Herbicides in the Environment. In Herbicides; Andrew, P., Jessica, K., Lina, S., Eds.; IntechOpen: Rijeka, Croatia, 2015; p. 4. [Google Scholar]
- Liu, Q.; Wang, L.; Chen, H.; Huang, B.; Xu, J.; Li, Y.; Héroux, P.; Zhu, X.; Wu, Y.; Xia, D. Prometryn induces apoptotic cell death through cell cycle arrest and oxidative DNA damage. Toxicol. Res. 2019, 8, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Almberg, K.S.; Turyk, M.E.; Jones, R.M.; Rankin, K.; Freels, S.; Stayner, L.T. Atrazine Contamination of Drinking Water and Adverse Birth Outcomes in Community Water Systems with Elevated Atrazine in Ohio, 2006–2008. Int. J. Environ. Res. Public Health 2018, 15, 1889. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, R.; Różalska, S.; Mironenka, J.; Bernat, P. Atrazine biodegradation by mycoinsecticide Metarhizium robertsii: Insights into its amino acids and lipids profile. J. Environ. Manag. 2020, 262, 110304. [Google Scholar] [CrossRef] [PubMed]
- Navarro, S.; Vela, N.; José Giménez, M.; Navarro, G. Persistence of four s-triazine herbicides in river, sea and groundwater samples exposed to sunlight and darkness under laboratory conditions. Sci. Total Environ. 2004, 329, 87–97. [Google Scholar] [CrossRef]
- Alavanja, M.C.R.; Ross, M.K.; Bonner, M.R. Increased cancer burden among pesticide applicators and others due to pesticide exposure. CA A Cancer J. Clin. 2013, 63, 120–142. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef]
- Rog-Zielinska, E.A.; O’Toole, E.T.; Hoenger, A.; Kohl, P. Mitochondrial Deformation During the Cardiac Mechanical Cycle. Anat. Rec. 2019, 302, 146–152. [Google Scholar] [CrossRef]
- Bisaccia, G.; Ricci, F.; Gallina, S.; Di Baldassarre, A.; Ghinassi, B. Mitochondrial Dysfunction and Heart Disease: Critical Appraisal of an Overlooked Association. Int. J. Mol. Sci. 2021, 22, 614. [Google Scholar] [CrossRef] [PubMed]
- Samreen; Zhang, X.; Wang, J.; Li, Y.; Li, X.; Zheng, Y.; Arif, M.; Ru, S. Environmental relevant herbicide prometryn induces developmental toxicity in the early life stages of marine medaka (Oryzias melastigma) and its potential mechanism. Aquat. Toxicol. 2022, 243, 106079. [Google Scholar] [CrossRef]
- Song, X.Y.; Li, J.N.; Wu, Y.P.; Zhang, B.; Li, B.X. Atrazine Causes Autophagy- and Apoptosis-Related Neurodegenerative Effects in Dopaminergic Neurons in the Rat Nigrostriatal Dopaminergic System. Int. J. Mol. Sci. 2015, 16, 13490–13506. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, C.P.; Langel, Ü. Effect of a Fusion Peptide by Covalent Conjugation of a Mitochondrial Cell-Penetrating Peptide and a Glutathione Analog Peptide. Mol. Ther. Methods Clin. Dev. 2017, 5, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Vetrivel, P.; Kim, S.M.; Ha, S.E.; Kim, H.H.; Bhosale, P.B.; Heo, J.D.; Lee, W.S.; Senthil, K.; Kim, G.S. Quantitative Proteomics Analysis for the Identification of Differential Protein Expression in Calf Muscles between Young and Old SD Rats Using Mass Spectrometry. ACS Omega 2021, 6, 7422–7433. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; Dias, I.H.K.; Debelec, B.; Vucetic, M.; Fladmark, K.E.; Basaga, H.; Ribaric, S.; Milisav, I.; Cuadrado, A. Redox control of protein degradation. Redox Biol. 2015, 6, 409–420. [Google Scholar] [CrossRef]
- Gilda, J.E.; Gomes, A.V. Proteasome dysfunction in cardiomyopathies. J. Physiol. 2017, 595, 4051–4071. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Cuanalo-Contreras, K.; Mukherjee, A.; Soto, C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013, 2013, 638083. [Google Scholar] [CrossRef]
- Aon, M.A.; Tocchetti, C.G.; Bhatt, N.; Paolocci, N.; Cortassa, S. Protective mechanisms of mitochondria and heart function in diabetes. Antioxid. Redox Signal. 2015, 22, 1563–1586. [Google Scholar] [CrossRef]
- Thai, P.N.; Ren, L.; Xu, W.; Overton, J.; Timofeyev, V.; Nader, C.E.; Haddad, M.; Yang, J.; Gomes, A.V.; Hammock, B.D.; et al. Chronic Diclofenac Exposure Increases Mitochondrial Oxidative Stress, Inflammatory Mediators, and Cardiac Dysfunction. Cardiovasc. Drugs Ther. 2021, 37, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Csordás, G.; Hajnóczky, G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle—Pivotal roles in Ca2+ and reactive oxygen species signaling. J. Cell Sci. 2013, 126, 2965–2978. [Google Scholar] [PubMed]
- Mostafalou, S.; Abdollahi, M. Pesticides and human chronic diseases: Evidences, mechanisms, and perspectives. Toxicol. Appl. Pharmacol. 2013, 268, 157–177. [Google Scholar] [CrossRef]
- Boulahia, K.; Carol, P.; Planchais, S.; Abrous-Belbachir, O. Phaseolus vulgaris L. Seedlings Exposed to Prometryn Herbicide Contaminated Soil Trigger an Oxidative Stress Response. J. Agric. Food Chem. 2016, 64, 3150–3160. [Google Scholar] [CrossRef] [PubMed]
- Stará, A.; Kouba, A.; Velíšek, J. Effect of chronic exposure to prometryne on oxidative stress and antioxidant response in red swamp crayfish (Procambarus clarkii). BioMed Res. Int. 2014, 2014, 680131. [Google Scholar] [CrossRef]
- Stara, A.; Kristan, J.; Zuskova, E.; Velisek, J. Effect of chronic exposure to prometryne on oxidative stress and antioxidant response in common carp (Cyprinus carpio L.). Pestic. Biochem. Physiol. 2013, 105, 18–23. [Google Scholar] [CrossRef]
- Stara, A.; Machova, J.; Velisek, J. Effect of chronic exposure to prometryne on oxidative stress and antioxidant response in early life stages of common carp (Cyprinus carpio L.). Neuro Endocrinol. Lett. 2012, 33 (Suppl. S3), 130–135. [Google Scholar]
- Đikić, D.; Židovec-Lepej, S.; Remenar, A.; Horvat-Knežević, A.; Benković, V.; Lisičić, D.; Sajli, L.; Springer, O. The Effects of Prometryne on Subchronically Treated Mice Evaluated by SCGE Assay. Acta Biol. Hung. 2009, 60, 35–43. [Google Scholar] [CrossRef]
- Fiveland, T.J. Residues of linuron and prometryne in carrots and decomposition in soil in the southern and northern parts of Norway. Forsk. Og Fors. I Landbruket 1977, 8, 345–363. [Google Scholar]
- Baldwin, F.L.; Santelmann, P.W.; Davidson, J.M. Prometryn Movement across and through the Soil. Weed Sci. 1975, 23, 285–288. [Google Scholar] [CrossRef]
- Reifenstein, H.; Pank, F. Triazine residues in medicinal plants. Die Pharm. 1975, 30, 391–393. [Google Scholar]
- Balduini, L.; Matoga, M.; Cavalli, E.; Seilles, E.; Riethmuller, D.; Thomassin, M.; Guillaume, Y.C. Triazinic herbicide determination by gas chromatography-mass spectrometry in breast milk. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 794, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Krivánkova, L.; Bocek, P.; Tekel, J.; Kovacicová, J. Isotachophoretic determination of herbicides prometryne, desmetryne, terbutryne and hydroxy-derivatives of atrazine and simazine in extracts of milk. Electrophoresis 1989, 10, 731–734. [Google Scholar] [CrossRef]
- Messow, A.; Benkwitz, F.; Hellwig, A. The toxicity of a prometryne/simazine combination in the lactation stage of rats. Z. Fur Die Gesamte Hyg. Und Ihre Grenzgeb. 1990, 36, 170–173. [Google Scholar]
- Allen, M.M.; Turnburke, A.C.; Lagace, E.A.; Steinback, K.E. Effects of Photosystem II Herbicides on the Photosynthetic Membranes of the Cyanobacterium Aphanocapsa 6308. Plant Physiol. 1983, 71, 388–392. [Google Scholar] [CrossRef]
- Đikić, D.; Židovec-Lepej, S.; Remenar, A.; Bendelja, K.; Benković, V.; Horvat-Knežević, A.; Brozović, G.; Oršolić, N. Effects of prometryne on apoptosis and necrosis in thymus, lymph node and spleen in mice. Environ. Toxicol. Pharmacol. 2009, 27, 182–186. [Google Scholar] [CrossRef]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef]
- Tiwari, S.; Mishra, M.; Salemi, M.R.; Phinney, B.S.; Newens, J.L.; Gomes, A.V. Gender-specific changes in energy metabolism and protein degradation as major pathways affected in livers of mice treated with ibuprofen. Sci. Rep. 2020, 10, 3386. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, Y.; Zhao, P.; Guo, Y.; Chen, S.; Wu, J.; Ren, Z. Glutathione S-transferase Mu 2 inhibits hepatic steatosis via ASK1 suppression. Commun. Biol. 2022, 5, 326. [Google Scholar] [CrossRef]
- Lei, X.G.; Zhu, J.H.; Cheng, W.H.; Bao, Y.; Ho, Y.S.; Reddi, A.R.; Holmgren, A.; Arner, E.S. Paradoxical Roles of Antioxidant Enzymes: Basic Mechanisms and Health Implications. Physiol. Rev. 2016, 96, 307–364. [Google Scholar] [CrossRef]
- Szyller, J.; Bil-Lula, I. Heat Shock Proteins in Oxidative Stress and Ischemia/Reperfusion Injury and Benefits from Physical Exercises: A Review to the Current Knowledge. Oxidative Med. Cell. Longev. 2021, 2021, 6678457. [Google Scholar] [CrossRef] [PubMed]
- Ikwegbue, P.C.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer. Pharmaceuticals 2017, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Academy. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.V.; Karpov, V.L. Proteasomes and Several Aspects of Their Heterogeneity Relevant to Cancer. Front. Oncol. 2019, 9, 761. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-F.; Li, S.; Chou, A.P.; Bronstein, J.M. Inhibitory effects of pesticides on proteasome activity: Implication in Parkinson’s disease. Neurobiol. Dis. 2006, 23, 198–205. [Google Scholar] [CrossRef]
- Rhodes, S.L.; Fitzmaurice, A.G.; Cockburn, M.; Bronstein, J.M.; Sinsheimer, J.S.; Ritz, B. Pesticides that inhibit the ubiquitin-proteasome system: Effect measure modification by genetic variation in SKP1 in Parkinson’s disease. Environ. Res. 2013, 126, 1–8. [Google Scholar] [CrossRef]
- Chou, A.P.; Maidment, N.; Klintenberg, R.; Casida, J.E.; Li, S.; Fitzmaurice, A.G.; Fernagut, P.O.; Mortazavi, F.; Chesselet, M.F.; Bronstein, J.M. Ziram causes dopaminergic cell damage by inhibiting E1 ligase of the proteasome. J. Biol. Chem. 2008, 283, 34696–34703. [Google Scholar] [CrossRef]
- Wills, J.; Credle, J.; Oaks, A.W.; Duka, V.; Lee, J.H.; Jones, J.; Sidhu, A. Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS ONE 2012, 7, e30745. [Google Scholar] [CrossRef]
- Kiyosawa, N.; Kwekel, J.C.; Burgoon, L.D.; Williams, K.J.; Tashiro, C.; Chittim, B.; Zacharewski, T.R. o,p′-DDT Elicits PXR/CAR-, Not ER-, Mediated Responses in the Immature Ovariectomized Rat Liver. Toxicol. Sci. 2007, 101, 350–363. [Google Scholar] [CrossRef]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes Preserve Protein Homeostasis upon Interferon-Induced Oxidative Stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef]
- Kaur, G.; Batra, S. Emerging role of immunoproteasomes in pathophysiology. Immunol. Cell Biol. 2016, 94, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Johnston-Carey, H.K.; Pomatto, L.C.; Davies, K.J. The Immunoproteasome in oxidative stress, aging, and disease. Crit. Rev. Biochem. Mol. Biol. 2015, 51, 268–281. [Google Scholar] [CrossRef]
- Mijanovic, O.; Petushkova, A.I.; Brankovic, A.; Turk, B.; Solovieva, A.B.; Nikitkina, A.I.; Bolevich, S.; Timashev, P.S.; Parodi, A.; Zamyatnin, A.A., Jr. Cathepsin D-Managing the Delicate Balance. Pharmaceutics 2021, 13, 837. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, S.; Yu, D. Protein kinase function of pyruvate kinase M2 and cancer. Cancer Cell Int. 2020, 20, 523. [Google Scholar] [CrossRef] [PubMed]
- Magadum, A.; Singh, N.; Kurian, A.A.; Munir, I.; Mehmood, T.; Brown, K.; Sharkar, M.T.K.; Chepurko, E.; Sassi, Y.; Oh, J.G.; et al. Pkm2 Regulates Cardiomyocyte Cell Cycle and Promotes Cardiac Regeneration. Circulation 2020, 141, 1249–1265. [Google Scholar] [CrossRef] [PubMed]
- Rajala, A.; Soni, K.; Rajala, R.V.S. Metabolic and Non-metabolic Roles of Pyruvate Kinase M2 Isoform in Diabetic Retinopathy. Sci. Rep. 2020, 10, 7456. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef]
- Fu, J.; Shinjo, T.; Li, Q.; St-Louis, R.; Park, K.; Yu, M.G.; Yokomizo, H.; Simao, F.; Huang, Q.; Wu, I.H.; et al. Regeneration of glomerular metabolism and function by podocyte pyruvate kinase M2 in diabetic nephropathy. JCI Insight 2022, 7, e155260. [Google Scholar] [CrossRef]
- Gao, J.; Zhao, Y.; Li, T.; Gan, X.; Yu, H. The Role of PKM2 in the Regulation of Mitochondrial Function: Focus on Mitochondrial Metabolism, Oxidative Stress, Dynamic, and Apoptosis. PKM2 in Mitochondrial Function. Oxidative Med. Cell. Longev. 2022, 2022, 7702681. [Google Scholar] [CrossRef]
- Song, L.; Guo, L.; Li, Z. Molecular mechanisms of 3,3′4,4′,5-pentachlorobiphenyl-induced epithelial-mesenchymal transition in human hepatocellular carcinoma cells. Toxicol. Appl. Pharmacol. 2017, 322, 75–88. [Google Scholar] [CrossRef]
- Morales-Prieto, N.; Abril, N. REDOX proteomics reveals energy metabolism alterations in the liver of M. spretus mice exposed to p, p′-DDE. Chemosphere 2017, 186, 848–863. [Google Scholar] [CrossRef] [PubMed]
- García, M.; Pujol, A.; Ruzo, A.; Riu, E.; Ruberte, J.; Arbós, A.; Serafín, A.; Albella, B.; Felíu, J.E.; Bosch, F. Phosphofructo-1-kinase deficiency leads to a severe cardiac and hematological disorder in addition to skeletal muscle glycogenosis. PLoS Genet. 2009, 5, e1000615. [Google Scholar] [CrossRef] [PubMed]
- Zanella, A.; Fermo, E.; Bianchi, P.; Valentini, G. Red cell pyruvate kinase deficiency: Molecular and clinical aspects. Br. J. Haematol. 2005, 130, 11–25. [Google Scholar] [CrossRef]
- Johnson, T.A.; Jinnah, H.A.; Kamatani, N. Shortage of Cellular ATP as a Cause of Diseases and Strategies to Enhance ATP. Front. Pharmacol. 2019, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Bolaños, J.P. Bioenergetics and redox adaptations of astrocytes to neuronal activity. J. Neurochem. 2016, 139 (Suppl. S2), 115–125. [Google Scholar] [CrossRef]
- Seo, E.; Kang, H.; Choi, H.; Choi, W.; Jun, H.S. Reactive oxygen species-induced changes in glucose and lipid metabolism contribute to the accumulation of cholesterol in the liver during aging. Aging Cell 2019, 18, e12895. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef]
- Chen, K.; Kobayashi, S.; Xu, X.; Viollet, B.; Liang, Q. AMP Activated Protein Kinase Is Indispensable for Myocardial Adaptation to Caloric Restriction in Mice. PLoS ONE 2013, 8, e59682. [Google Scholar] [CrossRef]
- Zheng, X.; Boyer, L.; Jin, M.; Kim, Y.; Fan, W.; Bardy, C.; Berggren, T.; Evans, R.M.; Gage, F.H.; Hunter, T. Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. eLife 2016, 5, e13378. [Google Scholar] [CrossRef]
- Min, N.; Park, H.; Hong, T.; An, G.; Song, G.; Lim, W. Developmental toxicity of prometryn induces mitochondrial dysfunction, oxidative stress, and failure of organogenesis in zebrafish (Danio rerio). J. Hazard. Mater. 2023, 443, 130202. [Google Scholar] [CrossRef]
- Pfister, K.; Radosevich, S.R.; Arntzen, C.J. Modification of Herbicide Binding to Photosystem II in Two Biotypes of Senecio vulgaris L. Plant Physiol. 1979, 64, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, Q.; Xu, C.; Shao, W.; Zhang, C.; Liu, H.; Jiang, Z.; Gu, A. Organochloride pesticides impaired mitochondrial function in hepatocytes and aggravated disorders of fatty acid metabolism. Sci. Rep. 2017, 7, 46339. [Google Scholar] [CrossRef] [PubMed]
- Chinen, Y.; Yanagi, K.; Nakamura, S.; Nakayama, N.; Kamiya, M.; Nakayashiro, M.; Kaname, T.; Naritomi, K.; Nakanishi, K. A novel homozygous missense SLC25A20 mutation in three CACT-deficient patients: Clinical and autopsy data. Hum. Genome Var. 2020, 7, 11. [Google Scholar] [CrossRef]
- Li, X.; Zhao, F.; Zhao, Z.; Zhao, X.; Meng, H.; Zhang, D.; Zhao, S.; Ding, M. Neonatal sudden death caused by a novel heterozygous mutation in SLC25A20 gene: A case report and brief literature review. Leg. Med. 2022, 54, 101990. [Google Scholar] [CrossRef]
- Chen, M.; Cai, Y.; Li, S.; Xiong, H.; Liu, M.; Ma, F.; Xiao, X.; Hao, H. Late-Onset Carnitine-Acylcarnitine Translocase Deficiency With SLC25A20 c.199-10T>G Variation: Case Report and Pathologic Analysis of Liver Biopsy. Front. Pediatr. 2020, 8, 585646. [Google Scholar] [CrossRef]
- Kankuri, E.; Finckenberg, P.; Leinonen, J.; Tarkia, M.; Björk, S.; Purhonen, J.; Kallijärvi, J.; Kankainen, M.; Soliymani, R.; Lalowski, M.; et al. Altered acylcarnitine metabolism and inflexible mitochondrial fuel utilization characterize the loss of neonatal myocardial regeneration capacity. Exp. Mol. Med. 2023, 55, 806–817. [Google Scholar] [CrossRef]
- Zhao, Q.; Yang, Y.; Wang, C.; Hou, Y.; Chen, H. ATP5B and ETFB metabolic markers in children with congenital hydronephrosis. Mol. Med. Rep. 2016, 14, 5111–5115. [Google Scholar] [CrossRef]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial Energetics in the Heart in Obesity-Related Diabetes: Direct Evidence for Increased Uncoupled Respiration and Activation of Uncoupling Proteins. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Sadayappan, S.; de Tombe, P.P. Cardiac myosin binding protein-C as a central target of cardiac sarcomere signaling: A special mini review series. Pflügers Arch.-Eur. J. Physiol. 2014, 466, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Vatner, D.E.; Sato, N.; Kiuchi, K.; Shannon, R.P.; Vatner, S.F. Decrease in myocardial ryanodine receptors and altered excitation-contraction coupling early in the development of heart failure. Circulation 1994, 90, 1423–1430. [Google Scholar] [CrossRef]
- Bround, M.J.; Wambolt, R.; Luciani, D.S.; Kulpa, J.E.; Rodrigues, B.; Brownsey, R.W.; Allard, M.F.; Johnson, J.D. Cardiomyocyte ATP production, metabolic flexibility, and survival require calcium flux through cardiac ryanodine receptors in vivo. J. Biol. Chem. 2013, 288, 18975–18986. [Google Scholar] [CrossRef] [PubMed]
- Dror, V.; Kalynyak, T.B.; Bychkivska, Y.; Frey, M.H.; Tee, M.; Jeffrey, K.D.; Nguyen, V.; Luciani, D.S.; Johnson, J.D. Glucose and endoplasmic reticulum calcium channels regulate HIF-1beta via presenilin in pancreatic beta-cells. J. Biol. Chem. 2008, 283, 9909–9916. [Google Scholar] [CrossRef]
- Tsuboi, T.; Silva Xavier, G.D.; Holz, G.G.; Jouaville, L.S.; Thomas, A.P.; Rutter, G.A. Glucagon-like peptide-1 mobilizes intracellular Ca2+ and stimulates mitochondrial ATP synthesis in pancreatic MIN6 beta-cells. Biochem. J. 2003, 369 Pt 2, 287–299. [Google Scholar] [CrossRef]
- Kho, C. Targeting calcium regulators as therapy for heart failure: Focus on the sarcoplasmic reticulum Ca-ATPase pump. Front. Cardiovasc. Med. 2023, 10, 1185261. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef]
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar]
- Wang, M.; Liu, J.; Wang, H.; Hu, T. Spiromesifen contributes vascular developmental toxicity via disrupting endothelial cell proliferation and migration in zebrafish embryos. Pestic. Biochem. Physiol. 2022, 188, 105242. [Google Scholar] [CrossRef]
- Su, J.B. Vascular endothelial dysfunction and pharmacological treatment. World J. Cardiol. 2015, 7, 719–741. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Siddharth, M.; Singh, N.; Kare, P.K.; Banerjee, B.D.; Wadhwa, N.; Tripathi, A.K. Organochlorine Pesticide-Mediated Induction of NADPH Oxidase and Nitric-Oxide Synthase in Endothelial Cell. J. Clin. Diagn. Res. JCDR 2017, 11, Bc09–Bc12. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiade, M.; Marti, C.N.; Sabbah, H.N.; Roessig, L.; Greene, S.J.; Bohm, M.; Burnett, J.C.; Campia, U.; Cleland, J.G.; Collins, S.P.; et al. Soluble guanylate cyclase: A potential therapeutic target for heart failure. Heart Fail. Rev. 2013, 18, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.M.; Ortiz-Ortiz, M.A.; Ruiz-Mesa, L.M.; Niso-Santano, M.; Bravosanpedro, J.M.; Sanchez, R.G.; Gonzalez-Polo, R.A.; Fuentes, J.M. Effect of paraquat exposure on nitric oxide-responsive genes in rat mesencephalic cells. Nitric Oxide 2010, 23, 51–59. [Google Scholar] [CrossRef]
- Télot, L.; Rousseau, E.; Lesuisse, E.; Garcia, C.; Morlet, B.; Léger, T.; Camadro, J.-M.; Serre, V. Quantitative proteomics in Friedreich’s ataxia B-lymphocytes: A valuable approach to decipher the biochemical events responsible for pathogenesis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 997–1009. [Google Scholar] [CrossRef]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Oberg, A.L.; Mahoney, D.W.; Eckel-Passow, J.E.; Malone, C.J.; Wolfinger, R.D.; Hill, E.G.; Cooper, L.T.; Onuma, O.K.; Spiro, C.; Therneau, T.M.; et al. Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J. Proteome Res. 2008, 7, 225–233. [Google Scholar] [CrossRef]
- Langer, H.T.; Mossakowski, A.A.; Sule, R.; Gomes, A.; Baar, K. Dominant-negative p53-overexpression in skeletal muscle induces cell death and fiber atrophy in rats. Cell Death Dis. 2022, 13, 716. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PANTHER GO-Slim Biological Process | Number of Differentially Expressed Proteins | Expected | +/− | Fold Enrichment | Raw p Value | False Discover Rate (FDR) |
|---|---|---|---|---|---|---|
| generation of precursor metabolites and energy (GO:0006091) | 24 | 1.47 | + | 16.28 | 1.33 × 10−20 | 2.96 × 10−17 |
| cellular respiration (GO:0045333) | 19 | 0.8 | + | 23.81 | 3.36 × 10−19 | 3.73 × 10−16 |
| energy derivation by oxidation of organic compounds (GO:0015980) | 20 | 1.01 | + | 19.87 | 7.96 × 10−19 | 5.89 × 10−16 |
| aerobic respiration (GO:0009060) | 17 | 0.7 | + | 24.3 | 1.98 × 10−17 | 1.10 × 10−14 |
| small molecule metabolic process (GO:0044281) | 40 | 8.51 | + | 4.7 | 1.48 × 10−15 | 6.58 × 10−13 |
| ATP metabolic process (GO:0046034) | 15 | 0.83 | + | 17.97 | 7.11 × 10−14 | 2.63 × 10−11 |
| ATP synthesis coupled proton transport (GO:0015986) | 12 | 0.5 | + | 23.84 | 1.36 × 10−12 | 4.30 × 10−10 |
| ATP biosynthetic process (GO:0006754) | 12 | 0.52 | + | 23.28 | 1.73 × 10−12 | 4.26 × 10−10 |
| electron transport chain (GO:0022900) | 11 | 0.39 | + | 28.01 | 2.77 × 10−12 | 4.74 × 10−10 |
| proton transmembrane transport (GO:1902600) | 13 | 0.71 | + | 18.26 | 2.85 × 10−12 | 4.52 × 10−10 |
| respiratory electron transport chain (GO:0022904) | 10 | 0.38 | + | 26.28 | 4.68 × 10−11 | 4.16 × 10−9 |
| oxidative phosphorylation (GO:0006119) | 10 | 0.38 | + | 26.28 | 4.68 × 10−11 | 4.00 × 10−9 |
| ribonucleotide biosynthetic process (GO:0009260) | 14 | 1.2 | + | 11.64 | 8.83 × 10−11 | 7.00 × 10−9 |
| protein folding (GO:0006457) | 14 | 1.23 | + | 11.41 | 1.12 × 10−10 | 8.61 × 10−9 |
| ATP synthesis coupled electron transport (GO:0042773) | 8 | 0.28 | + | 28.34 | 2.61 × 10−9 | 1.49 × 10−7 |
| organophosphate metabolic process (GO:0019637) | 22 | 4.84 | + | 4.55 | 8.84 × 10−9 | 4.90 × 10−7 |
| carboxylic acid metabolic process (GO:0019752) | 20 | 4.05 | + | 4.94 | 1.18 × 10−8 | 6.41 × 10−7 |
| organophosphate biosynthetic process (GO:0090407) | 17 | 2.86 | + | 5.94 | 1.21 × 10−8 | 6.37 × 10−7 |
| tricarboxylic acid cycle (GO:0006099) | 7 | 0.23 | + | 30.02 | 1.93 × 10−8 | 9.72 × 10−7 |
| mitochondrial ATP synthesis coupled electron transport (GO:0042775) | 7 | 0.27 | + | 25.92 | 4.43 × 10−8 | 2.19 × 10−6 |
| transmembrane transport (GO:0055085) | 25 | 6.87 | + | 3.64 | 5.69 × 10−8 | 2.75 × 10−6 |
| organic acid metabolic process (GO:0006082) | 20 | 4.59 | + | 4.36 | 8.37 × 10−8 | 3.95 × 10−6 |
| cellular amide metabolic process (GO:0043603) | 21 | 5.17 | + | 4.06 | 1.20 × 10−7 | 5.56 × 10−6 |
| ion transmembrane transport (GO:0034220) | 21 | 5.31 | + | 3.95 | 1.88 × 10−7 | 8.51 × 10−6 |
| peptide metabolic process (GO:0006518) | 18 | 4.01 | + | 4.48 | 2.53 × 10−7 | 1.12 × 10−5 |
| inorganic ion transmembrane transport (GO:0098660) | 18 | 4.11 | + | 4.38 | 3.54 × 10−7 | 1.54 × 10−5 |
| signaling (GO:0023052) | 8 | 31.66 | − | 0.25 | 4.32 × 10−7 | 1.84 × 10−5 |
| cell communication (GO:0007154) | 8 | 31.72 | − | 0.25 | 4.37 × 10−7 | 1.83 × 10−5 |
| signal transduction (GO:0007165) | 7 | 29.5 | − | 0.24 | 5.52 × 10−7 | 2.27 × 10−5 |
| aerobic electron transport chain (GO:0019646) | 6 | 0.25 | + | 24.44 | 5.65 × 10−7 | 2.28 × 10−5 |
| ion transport (GO:0006811) | 24 | 7.44 | + | 3.23 | 8.46 × 10−7 | 3.35 × 10−5 |
| carbohydrate derivative metabolic process (GO:1901135) | 20 | 5.38 | + | 3.72 | 9.12 × 10−7 | 3.55 × 10−5 |
| regulation of biological process (GO:0050789) | 34 | 66.36 | − | 0.51 | 1.50 × 10−6 | 5.73 × 10−5 |
| regulation of cellular process (GO:0050794) | 32 | 63.53 | − | 0.5 | 2.11 × 10−6 | 7.92 × 10−5 |
| cation transmembrane transport (GO:0098655) | 17 | 4.21 | + | 4.04 | 2.15 × 10−6 | 7.95 × 10−5 |
| cell surface receptor signaling pathway (GO:0007166) | 0 | 13.22 | − | <0.01 | 2.41 × 10−6 | 8.79 × 10−5 |
| mitochondrion organization (GO:0007005) | 11 | 1.73 | + | 6.36 | 2.60 × 10−6 | 9.32 × 10−5 |
| inorganic cation transmembrane transport (GO:0098662) | 16 | 3.81 | + | 4.2 | 2.62 × 10−6 | 9.25 × 10−5 |
| carbohydrate derivative biosynthetic process (GO:1901137) | 15 | 3.38 | + | 4.44 | 2.86 × 10−6 | 9.92 × 10−5 |
| amide biosynthetic process (GO:0043604) | 15 | 3.73 | + | 4.02 | 9.04 × 10−6 | 3.09 × 10−4 |
| cation transport (GO:0006812) | 18 | 5.27 | + | 3.42 | 9.85 × 10−6 | 3.31 × 10−4 |
| biological regulation (GO:0065007) | 41 | 71.93 | − | 0.57 | 1.08 × 10−5 | 3.58 × 10−4 |
| inner mitochondrial membrane organization (GO:0007007) | 4 | 0.15 | + | 27.16 | 3.43 × 10−5 | 1.02 × 10−3 |
| nucleic acid metabolic process (GO:0090304) | 12 | 32.07 | − | 0.37 | 3.79 × 10−5 | 1.11 × 10−3 |
| peptide biosynthetic process (GO:0043043) | 13 | 3.25 | + | 4 | 3.80 × 10−5 | 1.09 × 10−3 |
| cellular modified amino acid metabolic process (GO:0006575) | 7 | 0.86 | + | 8.15 | 4.16 × 10−5 | 1.18 × 10−3 |
| cellular response to stimulus (GO:0051716) | 15 | 35.84 | − | 0.42 | 5.89 × 10−5 | 1.65 × 10−3 |
| mitochondrial membrane organization (GO:0007006) | 4 | 0.2 | + | 20.37 | 8.79 × 10−5 | 2.30 × 10−3 |
| immune system process (GO:0002376) | 1 | 12.26 | − | 0.08 | 8.83 × 10−5 | 2.25 × 10−3 |
| cellular component assembly (GO:0022607) | 25 | 10.64 | + | 2.35 | 1.09 × 10−4 | 2.75 × 10−3 |
| carboxylic acid catabolic process (GO:0046395) | 7 | 1.06 | + | 6.63 | 1.39 × 10−4 | 3.47 × 10−3 |
| mitochondrial electron transport, ubiquinol to cytochrome c (GO:0006122) | 3 | 0.07 | + | 40.74 | 1.40 × 10−4 | 3.46 × 10−3 |
| regulation of nucleobase-containing compound metabolic process (GO:0019219) | 7 | 22.92 | − | 0.31 | 1.44 × 10−4 | 3.52 × 10−3 |
| organic acid catabolic process (GO:0016054) | 7 | 1.08 | + | 6.48 | 1.59 × 10−4 | 3.83 × 10−3 |
| small molecule catabolic process (GO:0044282) | 8 | 1.5 | + | 5.34 | 1.91 × 10−4 | 4.56 × 10−3 |
| monocarboxylic acid catabolic process (GO:0072329) | 5 | 0.49 | + | 10.18 | 2.08 × 10−4 | 4.90 × 10−3 |
| organonitrogen compound metabolic process (GO:1901564) | 55 | 33.61 | + | 1.64 | 2.76 × 10−4 | 6.38 × 10−3 |
| transport (GO:0006810) | 44 | 25.27 | + | 1.74 | 3.28 × 10−4 | 7.50 × 10−3 |
| fatty acid beta-oxidation (GO:0006635) | 4 | 0.29 | + | 13.58 | 3.44 × 10−4 | 7.80 × 10−3 |
| mitochondrial electron transport, cytochrome c to oxygen (GO:0006123) | 3 | 0.11 | + | 27.16 | 3.58 × 10−4 | 8.02 × 10−3 |
| localization (GO:0051179) | 52 | 31.53 | + | 1.65 | 3.88 × 10−4 | 8.62 × 10−3 |
| regulation of nitrogen compound metabolic process (GO:0051171) | 12 | 28.84 | − | 0.42 | 4.42 × 10−4 | 9.72 × 10−3 |
| protein-containing complex subunit organization (GO:0043933) | 15 | 5.41 | + | 2.77 | 4.88 × 10−4 | 1.06 × 10−2 |
| phosphate-containing compound metabolic process (GO:0006796) | 25 | 11.81 | + | 2.12 | 4.97 × 10−4 | 1.07 × 10−2 |
| establishment of localization (GO:0051234) | 44 | 25.65 | + | 1.72 | 5.22 × 10−4 | 1.11 × 10−2 |
| phosphorus metabolic process (GO:0006793) | 25 | 11.87 | + | 2.11 | 5.27 × 10−4 | 1.11 × 10−2 |
| sulfur compound metabolic process (GO:0006790) | 8 | 1.78 | + | 4.49 | 5.74 × 10−4 | 1.20 × 10−2 |
| regulation of metabolic process (GO:0019222) | 15 | 32.5 | − | 0.46 | 6.15 × 10−4 | 1.28 × 10−2 |
| regulation of gene expression (GO:0010468) | 10 | 25.2 | − | 0.4 | 6.55 × 10−4 | 1.35 × 10−2 |
| regulation of macromolecule metabolic process (GO:0060255) | 14 | 30.99 | − | 0.45 | 6.73 × 10−4 | 1.37 × 10−2 |
| cellular process (GO:0009987) | 156 | 127.69 | + | 1.22 | 7.18 × 10−4 | 1.45 × 10−2 |
| regulation of primary metabolic process (GO:0080090) | 13 | 29.21 | − | 0.45 | 7.20 × 10−4 | 1.44 × 10−2 |
| response to stimulus (GO:0050896) | 25 | 44.54 | − | 0.56 | 8.93 × 10−4 | 1.75 × 10−2 |
| immune response (GO:0006955) | 1 | 10.11 | − | 0.1 | 8.94 × 10−4 | 1.74 × 10−2 |
| lipid oxidation (GO:0034440) | 4 | 0.39 | + | 10.18 | 9.18 × 10−4 | 1.77 × 10−2 |
| cellular component biogenesis (GO:0044085) | 25 | 12.15 | + | 2.06 | 9.63 × 10−4 | 1.84 × 10−2 |
| positive regulation of response to stimulus (GO:0048584) | 0 | 7.48 | − | <0.01 | 1.03 × 10−3 | 1.95 × 10−2 |
| dicarboxylic acid transport (GO:0006835) | 3 | 0.17 | + | 17.46 | 1.06 × 10−3 | 1.99 × 10−2 |
| regulation of response to stimulus (GO:0048583) | 3 | 13.76 | − | 0.22 | 1.07 × 10−3 | 1.99 × 10−2 |
| fatty acid metabolic process (GO:0006631) | 7 | 1.52 | + | 4.6 | 1.11 × 10−3 | 2.05 × 10−2 |
| regulation of cellular metabolic process (GO:0031323) | 14 | 30.05 | − | 0.47 | 1.17 × 10−3 | 2.14 × 10−2 |
| glutathione metabolic process (GO:0006749) | 4 | 0.44 | + | 9.05 | 1.37 × 10−3 | 2.45 × 10−2 |
| fatty acid catabolic process (GO:0009062) | 4 | 0.44 | + | 9.05 | 1.37 × 10−3 | 2.43 × 10−2 |
| regulation of cellular biosynthetic process (GO:0031326) | 9 | 22.27 | − | 0.4 | 1.66 × 10−3 | 2.93 × 10−2 |
| regulation of biosynthetic process (GO:0009889) | 9 | 22.34 | − | 0.4 | 1.66 × 10−3 | 2.91 × 10−2 |
| cellular metabolic process (GO:0044237) | 89 | 65.85 | + | 1.35 | 1.67 × 10−3 | 2.90 × 10−2 |
| cellular component organization (GO:0016043) | 50 | 32.21 | + | 1.55 | 1.77 × 10−3 | 3.04 × 10−2 |
| mitochondrial transport (GO:0006839) | 4 | 0.53 | + | 7.58 | 2.50 × 10−3 | 4.18 × 10−2 |
| macromolecule metabolic process (GO:0043170) | 41 | 61.45 | − | 0.67 | 2.64 × 10−3 | 4.37 × 10−2 |
| Primary Antibodies | Dilution | Source | Identifier |
|---|---|---|---|
| GSTM1 | 1:2000 | Santa Cruz | Cat # sc-517262 |
| GSTA1/2 | 1:2000 | Santa Cruz | Cat # sc-398714 |
| PSMA6 | 1:20,000 | Abcam | Cat # 3109377 |
| PSMD12 | 1:2000 | Santa Cruz | Cat # sc-393401 |
| PFK-1 | 1:2000 | Santa Cruz | Cat # sc-166722 |
| PKM2 | 1:1000 | Santa Cruz | Cat # sc-135048 |
| LMP2 | 1:2000 | ThermoFisher Scientific | Prod # PA1-1960 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sule, R.O.; Phinney, B.S.; Salemi, M.R.; Gomes, A.V. Mitochondrial and Proteasome Dysfunction Occurs in the Hearts of Mice Treated with Triazine Herbicide Prometryn. Int. J. Mol. Sci. 2023, 24, 15266. https://doi.org/10.3390/ijms242015266
Sule RO, Phinney BS, Salemi MR, Gomes AV. Mitochondrial and Proteasome Dysfunction Occurs in the Hearts of Mice Treated with Triazine Herbicide Prometryn. International Journal of Molecular Sciences. 2023; 24(20):15266. https://doi.org/10.3390/ijms242015266
Chicago/Turabian StyleSule, Rasheed O., Brett S. Phinney, Michelle R. Salemi, and Aldrin V. Gomes. 2023. "Mitochondrial and Proteasome Dysfunction Occurs in the Hearts of Mice Treated with Triazine Herbicide Prometryn" International Journal of Molecular Sciences 24, no. 20: 15266. https://doi.org/10.3390/ijms242015266
APA StyleSule, R. O., Phinney, B. S., Salemi, M. R., & Gomes, A. V. (2023). Mitochondrial and Proteasome Dysfunction Occurs in the Hearts of Mice Treated with Triazine Herbicide Prometryn. International Journal of Molecular Sciences, 24(20), 15266. https://doi.org/10.3390/ijms242015266